

Abstract
Significant progress has been made in understanding the hematopoietic supportive capacity of both mesenchymal stem cells (MSCs) and osteogenic cells in maintaining hematopoietic stem and progenitor cells (HSPCs) in vitro. However the role of HSPCs in regulating their bone marrow niche environment through influencing the function of neighboring cell populations to complete this reciprocal relationship is not well understood. In this study, we investigated the influence of HSPCs on the osteogenic differentiation of MSCs in vitro, using a highly enriched population of hematopoietic cells with the phenotype c-Kit+Sca-1+Lineage− (KSL) and bone marrow derived mesenchymal stromal cells in direct contact co-culture in medium with or without the addition of the osteogenic supplement dexamethasone. The data suggest that a low dose of HSPCs in co-culture with MSCs in combination with dexamethasone treatment accelerates the osteogenic progression of MSCs, as evidenced in the earlier peak in alkaline phosphatase activity and enhanced calcium deposition compared to cultures of MSCs alone. We observed a longer persistence of functional primitive hematopoietic stem and progenitor cells in the population treated with dexamethasone, and this observation was positively correlated with enhanced osteogenic differentiation of MSCs. Therefore, our findings further support the concept that HSPCs are actively involved in regulating the development and maintenance of the stem cell niche environment in which they reside.
Keywords: Marrow stromal cell, Co-culture, Niche, Osteoblast, Dexamethasone
Introduction
Strategies to effect bone regeneration often seek to leverage select elements of native bone tissue, such as bone marrow derived stem cell populations or processed bone matrix components, to provide engines to drive tissue regeneration or structures to support tissue formation. Experimental approaches commonly employ mesenchymal stem cells (MSCs) harvested from the marrow to promote bone regeneration, yet other cellular components of the bone marrow may play an active role in promoting osteogenesis. Indeed, recent evidence suggests that MSCs together with hematopoietic stem cells form a unique niche in the bone marrow and function in strong cooperation with each other [1]. Accordingly, key elements of the marrow microenvironment present the potential to be harnessed in combination to promote bone osteogenesis. Understanding the bone marrow microenvironment and recreating key components or interactions in vitro may enable approaches utilizing culture expanded stem cells for bone regeneration.
To effectively engineer the marrow microenvironment, it is essential to understand the interactions between resident cell populations, as intimate contact between supporting cells, growth factors, and extracellular matrix cues provides a specific microenvironment that balances stem cell self-renewal versus differentiation and quiescence versus proliferation. The cellular components comprising the stem cell niche contain a heterogeneous population of cells, and in addition to hematopoietic progenitors, include multipotent mesenchymal progenitor cells and osteoblastic cells that may play an integral role within the stem cell niche. While significant progress has been made in understanding the hematopoietic supportive capacity of both MSCs and osteogenic cells [2–5], little is known about the ability of HSPCs to regulate the development and maintenance of their own niche environment by influencing neighboring cells. Since hematopoietic cells and mesenchymal populations reside in such close proximity [1], it is widely believed that there is substantial crosstalk between HSPCs and the other cellular components of the niche [6–8].
HSPCs and their primitive progeny are primarily located proximal to the endosteal surface of trabecular bone [9, 10]. The exact spatial relationship of HSPCs and stromal progenitor cells within the marrow is not well defined, but both cell populations coexist in close proximity within the marrow, suggesting that they play an interactive role in regulating their microenvironment and influencing the function of the other. HSPC development and localization is directly influenced by factors synthesized during the osteogenic program of MSCs. The differentiation of bone marrow stromal cells toward the osteogenic lineage results in a cascade of events, from the early expression of osteopontin to the development of a mineralized extracellular matrix. For example, osteopontin which is a potent regulator of mineralization and one of the most abundant non-collagenous proteins in bone [11], has been shown as a negative regulator of HSPC proliferation [12, 13], presumably facilitating the maintenance of a pool of hematopoietic progenitor cells within the marrow. Also, the mineral phase of bone is integral to the localization and adhesion of HSPCs within the endosteal niche, as HSPCs lacking the calcium-sensing receptor to detect the ionic content of the mineral phase do not function normally upon transplantation [14]. These examples support the concept that MSCs and osteoblastic cells actively regulate the function of HSPCs. The question remains whether HSPCs participate in completing this reciprocal relationship and how they influence the development and maintenance of the bone marrow niche.
Recent reports suggest that HSPCs regulate bone formation through the production of BMP-2 and BMP-6 [15, 16]. However, these studies emphasize the effect of soluble signaling as the cell populations were physically separated in culture. Here we investigate the role of HSPCs in regulating the osteogenic differentiation of MSCs in vitro by examining the progression of osteogenesis through incorporating direct cell-cell interactions. Specifically, we evaluated the osteogenic differentiation of MSCs induced by dexamethasone treatment, in order to direct cells toward the osteoblastic lineage prior to establishing experimental cultures as with our previous osteogenic studies using rat MSCs [17, 18], and hypothesized that both cell-cell interactions and paracrine signaling provided by HSPCs would augment the osteogenic response of MSCs. To investigate our hypothesis, MSCs were co-cultured in direct contact with HSPCs in medium with or without the addition of dexamethasone, in order to explore the progression of osteogenesis and examine how HSPCs participate in the physical development of a mineralized niche environment in vitro.
Materials and Methods
Mesenchymal stem cell isolation and expansion
MSCs were isolated from bone marrow collected and pooled from the femurs and tibias of twenty 8–10 week old C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) according to previously established methods [19] and approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine. Mice were anesthetized with isoflurane, euthanized via cervical dislocation, and then immersed in 70% ethanol. Femurs and tibias were excised and cleared of soft tissue. Bones were crushed using a mortar and pestle with Hanks Balanced Salt Solution (HBSS) (Invitrogen, Carlsbad, CA), supplemented with 2% fetal bovine serum (FBS) (Gemini Bio-Products, West Sacramento, CA), also with the addition of 1% antibiotics containing 10,000 U/mL penicillin and 10,000 µg/mL streptomycin (Invitrogen, Carlsbad, CA). The bone marrow suspension was filtered through a 100 µm cell strainer to remove bone debris, followed by a 40 µm cell strainer to obtain a single-cell suspension. Whole bone marrow was plated in tissue culture flasks with general expansion medium consisting of α-MEM, supplemented with 10% FBS, also with the addition of 1% antibiotics containing penicillin and streptomycin. Adherent cells were cultured for 7 days in general expansion medium with medium changes every 3 days. Following the primary culture period, MSCs were lifted with 0.25% trypsin and plated at low density for subculture expansion [20]. When confluent, MSCs were lifted and frozen in aliquots of medium containing 20% FBS and 10% dimethyl sulfoxide (DMSO). The adherent cells isolated from whole bone marrow and expanded through subculture will be referred to as the MSC population in subsequent co-cultures.
Hematopoietic stem and progenitor cell isolation
Following the same procedure described above to collect bone marrow from C57BL/6 mice, the marrow was alternatively suspended in phosphate buffered saline (PBS), supplemented with 2% FBS, 2 mM EDTA, and 10 mM HEPES, also with the addition of 1% antibiotics containing penicillin and streptomycin, then filtered through a 40 µm cell strainer to obtain a single-cell suspension. Whole bone marrow was enriched immunomagnetically for Sca-1+ cells using the EasySep Mouse SCA1 Positive Selection Kit (Stemcell Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions. In addition to labeling cells with phycoerythrin (PE) conjugated Sca-1 as part of the EasySep Kit, cells were incubated on ice for 20 min with the following antibodies all at 1:100 dilution; fluorescein isothiocyanate (FITC) conjugated c-Kit (BD Pharmingen, Franklin Lakes, NJ), PE-Cy5 conjugated Mac-1, Gr-1, CD4, CD8, B220, and Ter-119 (eBioscience, San Diego, CA, USA) as previously described [21]. Cells were sorted for the cell surface phenotype c-Kit+Sca-1+Lineage− (KSL), comprised of hematopoietic stem and progenitor cells, using a Cytomation MoFlo cell sorter (Dako, Carpinteria, CA). The hematopoietic stem and progenitor cells isolated and purified from whole bone marrow will be referred to as the HSPC population in subsequent co-cultures.
MSC-HSPC direct contact co-culture
Cryopreserved MSCs were thawed at 37 °C and plated in tissue culture flasks with general medium for 24 h, then changed to complete osteogenic medium for an additional 6 days with medium changes every 2 days for osteogenic pre-culture [17, 18]. Complete osteogenic medium for osteogenic pre-culture consisted of α-MEM, supplemented with 10% FBS, 10 nM dexamethasone, 10 mM β-glycerophosphate, and 50 mg/L ascorbic acid, also with the addition of 1% antibiotics containing penicillin and streptomycin. In preparation for cell seeding, individual wells of 12-well plates were filled with 1 mL of complete osteogenic medium either with or without the addition of 10 nM dexamethasone. Following the osteogenic pre-culture period, MSCs were lifted with 0.25% trypsin and seeded into 12-well plates at a density of 4 × 104 cells/well, where each well was 3.8 cm2. After allowing 24 h for MSCs to attach and form a monolayer, HSPCs were isolated as described above and seeded into wells designated for direct contact co-culture at either 400 cells/well or 1000 cells/well. The first medium change was performed after 4 days with subsequent medium changes every 2 days thereafter. Sixteen wells were cultured for each culture group (MSC, CC400, CC1000) and dexamethasone treatment (−DEX and +DEX) for each culture time (8, 16, 24 days), at the end of which wells were rinsed with PBS in preparation for analysis. Two wells were fixed for scanning electron microscopy, two wells were stained to visualize alkaline phosphatase activity, and two wells were stained to visualize calcium deposition. Four wells were prepared to quantitatively assess cellularity and alkaline phosphatase activity, four wells to assess calcium content, and two wells to assess colony-forming capacity in methylcellulose medium.
Scanning electron microscopy
Culture wells for scanning electron microscopy were fixed with 10% neutral-buffered formalin (Fisher Scientific, Pittsburgh, PA) then rinsed with ddH2O and air-dried. Wells were cut out from the culture plates using an X-660 Laser Platform laser cutter (Universal Laser Systems, Morningside, QLD, Australia) and mounted on aluminum stubs with conductive copper tape. Samples were sputter coated with gold for 1 min prior to imaging using a Quanta 400 SEM (FEI, Hillsboro, OR).
Staining and light microscopy
Alkaline phosphatase activity was visualized by staining culture wells using a Blue Alkaline Phosphatase Substrate Kit (Vector Laboratories, Burlingame, CA) according to the manufacturer’s instructions. Reagents provided with the kit were mixed in recommended proportions into 100 mM Tris-HCl buffer with pH adjusted to 8.2. Cells were incubated with 500 µL of the substrate solution and developed in the dark for 30 min at 37 °C. Following the staining procedure where cells expressing alkaline phosphatase were stained blue, wells were fixed with 10% neutral-buffered formalin then rinsed with ddH2O. Plates were placed at an angle to air-dry then stored at 4 °C. Cells were imaged using an Imager.Z2 light microscope with an AxioCam MRc 5 video camera attachment (Zeiss, Thornwood, New York).
Calcium deposition was visualized by staining culture wells with 40 mM Alizarin Red S (Sigma-Aldrich, St. Louis, MO) with pH adjusted to 4.1 using ammonium hydroxide [22]. Cells were fixed with 10% neutral-buffered formalin then rinsed with ddH2O. Wells were incubated with 500 µL of the Alizarin Red S solution for 30 min at room temperature. Wells were washed four times with 2 mL of ddH2O to remove any unincorporated dye. Calcium deposits indicative of matrix mineralization on differentiating cells were stained red. Plates were placed at an angle to air-dry then stored at 4 °C. Cells were imaged using an Imager.Z2 light microscope with an AxioCam MRc 5 video camera attachment.
Biochemical assays
Cells from individual culture wells were lifted with 0.25% trypsin and placed in separate microcentrifuge tubes. Cell pellets were washed with PBS then 500 µL of ddH2O was added. Cells were lysed via three repetitions of a freeze and thaw cycle, where samples were frozen in liquid nitrogen for 10 min, thawed in a 37 °C water bath for 10 min, and sonicated for 10 min.
As a measure of cellularity, double-stranded DNA was quantified using the fluorometric PicoGreen assay (Invitrogen, Carlsbad, CA) with DNA standards [23]. Fluorescence was measured on an FL ×800 plate reader (BioTek, Winooski, VT). DNA content is reported as µg of DNA per well to assess cellularity. Alkaline phosphatase activity was determined by quantifying the enzyme-mediated dephosphorylation of the substrate p-nitrophenol phosphate to p-nitrophenol in a colorimetric assay (Sigma-Aldrich, St. Louis, MO) with p-nitrophenol standards [24]. Absorbance was measured on a PowerWave ×340 plate reader (BioTek, Winooski, VT), with concentrated samples diluted as necessary to ensure absorbance readings within the linear range of the assay. Normalized alkaline phosphatase activity (ALP/DNA) was calculated by dividing alkaline phosphatase activity over DNA content for each sample and is reported as pmol per h per µg DNA as an early marker for osteogenic differentiation.
Calcium content was determined by quantifying free calcium ions in a colorimetric assay by first adding 500 µL of 1 N acetic acid directly into each culture well. After allowing calcium deposits to dissolve, samples were collected and wells were rinsed with an additional 200 µL of 1 N acetic acid. Calcium was quantified using the calcium assay (Genzyme, Cambridge, MA) with calcium chloride standards [25]. Absorbance was measured on a PowerWave ×340 plate reader, with concentrated samples diluted as necessary to ensure absorbance readings within the linear range of the assay. Fold change in calcium content at each time point was calculated by normalizing calcium content to that of MSCs alone within each respective dexamethasone treatment to assess matrix mineralization as a late marker for osteogenic differentiation.
Colony-forming assay
Cells from individual culture wells were lifted with 0.25% trypsin and placed in separate microcentrifuge tubes. The colony-forming capacity of HSPCs after each co-culture period was assessed by plating cells in Methocult GF M3434 methylcellulose-based medium (Stemcell Technologies, Vancouver, BC, Canada), then counting the number of colonies formed after 14 days [26]. Individual samples were first counted using a hemocytometer, aliquots of 104 total cells were plated in 35 mm low attachment culture dishes (Stemcell Technologies, Vancouver, BC, Canada) with 1.1 mL of Methocult GF M3434, and then incubated for 14 days. Following the incubation period, colonies were counted using gridded scoring transparencies on a Stemi 2000 C stereomicroscope (Zeiss, Thornwood, New York). Colony-forming unit counts are reported as colonies per 104 total cells to assess the number of functional hematopoietic stem and progenitor cells remaining within the total cell population after co-culture.
Statistical analysis
Biochemical assay results to assess cellularity, alkaline phosphatase activity, and calcium content are reported as mean ± standard deviation for n = 4. A three-factor ANOVA was first performed to determine significant main effects or interactions between culture group (MSC, CC400, CC1000), dexamethasone treatment (−DEX and +DEX), and culture time (8, 16, 24 days). Multiple pairwise comparisons were then made using the Tukey procedure to determine significant differences. All statistical analyses were performed at a significance level of 5%.
Colony-forming assay results are reported as mean ± standard deviation for n = 4. A two-factor ANOVA was first performed to determine significant main effects or interaction between dexamethasone treatment (−DEX and +DEX) and culture time (8, 16, 24 days). Multiple pairwise comparisons were then made using the Tukey procedure to determine significant differences. All statistical analyses were performed at a significance level of 5%.
Results
The influence of HSPCs on the osteogenic differentiation of MSCs in vitro was evaluated through direct contact co-culture with or without the addition of dexamethasone. Total DNA content per culture well was used to assess overall cellularity and proliferation throughout the culture period (Figure 1). Cellularity remained constant over time at approximately the initial seeding density for cultures of MSCs alone with dexamethasone (MSC+), whereas an increase in cellularity was observed from 8 to 16 days for cultures of MSCs alone without dexamethasone (MSC−). Unlike cultures of MSCs alone, cellularity increased over time for all co-cultures regardless of dexamethasone treatment, with significant differences compared to MSCs alone at 16 and 24 days within both dexamethasone treatments. Co-cultures with dexamethasone (CC400+ and CC1000+) resulted in lower cellularity at 16 and 24 days compared to those without dexamethasone (CC400− and CC1000−).
Figure 1.
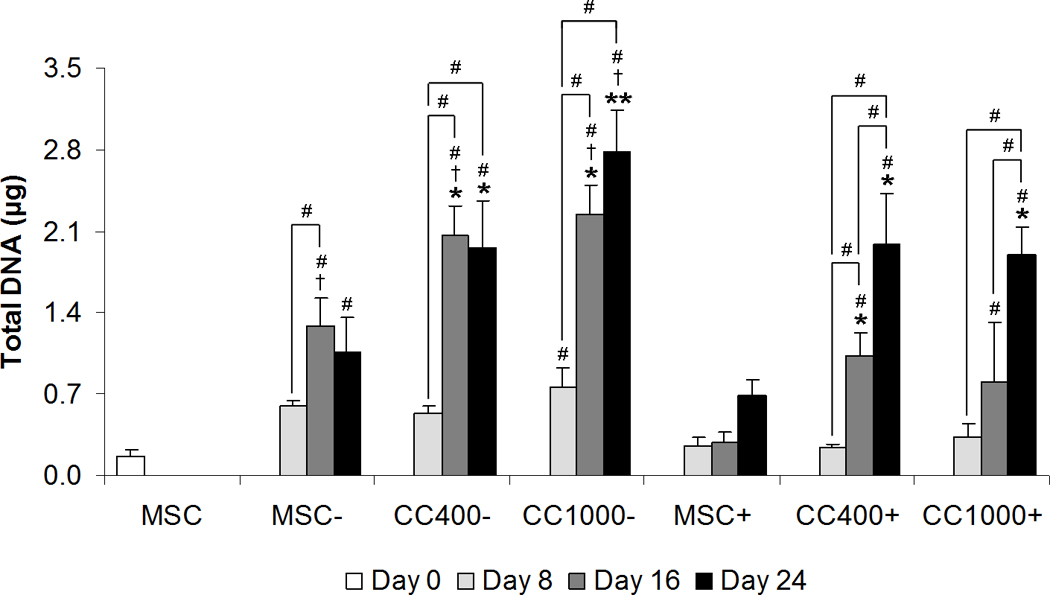
Total DNA content of wells cultured with MSCs alone (MSC) or MSCs and HSPCs in co-culture (CC) at specified seeding densities (400 or 1000 HSPCs seeded onto 40,000 MSCs) either with (+) or without (−) the addition of dexamethasone. Data are presented as mean ± standard deviation for n = 4. Within a specific treatment group, significant difference (p < 0.05) compared to MSCs at seeding and between time points is noted with (#). Within each culture group at a specific time point, significant difference (p < 0.05) between dexamethasone treatment is noted with (†). Within each dexamethasone group at a specific time point, significant difference (p < 0.05) compared to MSCs alone is noted with (*), with significant difference (p < 0.05) compared to all other groups noted with (**).
Scanning electron micrographs were taken to visualize the surface morphology of culture wells with MSCs alone and MSCs and HSPCs in co-culture, as well as changes in the overall topography over time (Figure 2). MSCs spread over the surface of culture wells forming a monolayer while HSPCs maintained a rounded phenotype. In short-term co-culture over 8 days, HSPCs appeared to grow on the surface of MSCs. In long-term co-culture over 24 days, HSPCs seemed to incorporate into the cell layer with MSCs. The cultures acquired a rough texture after 24 days with the development of mineralized extracellular matrix containing mineral nodules.
Figure 2.

Representative scanning electron micrographs of wells cultured with MSCs alone (MSC) or MSCs and HSPCs in co-culture (CC) either with (+) or without (−) the addition of dexamethasone after 8 days (A–D) and 24 days (E–H). Arrows indicate areas of mineralization showing mineral nodules. The scale bar represents 100 µm for all images with insets showing a 3× magnified view of HSPCs (B, D, F and H) and MSCs or mineral nodules (A, C, E and G) in more detail.
Alkaline phosphatase expression and activity was used to assess early osteogenic differentiation of cells in culture. Light micrographs were taken of culture wells stained blue to visualize qualitatively the ALP expression of MSCs alone and MSCs and HSPCs in co-culture after 8 days (Figure 3). All cultures showed positive expression of ALP with fairly even distribution within the culture wells overall. Microscopy images revealed that most of the spread MSCs express ALP with varying intensities of blue staining, while the rounded HSPCs did not appear to express ALP as evident in the lack of blue staining macroscopically. The alkaline phosphatase activity was also measured for each group and normalized to DNA content (ALP/DNA) to quantitatively reflect early osteogenic differentiation (Figure 4). ALP/DNA remained constant over time at approximately the initial level at seeding for cultures of MSCs alone without dexamethasone, whereas ALP/DNA increased significantly in the first 8 days and peaked at 16 days for cultures of MSCs alone with dexamethasone. Although co-cultures without dexamethasone showed the same trend and ALP/DNA levels as MSCs alone, those with dexamethasone resulted in a peak in ALP/DNA at 8 days.
Figure 3.

Alkaline phosphatase staining of wells after 8 days of culture with MSCs alone (MSC) or MSCs and HSPCs in co-culture (CC) at specified seeding densities (400 or 1000 HSPCs seeded onto 40,000 MSCs) either with (+) (D–F) or without (−) (A–C) the addition of dexamethasone. The scale bar represents 200 µm for all microscopy images, save insets. Bottom-left insets (A–F) show the overall alkaline phosphatase staining of the wells, and top-right insets (B, C, E and F) show a 3× magnified view (with respect to the respective main images) of co-cultures, with arrows indicating apparent HSPCs.
Figure 4.
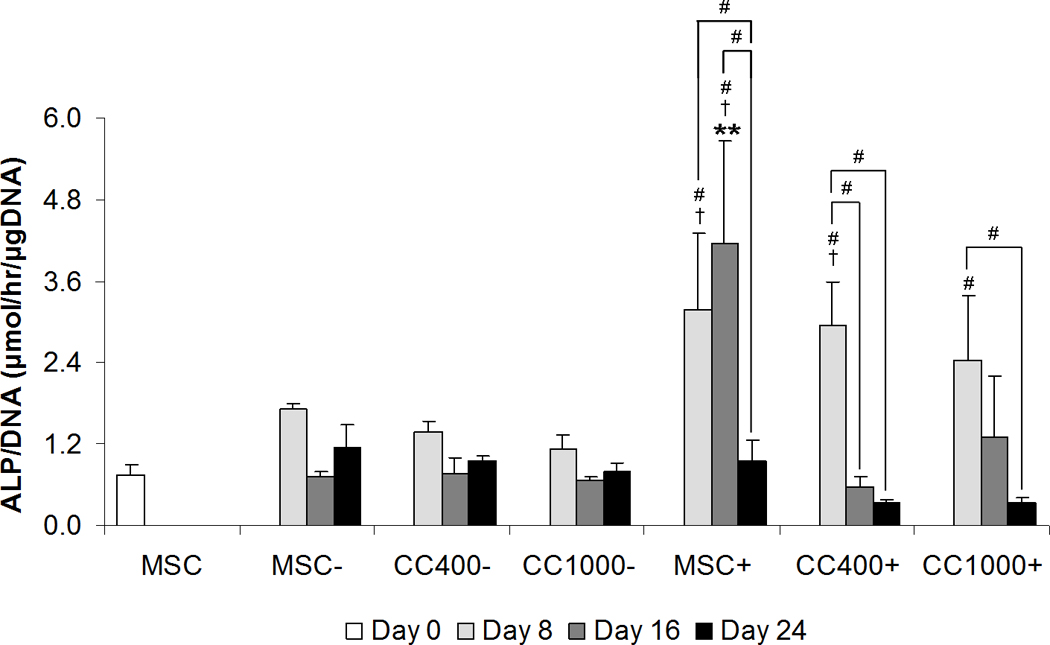
Alkaline phosphatase activity MSCs alone (MSC) or MSCs and HSPCs in co-culture (CC) at specified seeding densities (400 or 1000 HSPCs seeded onto 40,000 MSCs) either with (+) or without (−) the addition of dexamethasone. Plots show alkaline phosphatase activity normalized to DNA content. Data are presented as mean ± standard deviation for n = 4. Within a specific treatment group, significant difference (p < 0.05) compared to MSCs at seeding and between time points is noted with (#). Within each culture group at a specific time point, significant difference (p < 0.05) between dexamethasone treatment is noted with (†). Within each dexamethasone group at a specific time point, significant difference (p < 0.05) compared to MSCs alone is noted with (*), with significant difference (p < 0.05) compared to all other groups noted with (**).
Calcium content as compared to MSCs within each respective dexamethasone treatment was used to assess late osteogenic differentiation of cells in culture qualitatively by microscopy (Figure 5) and quantitatively via a calcium assay (Figure 6). Light micrographs were taken of culture wells stained red to visualize qualitatively the calcium deposition of MSCs alone and MSCs and HSPCs in co-culture after 24 days (Figure 5). Although all cultures showed calcium deposition with varying intensities of red staining, the distribution of calcium deposits within the culture wells varied overall. Blank regions lacking calcium deposits were most apparent for co-cultures without dexamethasone, whereas calcium deposition appeared more evenly distributed for co-cultures with dexamethasone. Microscopy images revealed that the blank regions indeed had functional cells growing which did not stain red for calcium deposits. Further, a calcium assay was applied to determine quantitatively the fold change in calcium content as compared to MSCs within each respective dexamethasone treatment (Figure 6). Only the low dose co-culture group with dexamethasone (CC400+) showed a significant difference in calcium content compared to MSCs alone with dexamethasone (MSC+). Fold change in calcium deposition for CC400+ was 5.8 ± 1.2 fold higher than MSC+ at 8 days and 5.5 ± 2.8 fold higher than MSC+ at 16 days. Interestingly, there was no significant difference at 24 days.
Figure 5.

Alizarin Red staining of wells after 24 days of culture with MSCs alone (MSC) or MSCs and HSPCs in co-culture (CC) at specified seeding densities (400 or 1000 HSPCs seeded onto 40,000 MSCs) either with (+) (D–F) or without (−) (A–C) the addition of dexamethasone. The scale bar represents 200 µm for all microscopy images, and arrows indicate apparent HSPCs. Insets show the overall Alizarin Red staining of the wells.
Figure 6.
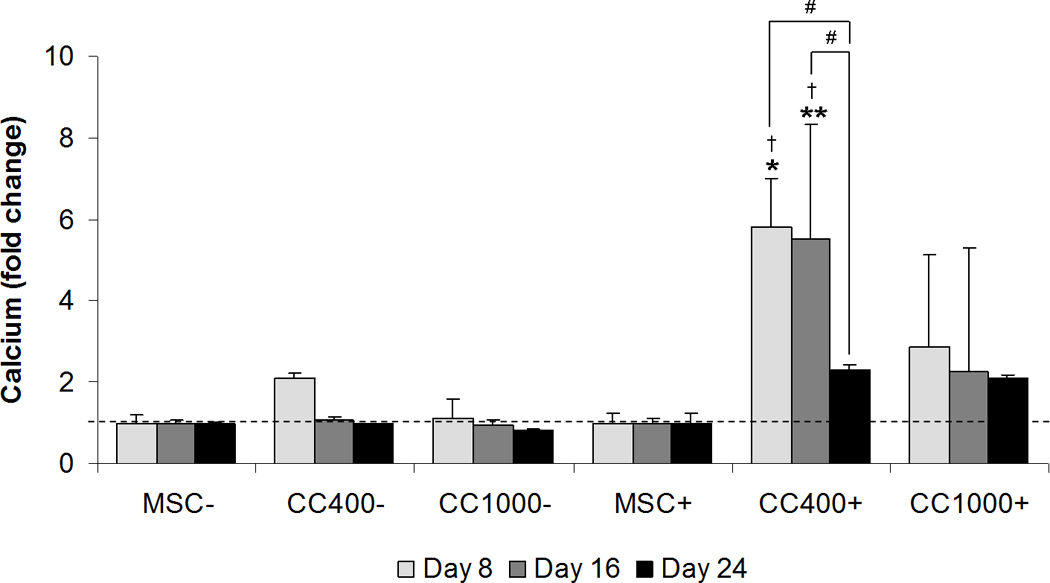
Calcium content of wells cultured with MSCs alone (MSC) or MSCs and HSPCs in co-culture (CC) at specified seeding densities (400 or 1000 HSPCs seeded onto 40,000 MSCs) either with (+) or without (−) the addition of dexamethasone. Plots show fold change in calcium content as compared to cultures with MSCs for each dexamethasone treatment at each time point. Data are presented as mean ± standard deviation for n = 4. Within a specific treatment group, significant difference (p < 0.05) between time points is noted with (#). Within each culture group at a specific time point, significant difference (p < 0.05) between dexamethasone treatment is noted with (†). Within each dexamethasone group at a specific time point, significant difference (p < 0.05) compared to MSCs alone is noted with (*), with significant difference (p < 0.05) compared to all other groups noted with (**).
Colony-forming cell growth in methylcellulose medium was used to assess the colony-forming capacity of HSPCs after co-culture (Figure 7). Although the number of hematopoietic stem and progenitor cells significantly decreased in the first week of co-culture as compared to the initial HSPC population following FACS analysis prior to seeding (99.4 ± 11.6 colony-forming units per 104 total cells), more colonies remained with dexamethasone treatment in short-term co-culture. After 8 days of co-culture, colony-forming unit counts per 104 total cells for CC400+ was 35.3 ± 12.4 and for C400− was 3.3 ± 1.3. While dexamethasone treatment resulted in this significant difference in colony counts at 8 days, the number of colonies decreased over extended culture periods.
Figure 7.
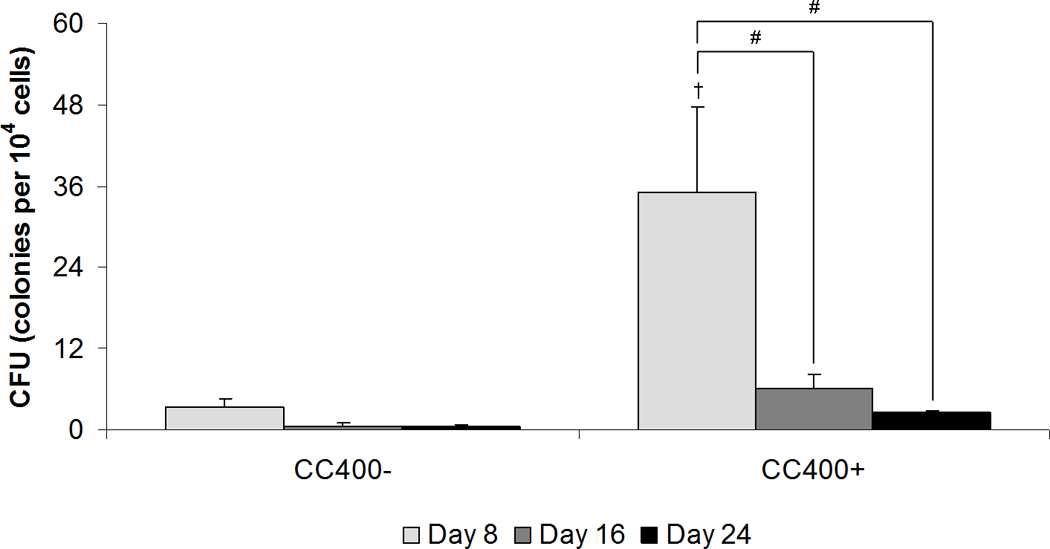
Colony-forming unit counts of colonies derived in methylcellulose medium from the total cell population after co-culture (CC) at the specified seeding density (400 HSPCs seeded onto 40,000 MSCs) either with (+) or without (−) the addition of dexamethasone. Data are presented as mean ± standard deviation for n = 4. Within a specific treatment group, significant difference (p < 0.05) between time points is noted with (#). Significant difference (p < 0.05) between dexamethasone treatment is noted with (†).
Discussion
The objective of this study was to investigate the influence of HSPCs on the osteogenic differentiation of MSCs through direct contact co-culture, to better understand the interactions of cellular components comprising the stem cell niche under in vitro culture conditions. This study was designed to evaluate the osteogenic differentiation of MSCs in vitro induced by dexamethasone treatment, and to examine how the inclusion of HSPCs in co-culture would augment this differentiation response by providing a niche microenvironment consisting of both direct cell-cell interactions and paracrine signaling.
Recent studies have reported that HSPCs actively participate in bone formation by producing BMP-2 and BMP-6 [15], especially when activated by elevated erythropoietin levels induced by acute bleeding [16]. Frequently in studies investigating the crosstalk between HSPCs and MSCs, the effect of soluble signaling is emphasized as HSPCs are cultured separately from MSCs in the top chambers of transwell plates then assessed for osteoblastic colony formation at the end of culture [15, 27]. Although inclusion in the present study of non-contact co-cultures of MSCs osteogenically induced by dexamethasone treatment and HSPCs may have provided a base-line regarding the effects of soluble signaling between HSPCs and MSCs in vitro, it is known that HSPCs and MSCs reside in close physical association to form a unique niche in the bone marrow [1]. Accordingly, the focus of the present study was to mimic the direct HSPC-MSC contact in the stem cell niche under in vitro conditions, and to examine osteogenic differentiation of MSCs in contact co-culture with HSPCs following induction via dexamethasone treatment. Indeed, here we investigated the progression of osteogenesis from induction to mineralized matrix production by incorporating direct cell-cell interactions in addition to paracrine signaling, which allowed us to examine the extent to which HSPCs participate in the physical development of a mineralized niche environment in vitro.
Our results showed that HSPCs influenced the osteogenic differentiation of MSCs under in vitro culture conditions with dexamethasone. We observed that low doses of HSPCs co-cultured in direct contact with MSCs and exposed to dexamethasone treatment, reduced overall cellular proliferation, stimulated early alkaline phosphatase activity, and enhanced calcium deposition, thus supporting the progression of osteogenic differentiation in vitro. Additionally, qualitative morphological observations from light microscopy and scanning electron microscopy suggest the presence of physical interactions between HSPCs and differentiating MSCs throughout the progression of osteogenesis in vitro, which may be examined in greater detail in future studies.
Cellularity and proliferation throughout the culture period was evaluated by quantifying total DNA content per culture well. Cultures of MSCs alone, particularly with dexamethasone treatment, maintained similar cellularity over 24 days of culture. Since cells were induced toward osteogenic differentiation in vitro with dexamethasone, we expect to see minimal proliferative activity as cells transition to an osteoblastic phenotype [28]. Although in all co-culture groups, the overall cell population rapidly proliferated, dexamethasone treatment significantly reduced cellularity after 16 and 24 days of culture. Qualitative morphological observations from light microscopy and scanning electron microscopy suggest that the HSPC population proliferated quickly in co-culture, while the MSC population maintained a confluent cell layer, which may have supported the growth and retention of HSPCs in vitro. Over an extended culture period, however, HSPCs appeared to incorporate into the cell layer with MSCs, where they could possibly outcompete MSCs for space and nutrients. While in this study, the contribution of each cell population to the overall change in cellularity over time was not specifically assessed, we consider that HSPCs proliferate much more rapidly than MSCs in co-culture, as MSCs and osteogenic cells are often used as feeder layers to expand hematopoietic cell numbers ex vivo due to their supportive role in the stem cell niche [2–5]. However in those applications, dexamethasone is not included as a culture supplement, and thus the effects of co-culture in combination with dexamethasone on HSPCs in vitro are not known. Studies investigating glucocorticoid treatment through intraperitoneal injections have shown hematoprotective effects of dexamethasone, promoting the quiescence of stem cells as seen in the maintenance of high colony-forming cell numbers even after cytotoxic chemotherapy [29, 30]. In exploring how HSPCs affect the progression of MSCs initiated toward osteogenic differentiation via dexamethasone exposure, we observed that dexamethasone may play a role in maintaining hematopoietic stem and progenitor cells in vitro. This is evidenced in the higher number of functional hematopoietic stem and progenitor cells within the total cell population that remain following short-term co-culture with dexamethasone, albeit those colony-forming cells decrease significantly in number over extended culture periods. Although the expansion of HSPCs ex vivo was not the focus of this current study, we found that dexamethasone as a culture supplement may be worth exploring in order to optimize co-culture conditions to permit the sustained expansion of HSPCs ex vivo.
Alkaline phosphatase activity was used as an early marker for osteogenic differentiation as enzyme levels peak during the onset of osteogenic differentiation then decrease as cells progress toward an osteoblastic phenotype [28]. Dexamethasone treatment induced a significant increase in ALP/DNA in the first 8 days for all culture groups. While MSCs alone showed a clear peak in ALP/DNA at 16 days, the data suggest that the peak in ALP/DNA for co-culture groups may have occurred sooner within the first 8 days of culture, since ALP/DNA levels were already declining after 8 days. Thus, these trends in ALP/DNA imply that dexamethasone indeed promotes osteogenic differentiation with a characteristic peak in the profile of alkaline phosphatase expression we typically observe in our osteogenic cultures [31–33], and that co-culture with HSPCs accelerates the osteogenic differentiation of MSCs in their transition to an osteoblastic phenotype.
Since cell populations were not separated following co-culture, total alkaline phosphatase activity of the entire cell population as a whole was evaluated qualitatively via microscopy. Through macroscopic inspection following the staining procedure to visualize ALP expression at 8 days, HSPCs did not appear to stain for ALP activity, as most of the staining was much more apparent and intense for the MSCs. While we do not know how the HSPC population contributes to quantitative ALP measurements, ALP expression has been documented for rare hematopoietic cells, particularly plasma cells as terminally differentiated B-cells [34]. This may account for the higher levels of total ALP detected qualitatively for co-cultures without dexamethasone, as the colony-forming assay revealed that cells rapidly differentiated into mature hematopoietic lineages within the first week of culture without dexamethasone treatment, as evident in the lower numbers of functional hematopoietic stem and progenitor cells remaining within the total cell population after co-culture.
Calcium deposition was used as a late marker for osteogenic differentiation, as cells with an osteoblastic phenotype deposit increasing amounts of extracellular matrix, which mineralizes over time [28]. Since mouse MSCs were expanded through a brief osteogenic pre-culture period with dexamethasone in order to direct cells toward the osteoblastic lineage prior to establishing experimental cultures as with our previous osteogenic studies using rat MSCs [17, 18], this transient exposure may have initiated osteogenic progression with sustained effects even after the removal of dexamethasone in subsequent experimental cultures, similar to what has been documented for human MSCs [35]. Thus, the sustained effects of dexamethasone in initiating a pre-osteoblastic phenotype likely contributed to the calcium deposition qualitatively observed in our experimental cultures without dexamethasone. Interestingly, there is a qualitative difference in the distribution of cell populations and calcium deposits within co-cultures not treated with dexamethasone. Mineralized extracellular matrix appears to be localized to the MSC population, with large regions of the cultures wells occupied by the HSPC population that did not stain for calcium, in contrast to the more even staining seen for co-cultures treated with dexamethasone.
When the quantitative calcium data for co-culture groups are normalized to that of MSCs alone within each respective dexamethasone treatment and considered as fold change in calcium content, it is apparent that, in combination with dexamethasone treatment, a low dose of HSPCs in fact enhance calcium deposition at early time points. Over an extended culture period, the signaling effects of HSPCs, which seem to accelerate osteogenic progression, dissipate as MSCs in all culture groups treated with dexamethasone converge to an osteoblastic phenotype. From the colony-forming assay, we see that it is the primitive hematopoietic stem and progenitor cells remaining in the total cell population following co-culture that exert this stimulatory effect on the osteogenic differentiation of MSCs. On the contrary, if HSPCs differentiate into mature hematopoietic lineages in co-culture, then those hematopoietic cells lose their ability to augment the osteogenic progression of MSCs. Our findings in this study support the concept that not only do osteoblastic cells play a supportive role in maintaining hematopoietic cells, but that there is a reciprocal relationship whereby hematopoietic cells regulate osteoblastic cell function as active participants in the maintenance and development of the stem cell niche [15, 27]. Interestingly, there appears to be an optimal cell density to achieve enhanced mineralization under co-culture conditions, as may be the case in the physiological environment where the balance between cell populations affects overall cell function and tissue morphology. Furthermore, in modeling the osteogenic development of MSCs through dexamethasone exposure, we observed that not only does dexamethasone assist in directing cells towards recreating a mineralized microenvironment in vitro via brief exposure in pre-culture, but dexamethasone may also promote the maintenance of functional hematopoietic stem and progenitor cells in short-term co-culture, perhaps through direct action on HSPCs or indirectly through promoting the osteogenic development of MSCs that in turn act upon HSPCs. However, further investigation is warranted to determine whether MSCs or osteogenically differentiated MSCs constitute a sufficient environment to support the hematopoietic function of HSPCs [2, 3, 36]. Nevertheless, our investigation into the reciprocal relationship between the two major cell populations comprising the bone marrow niche under in vitro culture conditions may facilitate the development of tissue engineering strategies further optimizing co-culture parameters to achieve expansion of hematopoietic cells ex vivo. Understanding the development of the bone marrow microenvironment and recreating key components or interactions in vitro brings us a step closer towards the realization of medical therapies utilizing culture expanded stem cells.
Conclusion
In this work, we demonstrated that primitive hematopoietic stem and progenitor cells enhance the osteogenic differentiation of mesenchymal stem cells through both cell-cell interactions and paracrine signaling as facilitated through dexamethasone treatment in vitro. We were able to examine how HSPCs participate in the physical development of a mineralized niche environment through direct contact co-culture with MSCs. This study further supports the concept that HSPCs actively regulate the development and maintenance of the stem cell niche environment in which they reside.
Acknowledgements
This work has been supported by the National Institutes of Health (R01 AR057083 and R01 EB005173). KE Hammerick was supported by an Alliance for NanoHealth Postdoctoral Fellowship. GA Challen was supported by the National Institutes of Health (K99 DK084259) and is an American Society of Hematology Scholar. We thank Christopher Threeton of the Texas Children’s Hospital Flow Cytometry Core Laboratory for cell sorting and analysis.
References
- 1.Mendez-Ferrer S, Michurina TV, Ferraro F, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 3.Jing D, Fonseca AV, Alakel N, et al. Hematopoietic stem cells in co-culture with mesenchymal stromal cells--modeling the niche compartments in vitro. Haematologica. 2010;95:542–550. doi: 10.3324/haematol.2009.010736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mishima S, Nagai A, Abdullah S, et al. Effective ex vivo expansion of hematopoietic stem cells using osteoblast-differentiated mesenchymal stem cells is cxcl12 dependent. Eur J Haematol. 2010;84:538–546. doi: 10.1111/j.1600-0609.2010.01419.x. [DOI] [PubMed] [Google Scholar]
- 5.Taichman RS, Reilly MJ, Emerson SG. Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood. 1996;87:518–524. [PubMed] [Google Scholar]
- 6.Li Z, Li L. Understanding hematopoietic stem-cell microenvironments. Trends Biochem Sci. 2006;31:589–595. doi: 10.1016/j.tibs.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6:93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- 8.Yin T, Li L. The stem cell niches in bone. J Clin Invest. 2006;116:1195–1201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balduino A, Hurtado SP, Frazao P, et al. Bone marrow subendosteal microenvironment harbours functionally distinct haemosupportive stromal cell populations. Cell Tissue Res. 2005;319:255–266. doi: 10.1007/s00441-004-1006-3. [DOI] [PubMed] [Google Scholar]
- 10.Taichman RS. Blood and bone: Two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood. 2005;105:2631–2639. doi: 10.1182/blood-2004-06-2480. [DOI] [PubMed] [Google Scholar]
- 11.Denhardt DT, Guo X. Osteopontin: A protein with diverse functions. Faseb J. 1993;7:1475–1482. [PubMed] [Google Scholar]
- 12.Nilsson SK, Johnston HM, Whitty GA, et al. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood. 2005;106:1232–1239. doi: 10.1182/blood-2004-11-4422. [DOI] [PubMed] [Google Scholar]
- 13.Stier S, Ko Y, Forkert R, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med. 2005;201:1781–1791. doi: 10.1084/jem.20041992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams GB, Chabner KT, Alley IR, et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature. 2006;439:599–603. doi: 10.1038/nature04247. [DOI] [PubMed] [Google Scholar]
- 15.Jung Y, Song J, Shiozawa Y, et al. Hematopoietic stem cells regulate mesenchymal stromal cell induction into osteoblasts thereby participating in the formation of the stem cell niche. Stem Cells. 2008;26:2042–2051. doi: 10.1634/stemcells.2008-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiozawa Y, Jung Y, Ziegler AM, et al. Erythropoietin couples hematopoiesis with bone formation. PLoS One. 2010;5:e10853. doi: 10.1371/journal.pone.0010853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao J, Guo X, Nelson D, et al. Modulation of osteogenic properties of biodegradable polymer/extracellular matrix scaffolds generated with a flow perfusion bioreactor. Acta Biomater. 2010;6:2386–2393. doi: 10.1016/j.actbio.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peter SJ, Liang CR, Kim DJ, et al. Osteoblastic phenotype of rat marrow stromal cells cultured in the presence of dexamethasone, beta-glycerolphosphate, and l-ascorbic acid. J Cell Biochem. 1998;71:55–62. doi: 10.1002/(sici)1097-4644(19981001)71:1<55::aid-jcb6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 19.Haylock DN, Williams B, Johnston HM, et al. Hemopoietic stem cells with higher hemopoietic potential reside at the bone marrow endosteum. Stem Cells. 2007;25:1062–1069. doi: 10.1634/stemcells.2006-0528. [DOI] [PubMed] [Google Scholar]
- 20.Kretlow JD, Jin YQ, Liu W, et al. Donor age and cell passage affects differentiation potential of murine bone marrow-derived stem cells. BMC Cell Biol. 2008;9:60. doi: 10.1186/1471-2121-9-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Challen GA, Goodell MA. Runx1 isoforms show differential expression patterns during hematopoietic development but have similar functional effects in adult hematopoietic stem cells. Exp Hematol. 2010;38:403–416. doi: 10.1016/j.exphem.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gregory CA, Gunn WG, Peister A, Prockop DJ. An alizarin red-based assay of mineralization by adherent cells in culture: Comparison with cetylpyridinium chloride extraction. Anal Biochem. 2004;329:77–84. doi: 10.1016/j.ab.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 23.Singer VL, Jones LJ, Yue ST, Haugland RP. Characterization of picogreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitation. Anal Biochem. 1997;249:228–238. doi: 10.1006/abio.1997.2177. [DOI] [PubMed] [Google Scholar]
- 24.Bretaudiere JP, Spillan T. Alkaline phosphatases. In: Bergmeyer HU, Bergmeyer J, Grassl M, editors. Methods of enzymatic analysis. Verlag Chemie: Deerfield Beach; 1984. pp. 75–92. [Google Scholar]
- 25.Holtorf HL, Jansen JA, Mikos AG. Flow perfusion culture induces the osteoblastic differentiation of marrow stroma cell-scaffold constructs in the absence of dexamethasone. J Biomed Mater Res A. 2005;72:326–334. doi: 10.1002/jbm.a.30251. [DOI] [PubMed] [Google Scholar]
- 26.Ogawa M, Livingston AG. Hematopoietic colony-forming cells. In: Klug CA, Jordan CT, editors. Hematopoietic stem cell protocols. Totowa: Humana Press; 2002. pp. 113–122. [Google Scholar]
- 27.Taichman RS, Reilly MJ, Verma RS, Emerson SG. Augmented production of interleukin-6 by normal human osteoblasts in response to cd34+ hematopoietic bone marrow cells in vitro. Blood. 1997;89:1165–1172. [PubMed] [Google Scholar]
- 28.Owen TA, Aronow M, Shalhoub V, et al. Progressive development of the rat osteoblast phenotype in vitro: Reciprocal relationships in expression of genes associated with osteoblast proliferation and differentiation during formation of the bone extracellular matrix. J Cell Physiol. 1990;143:420–430. doi: 10.1002/jcp.1041430304. [DOI] [PubMed] [Google Scholar]
- 29.Kim H, Choi JY, Lee JM, et al. Dexamethasone increases angiopoietin-1 and quiescent hematopoietic stem cells: A novel mechanism of dexamethasone-induced hematoprotection. FEBS Lett. 2008;582:3509–3514. doi: 10.1016/j.febslet.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 30.Kriegler AB, Bernardo D, Verschoor SM. Protection of murine bone marrow by dexamethasone during cytotoxic chemotherapy. Blood. 1994;83:65–71. [PubMed] [Google Scholar]
- 31.Datta N, Holtorf HL, Sikavitsas VI, et al. Effect of bone extracellular matrix synthesized in vitro on the osteoblastic differentiation of marrow stromal cells. Biomaterials. 2005;26:971–977. doi: 10.1016/j.biomaterials.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Holtorf HL, Datta N, Jansen JA, Mikos AG. Scaffold mesh size affects the osteoblastic differentiation of seeded marrow stromal cells cultured in a flow perfusion bioreactor. J Biomed Mater Res A. 2005;74:171–180. doi: 10.1002/jbm.a.30330. [DOI] [PubMed] [Google Scholar]
- 33.Sikavitsas VI, Bancroft GN, Holtorf HL, et al. Mineralized matrix deposition by marrow stromal osteoblasts in 3d perfusion culture increases with increasing fluid shear forces. Proc Natl Acad Sci U S A. 2003;100:14683–14688. doi: 10.1073/pnas.2434367100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borgen E, Beiske K, Trachsel S, et al. Immunocytochemical detection of isolated epithelial cells in bone marrow: Non-specific staining and contribution by plasma cells directly reactive to alkaline phosphatase. J Pathol. 1998;185:427–434. doi: 10.1002/(SICI)1096-9896(199808)185:4<427::AID-PATH127>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 35.Jaiswal N, Haynesworth SE, Caplan AI, Bruder SP. Osteogenic differentiation of purified, culture-expanded human mesenchymal stem cells in vitro. J Cell Biochem. 1997;64:295–312. [PubMed] [Google Scholar]
- 36.Askmyr M, Sims NA, Martin TJ, Purton LE. What is the true nature of the osteoblastic hematopoietic stem cell niche? Trends Endocrinol Metab. 2009;20:303–309. doi: 10.1016/j.tem.2009.03.004. [DOI] [PubMed] [Google Scholar]