

Abstract
Mitochondrial solute carriers are a family of membrane proteins that catalyze the transport of small molecules across the mitochondrial inner membrane1. We describe here a solution nuclear magnetic resonance (NMR) method for structural characterization of mitochondrial uncoupling protein 2 (UCP2), a member of the carrier family that translocates protons across the mitochonrial inner membrane2. The method, which overcomes some of the challenges associated with membrane-protein structure determination3, combines orientation restraints derived from NMR residual dipolar couplings (RDCs) and semi-quantitative distance restraints from paramagnetic relaxation enhancement (PRE) measurements. The local and secondary structures of the protein were determined by piecing together molecular fragments from the Protein Data Bank (PDB) that best fit experimental RDCs from samples weakly aligned in a DNA nanotube liquid crystal. The RDCs also determine the relative orientation of the secondary structural segments, and the PRE restraints provide their spatial arrangement in the tertiary fold. We determined the structure of UCP2 in the presence of GDP. We also mapped the interaction between UCP2 and GDP by measuring paramagnetic broadening of the protein NMR resonances from nitroxide-labeled GDP. UCP2 closely resembles the bovine ADP/ATP carrier (the only carrier protein of known structure4), but the relative orientations of the helical segments are different, resulting in a wider opening on the matrix side of the inner membrane. Moreover, the nitroxide-labeled GDP binds inside the channel and appears to be closer to transmembrane helix 1–4. We believe this biophysical approach, based on solution NMR spectroscopy, can be applied broadly to other membrane proteins and in particular to other mitochondrial carriers, not only for structure determination but also for characterizing various conformational states of these proteins linked to substrate transport.
The elucidation almost 50 years ago of the mechanism of oxidative phosphorylation provided the first example of the coupling of membrane transport to energy production5. The mitochondrial respiratory complex transfers electrons from nutrients to oxygen, pumping protons from the mitochondrial matrix to the intermembrane space as it builds up an electrochemical potential (ΔψH+) of ~200mV (Supplementary Fig. 1). While most of this potential is converted to work that drives ATP synthesis, a fraction is dissipated as heat, in a process involving leakage of protons back to the matrix. This leakage, or “uncoupling” of the proton electrochemical potential, is mediated primarily by a family of proteins in the mitochondrial inner membrane, collectively termed “uncoupling proteins” (UCPs)2. The proton translocation activity of UCPs requires fatty acids as cofactors; it is inhibited by GDP. UCP1 was first identified in the mitochondria of brown adipose tissue (BAT) as a protein responsible for BAT-mediated thermoregulation6. Unlike UCP1, the primary function of UCP2 may be related to lowering production of reactive oxygen species2. Expression of UCP2 in pancreatic β-cells is strongly correlated with decreased insulin secretion, and UCP2 inactivation restores the glucose-sensing pathway7. UCP2 also plays a role in glucagon secretion from pancreatic α-cells8, and in regulating of hunger through ghrelin in NPY/AGRP neurons9. More recently, UCP2 has been linked to chemotherapy resistance and metabolic reprogramming in cancer10,11.
NMR spectroscopy has yielded the structures of a number of membrane proteins, including channels12–14, a redox protein15, a kinase16, and the sensory rhodopsin17. For many of these studies, a major issue has been purifying the protein and reconstituting it in a model membrane medium that supports protein solubility and stability. In the case of UCP2, reconstitution in dodecylphosphocholine (DPC) micelles with a small amount of lipids (cardiolipin and dimyristoyl-phosphatidylcholine: DMPC) yielded workable NMR spectra (Fig. 1a&b). We prepared homogeneous UCP2 by passing detergent-solubilized protein through a GDP-analog column, which selects for native protein. Samples used for NMR measurements were at pH 6.5 and contained 5 mM GDP, 150 mM DPC, 1 mM cardiolipin and 2 mM DMPC. Under these conditions, a fluorescence resonance energy transfer (FRET) experiment showed that GDP binds to UCP2 with an apparent KD of ~5 µM (Fig. 1c). When the NMR sample was incorporated into liposomes, it showed fatty-acid activated and GDP-inhibited proton translocation activity (Supplementary Fig. 2). Negative-stain electron microscopy showed monodisperse, channel-like particles (Supplementary Fig. 3).
Figure 1. NMR spectra, GDP binding, and outline of RDC-based molecular fragment assignment.
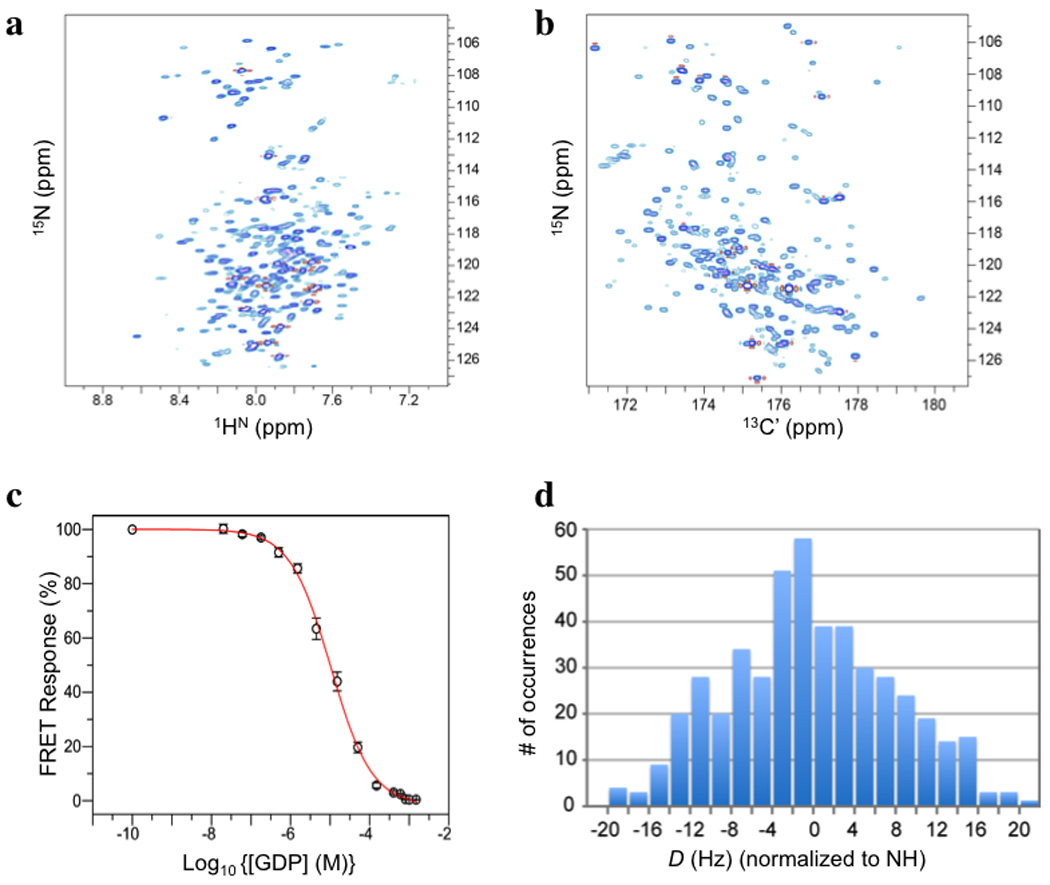
a, The 1HN-15N TROSY HSQC spectrum of 15N-,13C-,2H-labelled UCP2 reconstituted in DPC micelles (with 5 mM GDP) recorded at pH 6.5, 33 °C, and 1H frequency of 600 MHz. b, The (15N,13C’) projection of the 3D TROSY HNCO spectrum of the sample in a, recorded under the same conditions. Comparison of a and b, illustrates that most resonances can be resolved in the 3D HNCO spectrum. c, Specific binding of GDP to UCP2 under conditions of the NMR sample. The FRET Response (= %[FUMANT-GDP / GDP - FU0GDP]; see METHODS) follows displacement of fluorescent MANT-GDP by GDP as the concentration of the latter increases. d, Histogram of 470 unambiguously assigned 1DNH, 1DC’Cα or 1DNC’ (all normalized to 1DNH). The magnitude (Da) and rhombicity (Rh) of the alignment tensors are 10 Hz and 0.61, respectively.
Local structure determination by NMR typically involves assigning nuclear Overhauser effect (NOEs) between backbone amide protons and aliphatic protons. In the UCP2 case, it was difficult to collect a sufficient number of unambiguous NOEs because of severe overlap of backbone (1H,15N) resonances. The peaks in the 3D HNCO spectrum are mostly separated, however. The HNCO experiment, which correlates the chemical shifts of backbone 1HN, 15N, and 13C’ nuclides, has been widely used for measuring one-bond residual dipolar couplings (RDCs). For structure determination, we therefore relied on orientation restraints derived from RDCs. In a macromolecule marginally oriented in a magnetic field, RDC between a pair of spin-1/2 nuclides encodes orientations of the internuclear vector connecting the two nuclides. The most effective medium for weakly orienting protein in solution is a liquid crystal formed by large, elongated particles18; for membrane proteins, these particles must also resist a high concentration of detergent. We used DNA nanotubes, constructed by "DNA origami", which can form a stable nematic phase unaffected by detergent19 (Supplementary Fig. 4). The nanotube alignment medium allowed us to measure RDCs for internuclear vectors 1HN-15N (1DNH), 13C’-13Cα (1DC’Cα), and 15N-13C’ (1DNC’) (Fig. 1d; Supplementary Fig. 5). On average, there are 2.2 RDCs per residue for regions with confirmed resonance assignment.
With less than 3 RDCs per residue, calculating the local structures using the restrained Molecular Dynamics protocol presents a severe local minimum problem, because of the intrinsic orientation degeneracy of the dipolar coupling function. We used an approach that exhaustively searches the protein database for fragments of structure that agree with the experimental data. This method, commonly known as Molecular Fragment Replacement (MFR), was first applied in crystallography for building molecular fragments into crystallographically determined electron density20. A later NMR study demonstrated that with about 4 RDCs per residue, it was possible to determine the backbone structure of ubiquitin using molecular fragments fit to RDCs21. More recently, a fragment search method that combines experimental chemical shift and RDCs has been implemented in the CS-Rosetta structure modeling software and shown to be effective in determining water-soluble protein structures22,23.
We constructed a database containing 320,000 7-residue fragments extracted from structures of 1279 water-soluble and 235 membrane-associated proteins. For each 7-residue stretch along the UCP2 sequence, we fit the corresponding RDCs to all fragments in the database, using singular value decomposition (SVD)24. We evaluated the quality of fit with the parameter Qfree, which is a normalized r.m.s. difference between experimental RDCs and RDCs predicted independently by the structural model25. After this exhaustive search, we collected fragments with Qfree < 25%, and the magnitude (Da) and rhombicity (Rh) of the alignment tensor within 10% of their true values (Fig. 1d). These fragments were designated as candidate fragments and used to determine local backbone structure according to the following protocol. (1) Fragment assignment. Sort the candidate fragments, and assign the fragments in the list having the smallest Qfree to their corresponding protein segments, until no more fragments can be placed without overlap with already assigned segments (Fig. 2a). (2) Gap filling. For any of the gap regions (< 4 residues) not assigned in step (1), search for the candidate fragment that overlaps in ϕ and ψ angles with the two flanking segment and that provides the best Qfree for the merged segment consisting of the newly filled gap structure and the two original flanking fragments (Fig. 2b). (3) End extension. Extend the N- or C-terminal end of an assigned segment by searching for a fragment that overlaps best with the original segment in ϕand ψ angles, and that provides the best Qfree for the final extended segment (Fig. 2b). Operations (2) and (3) were repeated until no more structure could be assigned with confidence. Using the procedure just described, we identified 15 structured segments (Fig. 2c).
Figure 2. Conceptual illustration of the operations involved in RDC-based structural segment building.
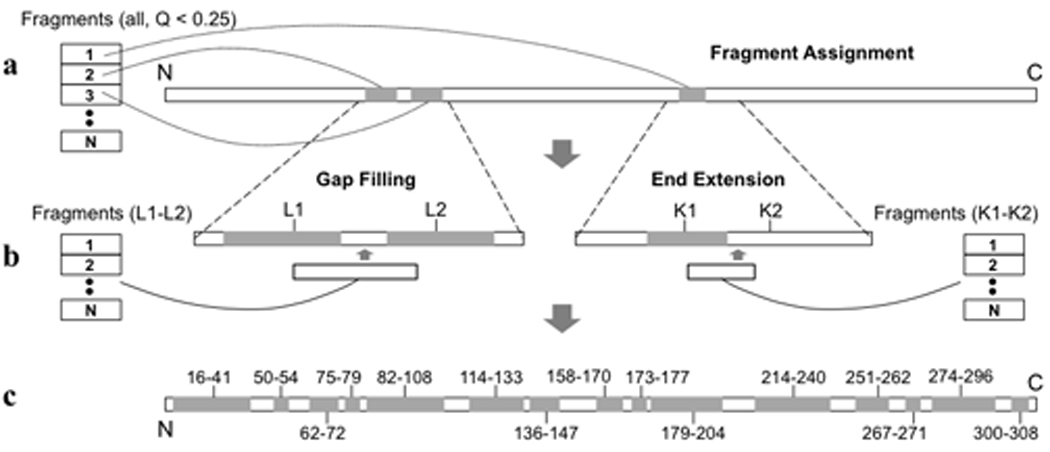
a, Initial fragment assignment. b, gap filling (left) and end extension (right). c, The 15 continuous structured segments of UCP2 determined by RDC-based MFR (shaded and labelled). Details of these operations are described in text and in METHODS.
In addition to RDCs, we obtained semi-quantitative distance restraints from PRE measurements. The paramagnetic moiety introduced for PRE measurement is a nitroxide spin-label in MTSL (see METHODS), which we covalently attached at cysteines. PRE restraints have been demonstrated in low-resolution structure determination of both water-soluble and membrane proteins16,26. Four UCP2 samples were produced, each with a spin label attached at a unique position (68, 105, 202, 255); they all showed similar NMR spectra (Supplementary Fig. 6). These samples together provided 452 PRE distance restraints (an important subset are shown in Fig. 3a).
Figure 3. Solution structure of UCP2 and region of GDP binding.
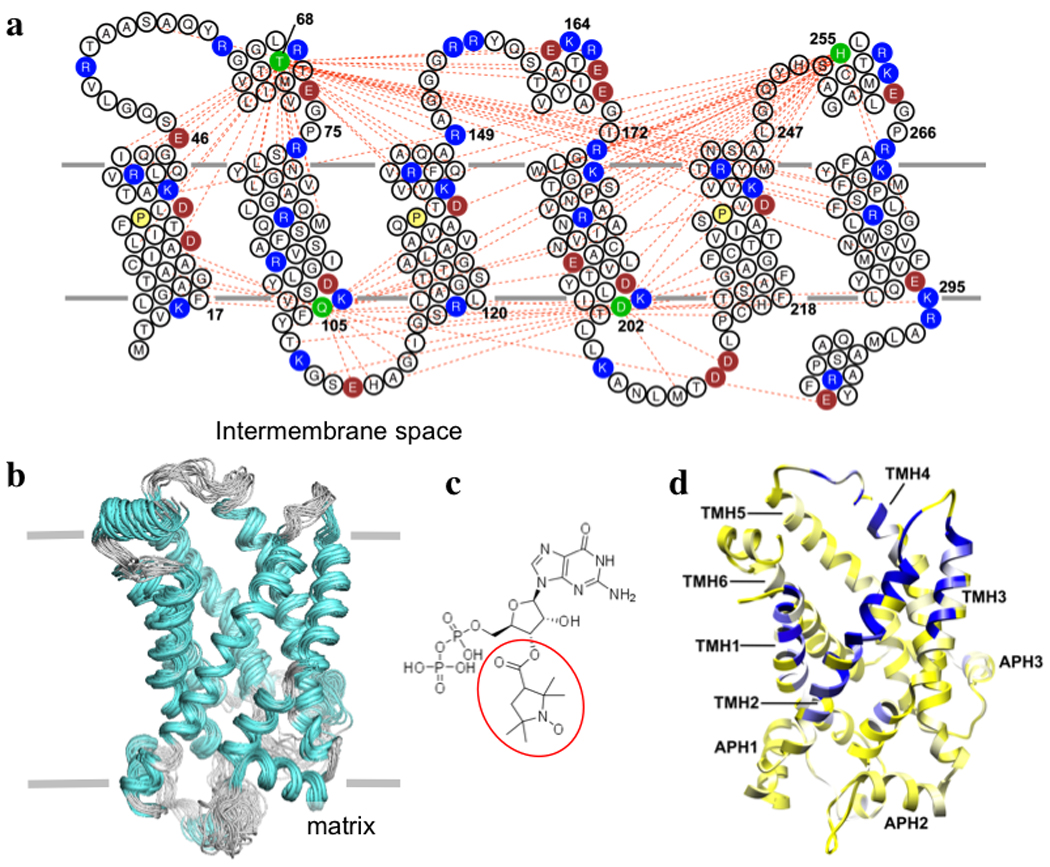
a, UCP2 sequence and membrane topology, with basic and acidic residues shown in blue and red, respectively. The conserved prolines at the proline kinks of TMH 1, 3, and 5 are shown in yellow. The spin-labeled positions are highlighted in green. The red dashed lines represent long-range or inter-helical PRE distances (< 19 Å) between the spin-label and backbone amide protons. b, An ensemble of 15 low-energy structures derived from NMR restraints. The backbone and heavy atom r.m.s. deviations for the structured segments in Fig. 2e are 1.2 Å and 1.8 Å, respectively. c, Chemical structure of the spin-labeled GDP, with the paramagnetic nitroxide moiety circled in red. d, Mapping the effect of spin-labeled GDP onto the ribbon drawing of UCP2. The color gradient is from yellow (resonance intensity ratio of broadened to non-broadened, ε = 1.0) to white (ε = 0.8) to blue (ε = 0.3).
Finally, to determine the tertiary structure, we strongly enforced the backbone ϕand ψ of the 15 MFR-derived structural segments while applying the PRE and RDC restraints. During the structure calculation, the MFR segments made up most of the local structure of the protein, while the PRE and RDC measurements provided spatial and orientational restraints, respectively, for these structured segments. The calculation generated an ensemble of 15 structures with backbone r.m.s. deviation of 1.3 Å (Fig. 3b). There are no experimental data on the sidechains. The structure was determined in the presence of GDP, a well-known inhibitor of UCP2 activity. Although sidechain resonance assignments, which are required for obtaining an atomic resolution view of GDP binding, were not available, we investigated GDP binding qualitatively with the PRE method. We used a paramagnetic nitroxide derivative of GDP (NO-GDP) (Fig. 3c) and measured broadening of protein resonances by the spin label. Mapping the magnitude of the PRE to the structure indicates that NO-GDP binds within the UCP2 channel, but the PRE appears to span a large area, covering residues of transmembrane helices (TMH) 1–4 (Fig. 3d). The distribution of PRE-sensitive residues indicates that the nitroxide radical is in the half of the channel facing the intermembrane space. Based on the PRE-derived position of the nitroxide moiety, we placed the GDP portion of the NO-GDP by maximizing electrostatic interactions between GDP and charged residues in the channel (Supplementary Fig. 7). The model shows that the PRE data are consistent with GDP binding deep within the UCP2 channel, similar to the proposed ADP binding site in ANT127,28
The overall conformation of UCP2 closely resembles that of the bovine ADP/ATP carrier (ANT1)4, despite their low (~20%) sequence identity. They are both channel-like structures, in which 3 pseudo-repeats (Fig. 3a) adopt similar folds (Fig. 4a). Each repeat consists of a TMH (odd-numbered), a loop, an amphipathic helix (APH), and another TMH (even-numbered). Moreover, as in ANT1, the TMHs have kinks at prolines that are conserved in the carrier-protein sequences (Fig. 3a). The structural similarity with ANT1 further supports the notion that the members of the large carrier family have a conserved structure and that small variations within the conserved fold govern specificity of substrate binding and translocation. The most striking differences between the UCP2 and ANT1 structures are in the third repeat. In each of the three repeats of ANT1, the APH packs against the segment of the odd-numbered TMH that follows the conserved proline, and the proline kink in the TMH closes the channel (Fig. 4b). Repeat 3 of the GDP-bound UCP2 breaks away from this pattern. The TMH5 appears to have shifted between TMH4 and TMH6 towards the intermembrane side of the carrier (Supplementary Fig. 8). The APH of the same repeat rotates away from the channel by ~45°, and its flanking regions are also substantially different from their counterparts in the other two repeats (Fig. 4b). Consequently, the matrix side of the channel is substantially more open in UCP2 than in ANT1. Although their functional relevance remains to be investigated, the differences we see could have implications for channel opening in proteins of the carrier family.
Figure 4. Comparison of UCP2 and ANT1.
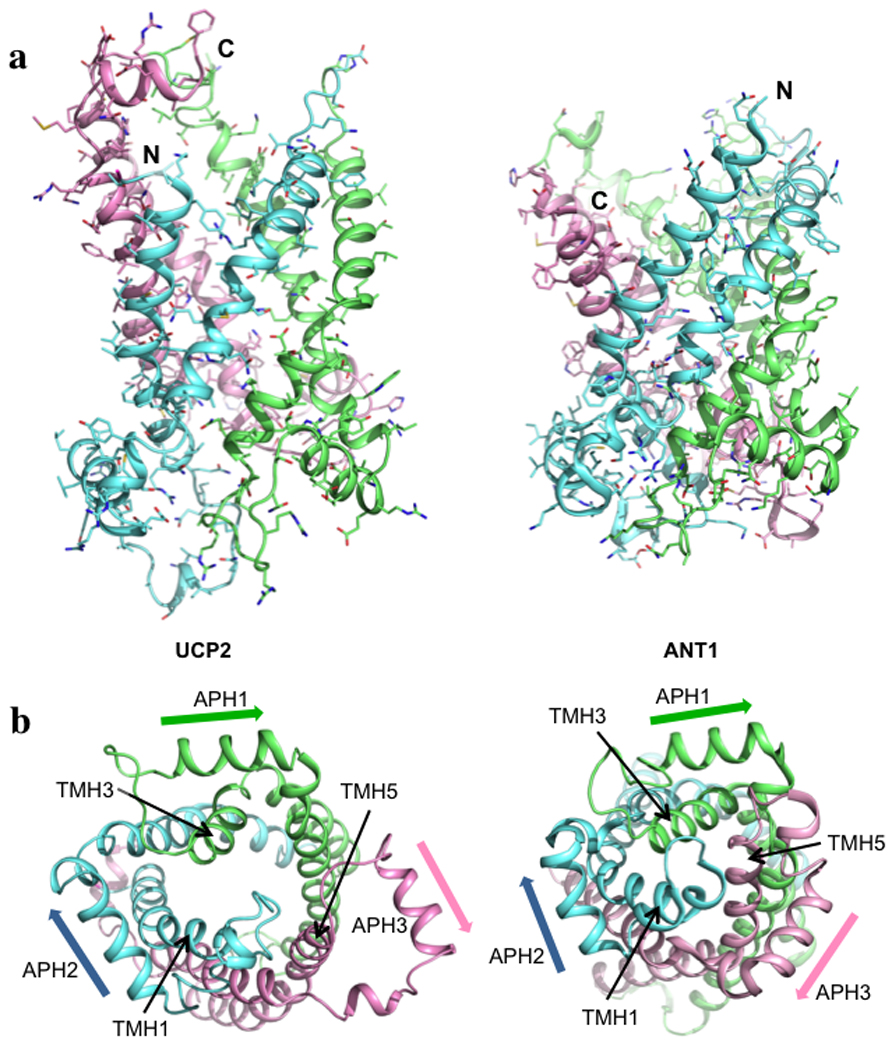
a, Side views of UCP2 and ANT1 (PDB code: 1OKC). Pseudo-repeats 1, 2, and 3 are in blue, green and pink, respectively. The three repeats are 1: 14–112, 2: 113–210, and 3: 211–309 (see Fig. 3a above for reference). b, Views of UCP2 and ANT1 from the matrix side of the carriers, showing loss of 3-fold pseudo-symmetry in UCP2 as a result of structural differences in repeat 3. The orientations of the amphipathic helices are emphasized by the arrows.
In summary, we have established a solution-NMR protocol for studying mitochondrial carriers. We have combined RDC-based fragment searching and PRE measurements to obtain a structure for UCP2 in its GDP-bound state. There are over 40 different carriers identified to date, which transport a variety of metabolites, nucleotides, ions and vitamins across the inner mitochondrial membrane. Although their overall folds are expected to be similar, obtaining structures of different carriers and different functional states of the same carrier will be important for understanding the mechanism and selectivity of substrate transport29. Use of RDCs is well suited for this purpose -- e.g. for describing concerted rearrangements of helices, coupled to opening or closing of the carrier on either side of the membrane -- as RDCs are sensitive to the relative orientation of structural segments. We emphasize that new methods for measuring sidechain constraints must be developed for providing higher resolution NMR structures of these carriers and of membrane proteins of comparable size.
METHOD SUMMARY
Mouse UCP2 (residues 14–309, with a C-terminal His6 tag) was expressed using a pET-21 vector in E. Coli Rossetta DE3 cells. After cell lysis, the lipid composition of the membrane fraction was adjusted by adding DMPC, cardiolipin, and phytanoyl lipids. The protein was extracted using 0.2% DPC in the presence of GDP. The solubilized UCP2 was then subjected to a series of purification steps including Ni-NTA affinity, MonoQ ion exchange, nucleotide-analog affinity, and size exclusion chromatography. The final NMR sample contained 0.8 mM UCP2, 5 mM GDP, 150 mM DPC, 2 mM DMPC, 1 mM cardiolipin, 5 mM β-mercatpoethanol, 30 mM potassium phosphate (pH 6.5), and 80 mM NaCl.
NMR experiments were conducted at 33 °C on spectrometers equipped with cryogenic probes. Sequence specific assignment of backbone chemical shifts was accomplished using three pairs of triple-resonance experiments and a double 15N-edited NOESY, recorded on (15N-, 13C, 2H) labeled protein. RDCs were obtained using an aligned sample containing 0.5 mM UCP2 and 20 mg/ml DNA nanotube19 (other components same as above). 1DNH was measured using the J-scaled TROSY-HNCO experiment. 1DC’Cα and 1DNC’ were measured using TROSY-HNCO with quantitative-JC’Cα and -JNC’ modulations, respectively. For obtaining PREs, we generated a Cys-less UCP2 mutant and introduced single cysteines at desired positions for labeling with MTSL (METHODS). Residue-specific broadening of protein resonances was measured with two TROSY-HNCO spectra, one recorded after nitroxide labeling and another after reducing the nitroxide free electron with ascorbic acid.
Structure determination had two stages: 1) determining local structural segments by RDC-based MFR protocol and 2) determining the spatial arrangement of the MFR-derived segments using PRE distance restraints. Structures were calculated using XPLOR-NIH30 with backbone ϕand ψ of the assigned structured segments, RDCs, and PRE-derived distances. A total of 30 structures were calculated using a simulated annealing protocol, and 15 low-energy structures were selected as the structural ensemble (statistics in Supplementary Table 1).
Supplementary Material
Acknowledgments
We thank Kirill Oxenoid and Remy Sounier for discussions, Mark McClintock for help with DNA nanotube preparation, Ian Stokes-Rees and Piotr Sliz of SBGrid for help with computation, and Niels Voigt for help with figures. The work was supported by NIH Grants 1U54GM094608 (to J.J.C.) and 1DP2OD004641 (to W.M.S.). S.C.H. is an Investigator in the Howard Hughes Medical Institute.
Footnotes
Author Contributions M.J.B., W.M.S., S.C.H. and J.J.C. conceived of the study; M.J.B. prepared NMR samples; M.J.B. and W.M.S. prepared DNA nanotubes; M.J.B. and J.J.C. designed experiments, collected and analyzed NMR data and determined the structure; M.J.B. and J.J.C. wrote the paper; and all authors contributed to editing of the manuscript.
Author Information The structure has been deposited in the Protein Data Bank under the accession number 2lck.
References
- 1.Palmieri F, et al. Identification of mitochondrial carriers in Saccharomyces cerevisiae by transport assay of reconstituted recombinant proteins. Biochim Biophys Acta. 2006;1757:1249–1262. doi: 10.1016/j.bbabio.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 2.Krauss S, Zhang CY, Lowell BB. The mitochondrial uncoupling-protein homologues. Nat Rev Mol Cell Biol. 2005;6:248–261. doi: 10.1038/nrm1592. [DOI] [PubMed] [Google Scholar]
- 3.Tate CG, Stevens RC. Growth and excitement in membrane protein structural biology. Curr Opin Struct Biol. 2010;20:399–400. doi: 10.1016/j.sbi.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 4.Pebay-Peyroula E, et al. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 6.Aquila H, Link TA, Klingenberg M. The uncoupling protein from brown fat mitochondria is related to the mitochondrial ADP/ATP carrier. Analysis of sequence homologies and of folding of the protein in the membrane. Embo J. 1985;4:2369–2376. doi: 10.1002/j.1460-2075.1985.tb03941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang CY, et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell. 2001;105:745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 8.Diao J, et al. UCP2 is highly expressed in pancreatic alpha-cells and influences secretion and survival. Proc Natl Acad Sci U S A. 2008;105:12057–12062. doi: 10.1073/pnas.0710434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andrews ZB, et al. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454:846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harper ME, et al. Characterization of a novel metabolic strategy used by drug-resistant tumor cells. FASEB J. 2002;16:1550–1557. doi: 10.1096/fj.02-0541com. [DOI] [PubMed] [Google Scholar]
- 11.Samudio I, Fiegl M, Andreeff M. Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009;69:2163–2166. doi: 10.1158/0008-5472.CAN-08-3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hiller S, et al. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science. 2008;321:1206–1210. doi: 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Pielak RM, McClintock MA, Chou JJ. Solution structure and functional analysis of the influenza B proton channel. Nat Struct Mol Biol. 2009;16:1267–1271. doi: 10.1038/nsmb.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Y, et al. NMR solution structure of the integral membrane enzyme DsbB: functional insights into DsbB-catalyzed disulfide bond formation. Mol Cell. 2008;31:896–908. doi: 10.1016/j.molcel.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Horn WD, et al. Solution nuclear magnetic resonance structure of membrane-integral diacylglycerol kinase. Science. 2009;324:1726–1729. doi: 10.1126/science.1171716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gautier A, Mott HR, Bostock MJ, Kirkpatrick JP, Nietlispach D. Structure determination of the seven-helix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nat Struct Mol Biol. 2010;17:768–774. doi: 10.1038/nsmb.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tjandra N, Bax A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- 19.Douglas SM, Chou JJ, Shih WM. DNA-nanotube-induced alignment of membrane proteins for NMR structure determination. Proc Natl Acad Sci U S A. 2007;104:6644–6648. doi: 10.1073/pnas.0700930104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones TA, Thirup S. Using known substructures in protein model building and crystallography. EMBO J. 1986;5:819–822. doi: 10.1002/j.1460-2075.1986.tb04287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delaglio F, Kontaxis G, Bax A. Protein structure determination using molecular fragment replacement and NMR dipolar couplings. J Am Chem Soc. 2000;122:2142–2143. [Google Scholar]
- 22.Shen Y, et al. Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci U S A. 2008;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raman S, et al. NMR structure determination for larger proteins using backbone-only data. Science. 2010;327:1014–1018. doi: 10.1126/science.1183649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Losonczi JA, Andrec M, Fischer MWF, Prestegard JH. Order matrix analysis of residual dipolar couplings using singular value decomposition. J. Magn. Reson. 1999;138:334–342. doi: 10.1006/jmre.1999.1754. [DOI] [PubMed] [Google Scholar]
- 25.Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]
- 26.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Tajkhorshid E. Electrostatic funneling of substrate in mitochondrial inner membrane carriers. Proc Natl Acad Sci U S A. 2008;105:9598–9603. doi: 10.1073/pnas.0801786105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dehez F, Pebay-Peyroula E, Chipot C. Binding of ADP in the mitochondrial ADP/ATP carrier is driven by an electrostatic funnel. J Am Chem Soc. 2008;130:12725–12733. doi: 10.1021/ja8033087. [DOI] [PubMed] [Google Scholar]
- 29.Kunji ER, Robinson AJ. Coupling of proton and substrate translocation in the transport cycle of mitochondrial carriers. Curr Opin Struct Biol. 2010;20:440–447. doi: 10.1016/j.sbi.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Schwieters CD, Kuszewski J, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2002;160:66–74. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.