

Abstract
Introduction:
Genetic effects contribute to individual differences in smoking behavior. Persistence to smoke despite known harmful health effects is mostly driven by nicotine addiction. As the physiological effects of nicotine are mediated by nicotinic acetylcholine receptors (nAChRs), we aimed at examining whether single nucleotide polymorphisms (SNPs) residing in nAChR subunit (CHRN) genes, other than CHRNA3/CHRNA5/CHRNB4 gene cluster previously showing association in our sample, are associated with smoking quantity or serum cotinine levels.
Methods:
The study sample consisted of 485 Finnish adult daily smokers (age 30–75 years, 59% men) assessed for the number of cigarettes smoked per day (CPD) and serum cotinine level. We first studied SNPs residing on selected nAChR subunit genes (CHRNA2, CHRNA4, CHRNA6/CHRNB3, CHRNA7, CHRNA9, CHRNA10, CHRNB2, CHRNG/CHRND) genotyped within a genome-wide association study for single SNP and multiple SNP associations by ordinal regression. Next, we explored individual haplotype associations using sliding window technique.
Results:
At one of the 8 loci studied, CHRNG/CHRND (chr2), single SNP (rs1190452), multiple SNP, and 2-SNP haplotype analyses (SNPs rs4973539–rs1190452) all showed statistically significant association with cotinine level. The median cotinine levels varied between the 2-SNP haplotypes from 220 ng/ml (AA haplotype) to 249 ng/ml (AG haplotype). We did not observe significant associations with CPD.
Conclusions:
These results provide further evidence that the γ−δ nAChR subunit gene region is associated with cotinine levels but not with the number of CPD, illustrating the usefulness of biomarkers in genetic analyses.
Introduction
Tobacco smoking is one of the major public health problems worldwide (World Health Organization Report on the Global Tobacco Epidemic, 2009). Genetic factors influence smoking-related traits such as smoking behavior and nicotine dependence (Rose, Broms, Korhonen, Dick, & Kaprio, 2009). Nicotine is the major addictive substance in tobacco keeping the smoker persistent to smoke by quickly evolving physiological, pharmacological, and social dependence. The nicotinic acetylcholine receptors (nAChRs) largely mediate the physiological effects of nicotine. Altogether nine nAChR subunits (α2–α7 and β2–β4) are expressed in the human brain. These subunits combine with each other in particular patterns to form functional pentameric receptors. The different receptor subtypes are distinguished by subunit composition and sensitivity to nicotine (Collins, Salminen, Marks, Whiteaker, & Grady, 2009).
Self-reported smoking quantity enquired with a questionnaire or in an interview is the most common method to assess the nicotine intake of a subject. However, this method may be compromised by misreporting, which may be either conscious underreporting, for example, due to social desirability or unconscious misreporting due to varying number of cigarettes smoked per day (CPD). In addition, subjects with similar smoking quantity may have differing nicotine intake as a result of varying smoking patterns (e.g., depth of inhalation and puff volume), type of cigarettes smoked, proportion of the cigarette smoked, and nicotine content of the cigarettes (Bernstein, 2004).
A more reliable measure of nicotine intake is the assessment of the cotinine level measured from either serum or saliva. Cotinine is the major metabolite of nicotine as approximately 80% of nicotine is metabolized to cotinine by CYP 2A6 enzyme. Cotinine has a half-life of 15–20 hr, and thus, its level reflects the cumulative nicotine intake in the past approximately seven days and the individual differences in nicotine metabolism (Hukkanen, Jacob, & Benowitz, 2005).
Several studies have targeted the role of genetic variants at nAChR subunit (CHRN) genes in smoking behavior. Many genome-wide association studies have identified a cluster of three nAChR subunit genes (CHRNA3/CHRNA5/CHRNB4) on chromosome 15 to be associated with nicotine dependence and the number of CPD (Bierut et al., 2008; Liu et al., 2010; The Tobacco and Genetics Consortium, 2010; Thorgeirsson et al., 2008, 2010). In our previous study, we showed that a single nucleotide polymorphisms (SNPs) residing within the cluster, rs1051730, accounted for nearly a fivefold larger proportion of the total variance of cotinine levels than that of CPD (Keskitalo et al., 2009). Genetic associations of smoking behaviors with other nAChR subunit genes have also been detected. For example, CHRNA4 (Feng et al., 2004; Li et al., 2005), CHRNA6/CHRNB3 (Hoft et al., 2009; Saccone et al., 2009), and CHRNG/CHRND (Saccone et al., 2009) have shown association with nicotine dependence, and CHRNB2 has shown association with initial subjective responses to nicotine (Ehringer et al., 2007) as well as with the ability to quit smoking (Perkins, Lerman, Mercincavage, Fonte, & Briski, 2009).
Our aim was to study whether genetic variation in neuronal nAChR subunit genes other than the previously reported CHRNA3/CHRNA5/CHRNB4 (Keskitalo et al., 2009) is associated with nicotine intake. In addition, we included in this study CHRNG/CHRND gene cluster not expressed in the brain but having previously shown linkage and association with smoking behavior (Saccone et al., 2009, Straub et al., 1999). We used two distinct methods of assessment of nicotine intake: number of CPD, based on the self-reporting of the subjects, and an immune-reactive measurement of serum cotinine level.
Materials and Methods
Subjects and Phenotypes
Subjects were drawn from the Health 2000 study, which includes a total of 8,028 subjects aged 30 years or over, and is a nationally representative sample of the adult Finnish population (Aromaa & Koskinen, 2004). The proportion of daily smokers was 29% among males and 18% among females, and the proportion of daily smokers decreased by age. Here, we studied a subcohort (n = 2,124, aged 30–75 years, 1,036 males) selected for a case–control genome-wide association study on metabolic syndrome. The metabolic syndrome is a cluster of the most dangerous heart attack risk factors: diabetes and prediabetes, abdominal obesity, high cholesterol, and high blood pressure. The metabolic syndrome cases were selected according to the International Diabetes Federation Worldwide Definition of the Metabolic Syndrome (http://www.idf.org/node/1271?node=1429), and the controls were subjects not carrying the trait. Diabetic subjects were not included in either case or control groups.
Interviewers asked the smoking quantity within the interview at the participants home by one question “How many of the following do you smoke each day currently or did prior to quitting? (a) factory-made cigarettes, (b) self-rolled cigarettes, (c), pipefuls of pipe tobacco, (d) cigars/cigarillos” with open-ended response to each. We summed the number of each tobacco product to create the CPD variable used in the analyses. The cotinine level (nanograms per milliliters) was determined from the serum using liquid-phase radioimmunoassay methodology (Nicotinic Metabolite DOUBLE ANTIBODY kit, Diagnostic Products Corporation). Details of the data collection are reported elsewhere (Aromaa & Koskinen, 2004, Keskitalo et al., 2009).
We included only current daily smokers in the statistical analyses as the serum cotinine level cannot be regarded as a reliable measure of the nicotine intake in former, occasional, or nonsmokers. This yielded a sample of 485 genotyped individuals including 201 females and 284 males. The mean age of the subjects was 47.7 years (SD: 9.7, range: 30–75 years). This sample of smokers included 209 subjects with metabolic syndrome (cases) and 276 healthy controls. The metabolic syndrome cases smoked slightly more (CPD mean 17.9 vs. 16.1, p = .04) and also had higher cotinine levels (mean: 524 vs. 463 ng/ml, p = .01) than controls.
Genotyping
Venous blood samples were drawn from the antecubital vein after an overnight fast. DNA was extracted from blood using standard procedures. The samples were genotyped by Illumina 610 Quad V1 BeadChip (Illumina, Inc.) at the Sanger Wellcome Trust Institute. This chip provides whole-genome SNP genotyping information with 598203 SNP markers per individual genotyped with a mean spacing of 4.7 kb. The data were checked for SNP clustering probability for each genotype (>95%), call rate (both SNPs and individuals >95%), minor allele frequency (>1%), Hardy–Weinberg equilibrium test p value (>1 × 10−6), heterozygosity, gender, and relatedness. Any discrepancies (altogether 10.6% of the markers and 0.4% of the samples) were removed from the data.
We selected SNPs within the gene regions and within ±10 kb flanking regions according to Entrez Gene database (http://ncbi.nlm.nih.gov/gene/). A total of 88 genotyped SNPs in CHRNA2 (chr 8), CHRNA4 (chr 20), CHRNA6-CHRNB3 cluster (chr8), CHRNA7 (chr 15), CHRNA9 (chr 4), CHRNA10 (chr 11), CHRNB2 (chr 1), and CHRNG-CHRND cluster (chr 2) passed the quality control and were included in the analyses. The regions and the number of SNPs in each locus are presented in Table 1.
Table 1.
Genetic Regions Included in the Analyses
| Gene | Chromosome | Location (bp) | # SNPs genotyped |
| CHRNA2 | 8 | 27,363,195–27,402,730 | 20 |
| CHRNA4 | 20 | 61,435,109–61,473,192 | 7 |
| CHRNA6/CHRNB3 | 8 | 42,661,719–42,752,776 | 10 |
| CHRNA7 | 15 | 30,109,018–30,258,541 | 24 |
| CHRNA9 | 4 | 40,022,226–40,061,730 | 8 |
| CHRNA10 | 11 | 3,633,393–3,659,190 | 8 |
| CHRNB2 | 1 | 152,796,881–152,828,978 | 4 |
| CHRNG/CHRND | 2 | 233,089,166–233,129,282 | 7 |
Statistical Methods
We used ordered categorized variables for both traits in the statistical analysis due to nonnormality of CPD and cotinine level (p < .001, Shapiro–Wilk test for normality) even after log or square root transformation. We classified CPD into four groups according to a question in the Fagerström Test for Nicotine Dependence (Heatherton, Kozlowski, Frecker, & Fagerström, 1991): (a) CPD = 1–10 (N = 167 subjects, 33%), (b) CPD = 11–20 (N = 258, 51%), (c) CPD = 21–30 (N = 55, 11%), and (d) CPD = 31–100 (N = 25, 5%). We categorized the cotinine level into quintiles with the following minimum and maximum values: 0–277, 278–417, 418–557, 558–698, and >698 ng/ml. Each quintile consisted of approximately 100 subjects.
We modeled the single SNP effects of the ordinal response (CPD or cotinine) by fitting univariate and multivariate proportional odds logistic regression models for each eight chromosomal region separately: , where we model the logit of the probability Y (n × 1 matrix) of the observed categorized values of CPD or cotinine being k or less (k = 1, … , 4 for CPD and k = 1, … , 5 for cotinine). Parameter αk is called the cutpoint parameter and is the logit of the cumulative distribution function. Regression coefficient β is (1 × 1) matrix (scalar) of the regression coefficients. In the single SNP, Model Z is (n × 1 matrix) with values corresponding to the number of minor alleles of the subject i at the SNP considered and therefore corresponding to a multiplicative dominance function of the SNP genotypes. In the multiple SNP model, all p SNPs at region of interest were modeled simultaneously; Z is (n × p matrix), β is (n × p matrix), and the model assumes multiplicative effects between SNPs. In the proportional odds model, the log of the ratio of the odds of cumulative probabilities of z1 and z2 is proportional to the distance between z1 and z2.; log (odds (j ≤ k|X = z1)/odds(j ≤ k|X = z2)) = β′(z1 – z2). This would imply that for a heterozygote genotype carrier, the log odds of the category k or less is increased/decreased by b compared with the log odds of k or less category of a common variant homozygote carrier.
We obtained the single SNP p values using likelihood ratio test from the above ordinal regression models including age and sex as covariates and comparing a model including a single SNP with a model without the SNP. We report the single SNP p values (single SNP p adjusted) corrected for multiple testing (number of SNPs in a region) based on the false discovery rate (Benjamini & Yekutieli, 2001) method. The multiple SNP likelihood ratio test compares model with all SNPs in a region with a model without any SNPs separately for eight candidate regions using standard likelihood ratio test. We considered SNPs interesting if both single SNP and multiple SNP p values were less than .05.
We imputed haplotypes for each subject using expectation maximation (EM)–based progressive iteration algorithm. We used a two-SNP sliding window method to assess the haplotype associations and report EM-based p values adjusted for gender and age from a global score test (global score p value) for the haplotype effects (Schaid, Rowland, Tines, Jacobson, & Poland, 2002). Due to nonnormality of the cotinine level phenotype, we report estimated medians of cotinine levels conditional on the haplotypes using a classical quantile regression method considering only the most probable haplotypes for each individual. In quantile regression, haplotype effects are modeled by the linear effects (Hm maternal haplotype and Hf paternal haplotype) on τth percentile (0.5) of cotinine level (y) using the following regression equation 0, where β is the (p + 1) × 1 matrix vector of haplotype effect coefficients and Z represents a n × (p + 1) matrix where each element codes for a haplotype of the individuals. We assumed additive mode of inheritance when estimating the haplotype effects. We obtained CIs using standard normal approximation method using estimated SE of the haplotype effect, which was obtained from quantile regression model.
All statistical analyses were performed using R-program version 2.10.0 (Ihaka & Gentleman, 1996) packages MASS for ordinal regression, haplo.stats (version 1.4.4) for haplotype analysis, quantreg-package for quantile regression, and design for calculating the effect size and the proportion of the variance explained by the SNP. Sex and age were included in all the models.
Results
Among our dataset of Finnish daily smokers, the median CPD was 16 (95% CI: 15.1–16.9, with interquartile range 10–20), and the median cotinine level was 476 ng/ml (95% CI: 444.51–507.49, with interquartile range 313–651). These measures correlated significantly (Spearman's ρ = 0.43, p < .001). Males smoked significantly more than females (median CPD males 20 vs. females 12, p < .001) and also had higher cotinine levels than females (median cotinine level males 503.0 vs. females 412.0, p = .004).
In the analyses, we observed a statistically significant association for cotinine level with CHRNG/CHRND gene cluster residing in chromosome 2 in both single SNP analysis with marker rs1190452 (single SNP p adjusted = .0006) and the multiple SNP analysis of the candidate region (multiple SNP test p = .002). This common variant (minor allele was G with a frequency in our dataset 37%) after adjusting for age and gender explained 3.4% of the variance in cotinine levels, while the variance on CPD was virtually zero (model adjusted for age and sex). The sex and gender-adjusted effect size of rs1190452 was 0.49 (SE = 0.12) on cotinine level and 0.001 (SE = 0.13) on CPD. The seven CHRNG/CHRND SNPs analyzed in this study are correlated at r2 ≥ .8 with 14 of 33 HapMap SNPs (42%) of the region in a Finnish population dataset. Linkage disequilibrium (LD) between the seven SNPs genotyped in this study is presented in the Supplementary Figure 5.
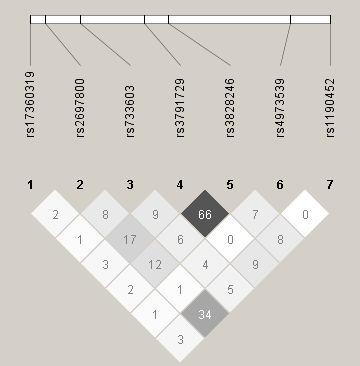
Haplotype analysis with 2-SNP sliding window showed the most prominent association in CHRNG/CHRND region (Figure 1) covering rs4973539 and rs1190452 (global score p value = .0006). Results from the haplotype and genotype analysis are presented in Figure 1 and the estimated haplotype-specific medians of cotinine level in Table 2. We present the univariate and multivariate SNP association results along with haplotype association plots of all candidate regions for CPD and cotinine level in the Supplementary Material.
Figure 1.

(a) Single SNP, multiple SNP, and (b) 2-SNP haplotype association test −log10 p values of CHRNG/CHRND region with serum cotinine level.
Table 2.
Estimated Haplotype-Specific Medians and 95% CIs of Cotinine Levels According to CHRNG/CHRND Region (rs4973539 and rs1190452) Estimated Most Likely 2-SNP Haplotypes
| Haplotype | N (%) | Median | 95 % CI |
| rs4973539–rs1190452 | |||
| G G | 33 (7.1) | 240 | 188–308 |
| G A | 163 (13.0) | 248 | 201–304 |
| A G | 329 (29.7) | 249 | 211–290 |
| A A | 531 (50.3) | 220 | 196–252 |
Discussion
In our analyses, we found significant association between SNPs in the region of CHRNG/CHRND on chromosome 2 and cotinine level. Despite the significant correlation between CPD and cotinine level, this region showed no association with CPD. The association between CHRNG/CHRND and cotinine level was somewhat unexpected in terms of biological plausibility as both subunits are part of the muscle-type nAChR. The γ subunit is known to be only fetally expressed and later replaced by the ϵ subunit. However, this gene cluster was found to be associated with nicotine dependence in an independent study by Saccone et al. (2009). An LD matrix of the CEU HapMap2 SNPs genotyped in our study and in the study by Saccone et al. is presented in the Supplementary Material (Supplementary Figure 6). It is noteworthy that our association signal emerges downstream (3′) of the CHRNG/CHRND gene cluster and the association could be due to adjacent genetic elements. The SNPs of the associating 2-SNP haplotype (rs4973539 and rs1190452) cover a region including TIGD1 (tigger transposable element–derived protein 1, chr2:233,121,023–233,123,470) and EIF4E2 (eukaryotic translation initiation factor 4E family member 2, chr2:233,123,601-233,142,164) in addition to CHRNG and CHRND. Our results do not reveal which of these genes contributes to the observed association.
Statistical association analyses of genetic traits that are not normally distributed, such as cotinine level in this study, suffer the lack of statistical methods suitable for comprehensive genetic association analysis. Therefore, we chose to treat cotinine level as an ordinal variable, despite its continuous nature, in our analysis and classified it into five categories. Bins were based on the 20, 40, 60, and 80 percentile of the trait distribution, and the results did not change when using cotinine variable based on deciles (data not shown). In addition, this study points out the importance of careful phenotyping and illustrates the usefulness of biomarkers in genetic analyses. The serum cotinine level likely includes less measurement error in terms of nicotine intake compared with the self-reported smoking quantity (Benowitz, Dains, Dempsey, Yu, & Jacob, 2010). Although an earlier Finnish population study has indicated that the current smokers do report their smoking status very accurately (Vartiainen, Seppala, Lillsunde, & Puska, 2002), the possibility of conscious or unconscious misreporting cannot be excluded. There are two methods available and commonly used to assess the cotinine concentration of a sample. The method used in this study, radioimmunoassay, provides slightly less accurate results compared with the chromatographic method (Byrd, Davis, & Ogden, 2005). Though the results obtained by these two widely used methods are highly correlated, they are not directly comparable.
Our results imply that the nAChR subunit genes apart from the chr 15 CHRNA3/CHRNA5/CHRNB4 complex do not generally have a strong impact on nicotine intake. These effects should be studied by specifically targeted studies with more smoking subjects phenotyped with several smoking-related traits and genotyped with a denser marker coverage of the genes. However, in our sample of smokers, we observed a promising association at the CHRNG/CHRND region with serum cotinine levels. This observation needs to be replicated in independent samples of smokers.
Supplementary Material
Supplementary Figures 1–6 can be found online at http://www.ntr.oxfordjournals.org
Funding
The study is funded by Center of Excellence in Disease Genetics, Academy of Finland to JK and the National Institutes of Health grant DA12854. The genome-wide association genotyping was funded by the Welcome Trust Sanger Institute. AL was supported by the Academy of Finland Post-Doctoral Fellowship. VS was supported by the Academy of Finland (129494), the Finnish Foundation for Cardiovascular Research, and the Sigrid Juselius Foundation. UB was supported by the Yrjö Jahnsson Foundation and the Juho Vainio Foundation for postdoctoral research.
Declaration of Interests
JK has served as a consultant to Pfizer in 2008 on pharmacogenetics of smoking cessation. UB has served as a consultant to Pfizer in 2008 on nicotine dependence measurements.
Supplementary Material
References
- Aromaa A, Koskinen S, editors. Health and functional capacity in Finland. Helsinki, Finland: Publications of the National Public Health Institute, KTL B12; 2004. Retrieved from http://www.terveys2000.fi/julkaisut/baseline.pdf. [Google Scholar]
- Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Annals of Statistics. 2001;29:1165–1188. doi:10.1214/aos/1013699998. [Google Scholar]
- Benowitz NL, Dains KM, Dempsey D, Yu L, Jacob P., 3rd Estimation of nicotine dose after low-level exposure using plasma and urine nicotine metabolites. Cancer Epidemiology, Biomarkers & Prevention. 2010;19:1160–1166. doi: 10.1158/1055-9965.EPI-09-1303. doi:10.1158/1055-9965.EPI-09-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein DM. A review of the influence of the particle size, puff volume, and inhalation pattern on the deposition of cigarette smoke particles in the respiratory tract. Inhalation Toxicology. 2004;6:675–689. doi: 10.1080/08958370490476587. doi:10.1080/08958370490476587. [DOI] [PubMed] [Google Scholar]
- Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, et al. Variants in nicotinic receptors and risk for nicotine dependence. American Journal of Psychiatry. 2008;165:1163–1171. doi: 10.1176/appi.ajp.2008.07111711. doi:10.1176/appi.ajp.2008.07111711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd GD, Davis RA, Ogden MW. A rapid LC-MS-MS method for determination of nicotine and cotinine in serum and saliva samples from smokers: Validation and comparison with a radioimmunoassay method. Journal of Chromatographic Science. 2005;43:133–140. doi: 10.1093/chromsci/43.3.133. Retrieved from http://www.j-chrom-sci.com/abstracts/2005/march/133-byrd.html. [DOI] [PubMed] [Google Scholar]
- Collins AC, Salminen O, Marks MJ, Whiteaker P, Grady SR. The road to discovery of neuronal nicotinic cholinergic receptor subtypes. In: Henningfield JE, London ED, Pogun S, editors. Nicotine psychopharmacology. Heidelberg, Germany: Springer-Verlag; 2009. pp. 85–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehringer MA, Clegg HV, Collins AC, Corley RP, Crowley T, Hewitt JK, et al. Association of the neuronal nicotinic receptor beta2 subunit gene (CHRNB2) with subjective responses to alcohol and nicotine. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2007;144B:596–604. doi: 10.1002/ajmg.b.30464. doi:10.1002/ajmg.b.30464. [DOI] [PubMed] [Google Scholar]
- Feng Y, Niu T, Xing H, Xu X, Chen C, Peng S, et al. A common haplotype of the nicotine acetylcholine receptor alpha-4 subunit gene is associated with vulnerability to nicotine addiction in men. American Journal of Human Genetics. 2004;75:112–121. doi: 10.1086/422194. doi:10.1086/422194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heatherton TF, Kozlowski LT, Frecker RC, Fagerström K-O. The Fagerström Test for Nicotine Dependence: A revision of the Fagerström Tolerance Questionnaire. British Journal of Addiction. 1991;86:1119–1127. doi: 10.1111/j.1360-0443.1991.tb01879.x. Retrieved from http://www.musc.edu/psychiatry/research/cns/upadhyayareferences/Heatherton_1991.pdf. [DOI] [PubMed] [Google Scholar]
- Hoft NR, Corley RP, McQueen MB, Schlaepfer IR, Huizinga D, Ehringer MA. Genetic association of the CHRNA6 and CHRNB3 genes with tobacco dependence in a nationally representative sample. Neuropsychopharmacology. 2009;34:698–706. doi: 10.1038/npp.2008.122. doi:10.1038/npp.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukkanen J, Jacob P, III, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacological Reviews. 2005;57:79–115. doi: 10.1124/pr.57.1.3. doi:10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- Ihaka R, Gentleman RR. A Language for data analysis and graphics. Journal of Computational and Graphical Statistics. 1996;5:299–314. Retrieved from http://www.jstor.org/pss/1390807. [Google Scholar]
- Keskitalo K, Broms U, Heliövaara M, Ripatti S, Surakka I, Perola M, et al. Association of serum cotinine level with a cluster of three nicotinic acetylcholine receptor genes (CHRNA3/CHRNA5/CHRNB4) on chromosome 15. Human Molecular Genetics. 2009;18:4007–4012. doi: 10.1093/hmg/ddp322. doi:10.1093/hmg/ddp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MD, Beuten J, Ma JZ, Payne TJ, Lou X-Y, Garcia V, et al. Ethnic- and gender-specific association of the nicotinic acetylcholine receptor alpha-4 subunit gene (CHRNA4) with nicotine dependence. Human Molecular Genetics. 2005;14:1211–1219. doi: 10.1093/hmg/ddi132. doi:10.1093/hmg/ddi132. [DOI] [PubMed] [Google Scholar]
- Liu JZ, Tozzi F, Waterworth DM, Pillai SG, Muglia P, Middleton L, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nature Genetics. 2010;42:436–440. doi: 10.1038/ng.572. doi:10.1038/ng.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins KA, Lerman C, Mercincavage M, Fonte CA, Briski JL. Nicotinic acetylcholine receptor beta2 subunit (CHRNB2) gene and short-term ability to quit smoking in response to nicotine patch. Cancer Epidemiology, Biomarkers & Prevention. 2009;18:2608–2612. doi: 10.1158/1055-9965.EPI-09-0166. doi:10.1158/1055-9965.EPI-09-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose RJ, Broms U, Korhonen T, Dick D, Kaprio J. Genetics of smoking behavior. In: Kim YK, editor. Handbook of behavior genetics. New York: Springer; 2009. [Google Scholar]
- Saccone NL, Saccone SF, Hinrichs AL, Stitzel JA, Duan W, Pergadia ML, et al. Multiple distinct risk loci for nicotine dependence identified by dense coverage of the complete family of nicotinic receptor subunit (CHRN) genes. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2009;150B:453–466. doi: 10.1002/ajmg.b.30828. doi:10.1002/ajmg.b.30828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. American Journal of Human Genetics. 2002;70:425–434. doi: 10.1086/338688. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC384917/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub RE, Sullivan PF, Ma Y, Myakishev MV, Harris-Kerr C, Wormley B, et al. Susceptibility genes for nicotine dependence: a genome scan and followup in an independent sample suggest that regions on chromosomes 2, 4, 10, 16, 17 and 18 merit further study. Molecular Psychiatry. 1999;4:129–144. doi: 10.1038/sj.mp.4000518. Retrieved from http://www.nature.com/mp/journal/v4/n2/pdf/4000518a.pdf. [DOI] [PubMed] [Google Scholar]
- Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. doi:10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F, et al. Sequence variants at CHRNB3–CHRNA6 and CYP2A6 affect smoking behavior. Nature Genetics. 2010;42:448–453. doi: 10.1038/ng.573. doi:10.1038/ng.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Tobacco and Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nature Genetics. 2010;42:441–447. doi: 10.1038/ng.571. doi:10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartiainen E, Seppala T, Lillsunde P, Puska P. Validation of self reported smoking by serum cotinine measurement in a community-based study. See comment. Journal of Epidemiology and Community Health. 2002;56:167–170. doi: 10.1136/jech.56.3.167. doi:10.1136/jech.56.3.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization Report on the Global Tobacco Epidemic. Implementing smoke-free environments. World Health Organization; 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}