

Abstract
Calcific aortic valve stenosis (CAVS) is a major health problem facing aging societies. The identification of osteoblast-like and osteoclast-like cells in human tissue has led to a major paradigm shift in the field. CAVS was thought to be a passive, degenerative process, whereas now the progression of calcification in CAVS is considered to be actively regulated. Mechanistic studies examining the contributions of true ectopic osteogenesis, non-osseous calcification, and ectopic osteoblast-like cells (that appear to function differently from skeletal osteoblasts) to valvular dysfunction have been facilitated by the development of mouse models of CAVS. Recent studies also suggest that valvular fibrosis, as well as calcification, may play an important role in restricting cusp movement, and CAVS may be more appropriately viewed as a fibrocalcific disease. High resolution echocardiography and magnetic resonance imaging have emerged as useful tools for testing the efficacy of pharmacological and genetic interventions in vivo. Key studies in humans and animals are reviewed that have shaped current paradigms in the field of CAVS, and suggest promising future areas for research.
Keywords: calcification, fibrosis, animal models, phenotyping, echocardiography
Calcific aortic valve stenosis (CAVS) is an important clinical problem: 2.8% of adults over 75 years old have some degree of CAVS.1, 2, and as many as 25% of adults over 65 have valvular sclerosis3. Although risk factors and downstream mediators appear similar for CAVS and atherosclerosis (older age, male sex, hypertension, smoking, hypercholesterolemia, and diabetes4, 5, see Figure 1), as many as 50% of patients with CAVS do not have clinically significant atherosclerosis 6, 7.
Figure 1.

Overview of risk factors and potential mechanisms that contribute to calcification and fibrosis of the aortic valve. For clarity, effects of potential mediators on various cell types in the valve have been omitted. Abbreviations: ANG II = angiotensin II; RAGE = receptor for advanced glycosylation end products; LDL = low density lipoproteins; ROS = reactive oxygen species. Grey ovals depict endothelial cells.
Studies of valves from humans and experimental animals have begun to clarify mechanisms that lead to CAVS8–10. A major obstacle to research in this area is that although several experimental models of CAVS develop valvular sclerosis, few develop hemodynamically significant stenosis. Two experimental models of CAVS have now been identified in mice, which consistently develop hemodynamically significant CAVS11–14. These models will allow studies of mechanisms contributing to valve calcification, the cardiac and systemic consequences of CAVS, and the efficacy of interventions.
In this review, we will summarize 1) methods to evaluate the normal and stenotic aortic valve in mice, by histology and imaging, 2) mechanisms that may contribute to valve calcification and fibrosis in humans and animal models of CAVS, and 3) mechanisms that be useful therapeutic targets to inhibit development and/or progression of CAVS. Finally, we will speculate about future directions of this area of research.
Assessment of Aortic Valve Function in Mice
Imaging approaches have evolved from techniques that were introduced first in the clinical setting, and later scaled to studies in mice. Three approaches are summarized below to evaluate cardiac function in mice: echocardiography, magnetic resonance imaging, and invasive hemodynamic assessment.
Echocardiography
Echocardiography is a mainstay of clinical evaluation of aortic valve disease2. Echocardiographic imaging techniques are non-injurious, which facilitates longitudinal studies. These techniques can readily be performed in minimally sedated mice, avoiding the risks and physiologic perturbations associated with general anesthesia.
Continuous- and pulse-wave Doppler evaluation of blood velocities are useful for estimation of transvalvular pressure gradients and valve areas (see Figure 2). This approach has been useful for quantitation of aortic valve function across a very broad range, from normal 15 to “sclerotic”16 to severely stenotic11.
Figure 2. Assessment of aortic valve function in mice.
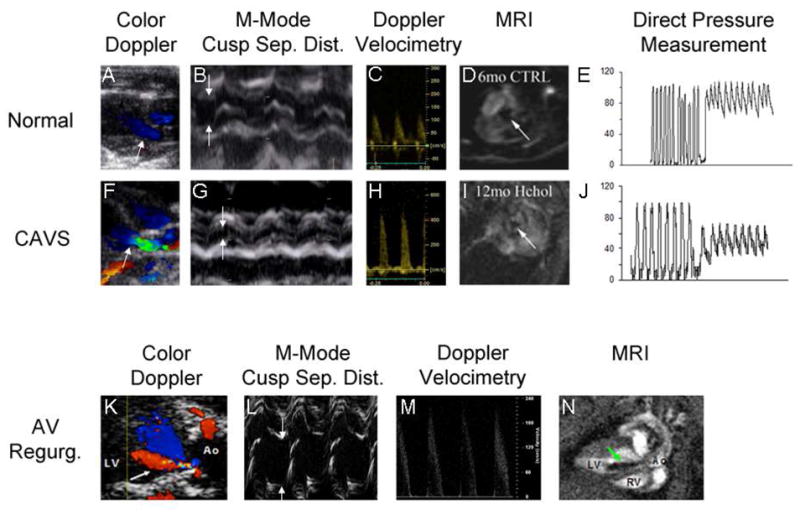
Two-dimensional color Doppler images (Panels A,F,K) are used to target M-mode imaging of the aortic valve and Doppler velocimetry, and to assess the presence (arrow, panel K) or absence of aortic regurgitation. Direction of blood flow is indicated by pseudocolors (red = blood flow towards probe, blue = blood flow away from probe). In CAVS, the irregular stenotic valve orifice causes flow acceleration and turbulence (green). The superior spatial and temporal resolution of M-mode echocardiography facilitates quantitation of systolic cusp separation (arrows, panels B,G,L). Doppler velocimetry (Panels C, H, M) allows for estimation of the transvalvular systolic pressure gradient via the Bernoulli Equation. Aortic regurgitation can cause a modest transvalvular systolic pressure gradient even in the absence of reductions in cusp separation (Panel L, green arrow points to regurgitant jet), by virtue of pre-load dependent increases in contractile force and stroke volume and subsequent increases in velocity (Panel M). Magnetic resonance imaging provides temporal and spatial resolution sufficient to portray the aortic valve orifice in two dimensions (arrows, Panels D,I), by virtue of magnetic dephasing of ejected blood. The same principle is employed to depict retrograde flow when aortic regurgitation is present (arrow, Panel N, cine images can be viewed in the Supplemental Movie). Direct pressure measurements can provide incontrovertible evidence of a transvalvular gradient (Panels E and J), but are influenced by the potential for cardiodepression and vasodilation caused by deep general anesthesia, especially in the presence of CAVS. CAVS calcific aortic stenosis; AV Regurg. aortic valve regurgitation; Sep. Dist. separation distance; MRI magnetic resonance imaging.
This approach also has some disadvantages. It is not always possible to register the line of Doppler interrogation parallel to the direction of blood flow, which can result in underestimation of valve gradients. The region of interrogation needs to be small, to avoid contamination of velocity profiles by adjacent tissue motion, especially during active respiration, which challenges the limits of commercially available Doppler equipment. The requirement for continuous-wave Doppler limits the choice of transducers to those that image at frequencies of < 12 MHz, when commercially available equipment is used, resulting in suboptimal visualization of valve structures. Doppler velocities can be affected by factors other than effective valve orifice area. Reduced left ventricular contractility, for example, can result in underestimation of the severity of aortic stenosis in humans2, and probably in mice. When alterations in valve tissue produce valvular regurgitation, the increased stroke volume and ventricular preload recruitment may increase transvalvular gradients, even in the absence of valve stenosis15(Figure 2). As in clinical studies, those findings invoke a note of caution when Doppler velocities alone are used as evidence of aortic valve stenosis.
The superior spatial and temporal resolution of M-mode echocardiography are useful for quantitative assessments of aortic valve function12–14, which correlate well with invasive hemodynamic measurements14 (Figure 2).
M-mode echocardiography has several advantages for assessment of aortic valve function in mice. Images are readily obtained from parasternal short- and long-axis views, and do not require co-registration with a Doppler line of interrogation. Variance of valve orifice dimensions is relatively low, achieving statistical power in between-group comparisons, with manageable sample sizes12. M-mode derived valve orifice dimension, and by extrapolation valve area, are not appreciably affected by left ventricular contractility or the presence of aortic regurgitation.
Disadvantages of M-mode echo techniques arise from reliance on a unidimensional measurement to portray valve function. Thus, in the presence of eccentric valve remodeling, e.g. partial or complete cusp fusion, M-mode methods are susceptible to both under- and overestimation of the severity of valve dysfunction.
2-D echocardiography using clinical equipment does not reliably provide sufficient spatial resolution for visualization of valve motion in normal mice. A newer generation of ultrasound devices, developed solely for use in small experimental animals, employs transducers capable of imaging at very high frequencies. These new devices hold promise for quantitative assessment of valve function in 2-D17.
2-D echocardiography is a powerful technique for characterization of the impact of aortic valve disease upon left ventricular structure and function14, 17. The presence and severity of left ventricular hypertrophy and systolic dysfunction can be ascertained rapidly and reproducibly in conscious minimally sedated mice, in longitudinal studies.
Magnetic Resonance Imaging
Magnetic Resonance Imaging (MRI) provides useful information about aortic valve function in patients18. In mice, MRI at field strengths ≥ 4.7T provides sufficient spatial and temporal resolution to assess aortic valve function in two dimensions12 (see Figure 2). Flow turbulence causes “dephasing” of the blood signal, which facilitates visualization of aortic regurgitation 19 (Figure 2). MRI affords the added benefit of precise quantitative assessment of the structure and function of both the right and left ventricles and, consequently, precise measurement of regurgitant volume in mice with aortic regurgitation20.
The advantages of MRI are balanced by distinct disadvantages, notably limited availability. Relatively long imaging times, on the order of 20 minutes per study, require deep sedation or general anesthesia, which places mice with severe aortic valve disease at a higher mortality risk21.
Invasive hemodynamic techniques
In clinical studies of valvular and left ventricular function, invasive hemodynamic techniques have been employed as a “gold standard”2. In mice, microtransducer-tipped catheters provide high-fidelity assessments, by virtue of sampling rates on the order of 1000 Hz. The small caliber, e.g. 1.4Fr (MillarR, Houston, TX), allows retrograde introduction into the left ventricle via the carotid artery11, 14 (see representative traces in Figure 2). Advantages of invasive techniques include precise ascertainment of transvalvular gradients, and left ventricular systolic and diastolic function 22. As is the case in the clinical setting, invasive hemodynamic techniques in mice can serve as a validation standard for more convenient non-invasive methods11, 14.
Disadvantages of invasive methods include the need for arterial access, risking blood loss and rendering longitudinal studies very difficult. General anesthesia can result in cardiac depression, especially in mice with severe aortic valve disease – resulting in discordance of findings obtained by invasive studies and those acquired by echocardiography in minimally sedated mice11, 14. Catheter-induced valve trauma is also likely to introduce artefactual cellular and molecular changes in valve tissue.
Summary of findings from studies of aortic valve function in mice
In adult C57BL/6 mice, the systolic aortic valve dimension is approximately 1.2 mm14. Assuming that the orifice is roughly circular, anatomic estimates of normal adult aortic valve area from various strains of mice are about 0.8 – 1.3 mm2. Estimates of normal aortic valve area are somewhat higher (~1.60 mm2), when Doppler methods are employed11.
Normal peak systolic velocity of blood flow across the aortic valve in mice is < 1.5 m/sec11, 15–17, predicting peak transvalvular gradients of < 10 mmHg, findings corroborated by invasive hemodynamic studies11, 14. Reduction of systolic aortic valve dimension by > 50%, corresponding to reduction of valve area by > 75%, is sufficient to induce hemodynamically important transvalvular pressure gradients of > 50 mmHg, a finding that recapitulates seminal findings in humans with aortic valve disease2. Hemodynamically significant aortic valve stenosis causes left ventricular hypertrophy and reduced systolic function in mice11, 14.
Aortic transvalvular systolic gradients are increased in mice with aortic valve regurgitation22. Thus, it is advantageous to evaluate valvular function with at least two imaging techniques (e.g., cusp separation distance by m-mode echocardiography and transvalvular velocity with Doppler), in addition to determining whether there is aortic valve regurgitation (e.g., with color Doppler imaging or MRI imaging). This is particularly important when evaluating the myocardial consequences of CAVS, as mice with moderate or severe aortic valve regurgitation develop left ventricular hypertrophy, bi-ventricular enlargement, and decreased systolic function of both ventricles, even in the absence of aortic stenosis20.
These findings were culled from studies in relatively few mice, and may not account for differences between “normal” mouse strains or differences between sexes. Thus, they may be useful for planning future studies, but do not supplant the need to address possible strain or sex differences in the development of CAVS.
Noninvasive imaging techniques are useful for longitudinal studies to characterize the evolution of aortic valve dysfunction, and responses to therapeutic interventions. Characterization of events at the cellular and molecular levels, however, generally require studies of tissue ex vivo or cells in vitro, which greatly increases the complexity of long term investigations. Development of multimodality imaging methods suitable for long-term, serial imaging studies of the aortic valve (similar to what has been accomplished in blood vessels, where movement artifact and sampling rate are less 23) will undoubtedly provide significant insight into mechanisms contributing to the development of aortic valve stenosis and biological responses to therapeutic interventions.
Assessment of histological, structural, and biological changes in mouse aortic valves
Histological changes
Histological examination of the aortic valve is useful to quantify calcium deposition in sections of the valve. Staining with alizarin red is preferable to von Kossa, not only because of its specificity for calcium, but also because mice with a C57BL/6 background often have artefactual deposits of black pigment (perhaps lipofuscin) in the aortic valve that resemble the black stain of calcium with von Kossa24. Masson’s trichrome stain and picrosirius red staining are useful for detection of gross changes in collagen12, 25–27, and Movat’s pentachrome staining is useful for evaluation of changes in content of collagen, elastin, and proteoglycans28 Oil red O is commonly used for assessing lipid deposition in the valve12, 13, 24. It is important to evaluate histological changes not only in the cusps of the valve, but also at the attachment points of the valve cusps (where calcification often begins).
Gene expression, protein levels, and enzyme activity
In studies of aortic valve from humans, the relatively large amount of tissue facilitates evaluation of DNA (e.g., genome sequencing), mRNA (e.g., using quantitative real-time RT-PCR), and protein (e.g., western blots, ChIP assays, etc.), often from the same patient or sample.
In mice, the amount of tissue in aortic valve from one mouse is sufficient for measurement of gene expression with quantitative real-time RT-PCR29–31. To examine changes in protein levels during various stages of valve disease, immunohistochemistry is useful 12, 13, 15, 30 but is limited because it is semi-quantitative. High levels of tissue autofluorescence in calcified tissue require careful correction for background fluorescence with adjacent sections.
Although valve tissue could be pooled from a cohort of animals to use in more quantitative assays (e.g., Western blotting), the amount of time required to generate animals with hemodynamically significant CAVS (9–12 months or longer) and number of animals required for pooling (>5) make it logistically and financially difficult to use such techniques.
Evaluation of enzymatic activity in mouse valve tissue is extremely challenging when isolated protein is required (for the sample size limitations listed above). Indirect assays of enzyme activity are frequently used in frozen histological sections. For example, we have used PEG-superoxide dismutase-inhibitable fractions of dihydroethidium to evaluate superoxide levels in mouse valves12, 13, and similar approaches could be used with enzymatic inhibitors (e.g., oxidase inhibitors, etc.). Recent development of high-sensitivity chemiluminescent compounds (e.g., L-012) have been used to measure superoxide levels in mouse basilar arteries32, providing hope for a more quantitative assay for use on micro-samples.
Finally, the emerging field of molecular imaging may be useful for valvular and vascular biology. Of particular interest are compounds that emit fluorescence after they are cleaved by specific enzymes. These molecules have been used to demonstrate that MMP activity19, cathepsin activity33, inflammatory cell infiltrate34, and osteoblast-like cell activity19, 33, 34 are substantially increased in aortic valves from hypercholesterolemic mice. These compounds are available with different excitation/emission wavelengths, making them a powerful tool to understand valvular biology when they are combined with each other or with standard fluorescent immunohistochemical methods.
Limitations and future directions
Limitations
One major advantage of studying CAVS in mice is that they are the only species, other than humans, that have been shown to develop hemodynamically important stenosis11–14. Other advantages, like other studies in mice, are that genetic alterations are readily available, and new strains and colonies can be expanded rapidly. Perhaps more importantly, the relatively short lifespan of the mouse makes it an attractive model for the study of time and/or age-dependent diseases such as CAVS. There are, however, significant limitations associated with using mouse models of CAVS.
The major disadvantages of studying CAVS in mice relate largely to their size. The hemodynamic evaluation of severity of stenosis has been challenging. However, echocardiographic evaluation of severity of aortic valve stenosis in mice has been refined, and high resolution imaging systems are commercially available. Direct measurements of transvalvular pressures have been used to validate the use of echocardiographic measurements of cusp separation distance, which correlate well with peak transvalvular blood velocity (in the absence of aortic valve regurgitation)11, 14.
Histological assessment of valvular structure and calcium deposition has inherent limitations. A great limitation lies in the methods required to reconstruct serial sections into a three-dimensional image that can be quantitated. Although advances in quantitative stereology and associated software packages have made some advances possible35, 36, it is still extremely challenging to accurately quantitate valvular collagen, cellular composition, and other variables. Accurate quantitation of valve structure from 3-dimensional confocal/multiphoton images from intact tissues is difficult due to image distortion, secondary to differing refractive indices37, 38, which also vary at different excitation/emission wavelengths (making image registration across wavelengths and imaging modalities challenging).
Quantitative analysis of protein levels in valves from mice is challenging. Semi-quantitative analyses of immunohistochemically stained tissues are the predominant tool to evaluate changes in protein levels and post-translational modifications. Fluorescent immunohistochemical techniques provide the distinct advantage of analyzing spatial distribution of changes in protein levels (e.g., base versus tip of valve) and double- or triple-staining methods allow for evaluation of co-expression/co-localization of specific molecules. Pooling of tissue from multiple animals for Western blotting is theoretically feasible for young animals/cohorts, but routine quantitative analysis of proteins in valves from mice with severe CAVS will require the refinement of microdissection, micropurification, and microanalysis techniques.
Perhaps the greatest challenge in using mice is examination of molecular mechanisms underlying CAVS. The small size of the valve cusps and base, and limited amount of tissue, makes isolation of pure tissue and examination of gene expression (by qRT-PCR) challenging. Yields of RNA are sufficient for use in such applications, however, when combined with high fidelity, high efficiency RT enzymes.
Future directions
The resolution available with echocardiographic imaging of the aortic valve is likely to continue to improve, and provide greater accuracy and precision to analysis of aortic valve structure and function. Improvements in imaging systems, however, will further our understanding of CAVS only if measurements are taken under reasonably physiologically meaningful conditions (e.g., heart rate >500, EF > 75%). Furthermore, great care must be taken to evaluate not only the severity of aortic valve stenosis, but also the presence and severity of aortic valve regurgitation, which can alter the transvalvular systolic gradient.
Advances have been made in micro computed tomography imaging, which does not have distortion or registration issues associated with laser-/fluorescence-based, three-dimensional microscopy techniques. Thus, in complex structures such as the aortic valve, three-dimensional micro-CT imaging may prove to be useful for understanding the spatial distribution of calcium deposition during progression of CAVS.
Several echocardiographic and MRI systems can combine high resolution imaging with molecular probes targeting surface molecules of cells. Multimodality imaging methods suitable for long-term, serial imaging studies have already been applied to the study of atherosclerosis in mice23, 39–41 and aortic valve sclerosis in rabbits42, and will undoubtedly provide significant insight into mechanisms contributing to the development of aortic valve stenosis and biological responses to therapeutic interventions.
Pro-calcific and anti-calcific signaling during the progression of CAVS
Activation of pro-osteogenic signaling cascades is thought to be a central mechanism contributing to the initiation and progression of calcific aortic valve stenosis. Osteogenic signaling cascades, especially bone morphogenetic protein and Wnt/β-catenin signaling, are activated in calcifying valves. Because other papers in this series will focus on these signaling cascades in detail, we will discuss them only briefly.
Bone morphogenetic protein (BMP) signaling
Increased levels of phospho-smad1/5/8, a hallmark of canonical BMP signaling, occur in stenotic valves43. Mechanisms that contribute to increased BMP elaboration are not clear, but recent data suggest that non-laminar flow patterns on the aortic side of the valve may be a key initiator of BMP2/4 secretion from the valvular endothelium44–46. In hypercholesterolemic mice, phospho-smad1/5/8 levels increase prior to reduction of valve opening, and increase further as valvular calcification progresses and valve function becomes impaired12, 13. Tonic suppression of BMP signaling by inhibitory Smads is important in preventing cardiovascular calcification, as Smad6-null mice develop cardiovascular calcification and have evidence of aortic ossification at only 2 weeks of age47.
Wnt/β-catenin signaling
Increases in levels of low-density lipoprotein receptor-related protein 5 (Lrp5) and associated increases in nuclear accumulation of β-catenin have been reported in valves from humans with CAVS48. β-catenin immunofluorescence also increases in calcified valves from hypercholesterolemic rabbits49 and in hypercholesterolemic mice with advanced CAVS13.
TGF-β signaling and calcification
The role of TGF-β in the initiation and progression of aortic valve calcification is not clear. Data from valvular interstitial cells plated directly on plastic or glass culture dishes show convincingly that TGF-β1 induces cell apoptosis, cellular aggregation, and calcified nodule formation50–57, but administration of TGF-β1 to cells plated on a less stiff collagen matrix does not induce osteogenic differentiation and calcification58. Furthermore, there is a clear dissociation between TGF-β signaling and osteogenic protein levels with lipid lowering in vivo13, which suggests that TGF-β is not a primary inducer of pro-osteogenic signaling in hypercholesterolemic mice with advanced valve disease. Although some data suggest that TGFβ may actively suppress pro-osteogenic signaling in skeletal osteoblasts in vivo59, the role of TGF-β in suppression or activation of osteogenic signaling in vivo is not clear.
Identifying the origin of cells that redifferentiate to osteoblast-like cells
The conventional wisdom is that osteogenesis in stenotic valves results from activation of maladaptive signaling, which drives the redifferentiation of resident valvular interstitial cells to an osteoblast-like phenotype60 (Figure 3). There are, however, several lines of evidence which suggest that cells other than the resident valvular interstitial cell can contribute to osteogenesis in the valve. First, a subset of valvular endothelial cells appears to undergo endothelial-mesenchymal transformation61, 62, which may provide a sub-population of cells with a propensity for activation and calcification (Figure 3). Second, circulating progenitor cells may contribute to vascular and valvular calcification by either redifferentiating to an osteoblast-like cell or by promoting interstitial cell calcification through paracrine signaling63–66 (Figure 3). Studies from lethally-irradiated mice, which undergo green fluorescent protein marrow transplantation, suggest that ~15% or more of the valvular cell population (endothelium, α-smooth muscle actin positive cells, and inflammatory cell infiltrate) may be comprised of cells that originated from bone marrow 15, 67.
Figure 3. Potential origins of cells that contribute to valvular calcification and fibrosis.

Possible origin of osteoblast-like and osteoclast-like cells in aortic valves in human and murine CAVS. Activated myofibroblasts are likely to come from either quiescent valvular interstitial cells (VICs) or from a sub-population of endothelial cells that undergo endothelial to mesenchymal transformation (EMT). Osteoclast-like cells may originate from circulating monocytes. (Illustration Credit: Cosmocyte/Ben Smith).
Endogenous mechanisms that may modulate pro-calcific signaling in CAVS
Reactive oxygen species
Reactive oxygen species (ROS) appear to be a central pathophysiological component of a number of cardiovascular diseases, including atherosclerosis68–70, hypertension71–73, and thrombosis74–77, and can originate from a number of enzymatic sources 68, 76. Superoxide and hydrogen peroxide are significantly increased in the calcified and peri-calcific regions of stenotic aortic valves 78. Uncoupled nitric oxide synthase and reductions in antioxidant enzyme expression and activity appear to be major contributors to increased ROS in stenotic valves78. Although increases in global NAD(P)H oxidase activity do not appear to be major contributors to increased ROS in stenotic valves78, ROS derived from p47phox-dependent oxidases may be generated in peri-calcified microenvironments79.
There are several lines of evidence supporting the concept that ROS play an important role in progression of CAVS (Figure 4). First, ROS are increased prior to valve dysfunction in mice, which suggests that increased ROS are not merely the consequence of increased cusp stress associated with valve calcification 12. Second, ROS have been implicated as a critical link in the transduction of pro-osteogenic and pro-fibrotic signaling cascades (see sections on TGF-β signaling 80 and Figure 4). Third, addition of exogenous ROS accelerates calcification of VSMC’s in vitro(81, 82). Finally, administration of lipoic acid (which reduces superoxide and H2O2), but not tempol (which only reduces superoxide), attenuates calcification in rabbit model of valvular sclerosis79.
Figure 4.

Mechanisms whereby reactive oxygen species (ROS) may modulate pro-calcific and pro-fibrotic signaling in CAVS. Nox4-derived ROS may play an obligatory role in TGFβ signaling and induction of fibrosis. In contrast, ROS may play a modulatory role in promoting aortic valve calcification.
Although we have a general understanding of ROS that are increased and general enzymatic sources of ROS in stenotic valves, there are major gaps in our understanding of the role of ROS in valvular calcification. Specifically, it is not known which nitric oxide synthase isoforms and/or NAD(P)H oxidase isoforms contribute to increased generation of ROS, nor do we know the relative contributions of different antioxidant mechanisms (e.g., catalase, superoxide dismutases, and peroxidases) in their respective subcellular compartments. Genetically altered mice will allow elucidation of the role of specific ROS-related enzymes in the pathogenesis of CAVS.
Nitric oxide bioavailability
Reduction of nitric oxide bioavailability is strongly associated with a number of cardiovascular diseases, and NO bioavailability is often inversely correlated with increases in ROS. Expression of endothelial nitric oxide synthase is increased on the aortic side of the valve in early stages of valve disease83, and increased eNOS immunofluorescence is evident in neovessels in advanced stages of valve disease84, 85. One might anticipate that increased expression of eNOS would protect against CAVS and suppress interstitial cell proliferation (Figure 4). Overexpression of eNOS in hypercholesterolemic mice, however, accelerates atherosclerosis due to NOS uncoupling86, and NOS uncoupling may contribute to increases in reactive oxygen species in human CAVS 78.
Nevertheless, there are several observations which together suggest that increases in NO bioavailability may be a useful strategy to slow progression of aortic valve calcification (see Figure 4). First, endogenous inhibitors of nitric oxide synthase—such as asymmetric dimethylarginine—are significantly increased in patients with CAVS87, 88. Second, addition of exogenous NO slows calcium nodule formation in valve interstitial cells in vitro51. Third, administration of statins to hypercholesterolemic rabbits is associated with robust increases in eNOS levels89 and attenuation of valvular calcium deposition. Finally, administration of NOS cofactors slows progression of atherosclerosis in mice90.
Renin-angiotensin system (RAS)
Several findings suggest that angiotensin II may lead to oxidative stress, inflammation, and accelerated development of CAVS. First, hypertension, which often involves the RAS, is a risk factor for vascular calcification91 and CAVS1. Second, monocytes92 and macrophages in atherosclerotic lesions93 contain Ang II, and there are many macrophages in stenotic aortic valves.12, 94, 95 Third, expression of angiotensin-converting enzyme (ACE) and angiotensin type I receptors (AT1r) is increased in stenotic valves, and co-localizes with macrophages and mast cells primarily in peri-calcific regions of stenotic valves95. Fourth, Ang II promotes oxidative stress and inflammation96–98, which are associated with CAVS.
In animal models of atherosclerosis and hyperlipidemia-induced valve disease, AT1R blockade prevented inflammatory cell infiltration and myofibroblast activation in early stages of valve disease99. A retrospective clinical study also suggested that an AT1R blocker, but not an ACE inhibitor, protected against progression of valve disease in early (but not late) stages of CAVS100. Other retrospective clinical studies, however, have yielded conflicting results with regards to the effects of ACE inhibitors on calcium and progression of CAVS101, 102. The efficacy of ACE inhibitors in reducing Ang II levels in patients with CAVS may be limited by high levels of chymase (which can convert Ang I to Ang II) in human valves95. Thus, although there is a strong biological rationale that implicates the RAS in progression of CAVS, the lack of prospective, experimental data (in studies in animals or humans) prevents a firm conclusion.
RANK/RANKL/OPG
There is a complex interaction between receptor activator of NFκB (RANK), RANK ligand (RANKL), and osteoprotegerin (OPG) in relation to oxidative stress and inflammation, through effects on NFκB103, 104. This interaction has important consequences for calcification of bone, arteries, and perhaps the aortic valve.
Increased RANK activation/RANKL levels may influence cardiovascular calcification through effects on both circulating cells and resident cells. RANKL elaborated from calcifying vascular smooth muscle cells is Runx2-dependent and sufficient to induce monocyte recruitment and osteoclast-differentiation in calcifying atherosclerotic lesions in mice105. This mechanism may modulate intimal plaque calcification. This RANKL-stimulated osteoclast differentiation may be mediated in part by ROS, and RANKL in turn induces further ROS generation106, 107. In contrast, RANKL increases calcification of vascular smooth muscle cells, perhaps through a BMP4 pathway108. Finally, OPG (which is an endogenous decoy receptor for RANKL, and inhibits inflammation and the pro-osteogenic pathway) inhibits aortic calcification in OPG−/−mice109.
Based on seminal studies in blood vessels110, 111, we speculate that OPG may inhibit calcification of the aortic valve. First, RANKL is greater in stenotic than normal aortic valves from humans112, 113, and promotes calcification of myofibroblasts in vitro112. Second, calcification of atherosclerotic lesions in innominate artery is accelerated in apoE-deficient mice that are OPG-deficient.110 Third, injections of OPG prevented calcification of the aorta in ldlr−/− mice but had no impact on extent of total atherosclerosis. This finding suggests that intimal calcification can be dissociated from lipid deposition and tissue fibrosis in atherosclerosis111. It is not known whether similar phenomena occur in calcifying valves.
Peroxisome proliferator-activated receptor gamma (PPARγ)
PPARγ is a member of the nuclear hormone receptor superfamily of ligand-dependent transcription factors114, 115. Several findings imply that PPAR may protect against CAVS. First, increasing PPARγ impairs differentiation of progenitor cells and calcifying vascular cells into an osteoblast-like lineage in vitro116. Second, inhibition of PPARγ (pharmacologically or with siRNA) increases differentiation of embryonic stem cells to osteoblasts117. Third, PPARγ ligands promote antioxidant and anti-inflammatory gene expression profiles118.
The role of PPARγ in valve calcification is not known. Interestingly, PPARγ-related pathways, however, are increased in early stages of hypercholesterolemia-induced valve disease, which may contribute to protection of the endothelium83.
Thus, PPARγ regulates expression of genes that modulate expression of osteoblasts, are antioxidant and antiinflammatory, and may thereby protect against CAVS. Multiple signaling pathways appear to be important in the pathophysiology of CAVS. Activation of PPARγ is attractive as a potential treatment for CAVS because, instead of targeting a single mechanism, PPARγ affects a large clusters of genes119, and thus may protect the valve through multiple pathways.
Direct inhibitors of osteogenic signaling
Downregulation of inhibitors of osteogenic signaling have been implicated in the progression of CAVS. Matrix Gla protein (MGP) binds bone morphogenetic proteins, rendering them inactive120, and enhances fetuin-dependent uptake of mineralizing matrix vesicles121. In the setting of cardiovascular calcification, however, it appears that MGP may be inactivated by undercarboxylation or physical interactions with inflammatory proteins (e.g., HSP70120). A retrospective clinical study suggested that warfarin use, which inhibits gamma-carboxylase (thereby impairing MGP), is a risk factor for progression of valve disease in patients with early CAVS100. MGP-deficient mice develop massive medial vascular calcification and aortic valve calcification early in life122, and hypercholesterlemic MGP-transgenic mice are protected against cardiovascular calcification123. Thus, inactivation of MGP may be a key permissive event in initiation of osteogenic signaling in cardiovascular tissue.
Notch signaling
Loss-of-function polymorphisms in Notch1 are strongly associated with development and early calcification of bicuspid aortic valves in humans124. Mice that are haploinsufficient in Notch1 do not develop bicuspid aortic valves, but develop calcific aortic valve disease due to the derepression of BMP2 expression125, 126. The mice, however, do not develop significant abnormalities in valve cusp function/aortic valve stenosis125, 126. An intriguing possibility is that reductions in Notch1 may play a dual role in CAVS, being permissive for both BMP2/4 elaboration in the endothelium and for osteogenic differentiation in interstitial cells127. Interestingly, Notch1 activation induces Msx2-dependent osteogenic differentiation in vascular smooth muscle cells128, which implies that effects of Notch1 activation are highly context dependent.
Matrix metalloproteinases (MMP), cathepsins, and valvular calcification
MMP-1129–131, MMP-2132, MMP-3133, MMP-9134, and cathepsins S135, K135, V135, and G136 are increased in stenotic human valves. The functional significance of alterations in MMPs in valve calcification remains unclear, although matrix remodeling is likely to play an important role in permitting the expansion of calcified plaques and in the generation of pro-inflammatory collagen fragments137, 138. Elastin fragments produced by active cathepsin S are a major contributor to valvular and vascular calcification in hypercholesterolemic mice with chronic renal failure33. Genetically altered mice will be useful in determining whether inhibitors of MMPs or cathepsins are a viable therapeutic target to slow the progression of CAVS.
Pro-inflammatory cytokines
Inflammatory cell infiltrate and production of pro-inflammatory cytokines are markedly elevated in valves from both humans and mice with CAVS (Figure 1). Effects of pro- and anti-inflammatory cytokines on valve biology have not been thoroughly examined, but two lines of evidence suggest that TNFα may be a critical downstream mediator of inflammation-induced calcification. First, mice that are deficient in interleukin-1 receptor antagonist (IL-1rn) have pronounced valve thickening, calcification, and modest sclerosis/stenosis (peak velocity = ~2m/sec); this valvular phenotype is abrogated in IL-1rn/TNFα double knockout mice139. Second, TNF-α appears to be a critical intermediary in the induction of vascular calcification and MMP activation in diabetic mice, as administration of infliximab (a TNF-α neutralizing antibody) inhibits BMP2-Msx-Wnt signaling in aorta140. We speculate that isoform-specific receptor blockers may be useful when targeting TNF-α signaling, because activation of TNF-α receptor 1a is responsible for many of the deleterious effects of TNF-α, and activation of TNF-α receptor 1b may confer some beneficial/protective effects141.
Activation of receptors of advanced glycosylation end products (RAGE) can accelerate VSMC calcification both in vitro142–145 and in vivo142, 146, 147. The contribution of this pathway to development of CAVS has not been tested experimentally, but several observations in patients suggest that RAGE activation may contribute to progression of CAVS. First, metabolic syndrome and diabetes are risk factors for development of CAVS148, and such patients have marked increases in plasma and tissue AGE levels149. Second, circulating soluble RAGEs, which prevent AGEs from binding to tissue RAGEs, are reduced in patients with CAVS150. Thus, reducing AGE levels and RAGE activation may prove to be useful in slowing progression of CAVS in some patients.
Is valvular calcification always an osteogenic process?
Recent studies of mechanisms that contribute to CAVS have assumed that ectopic calcification is primarily or exclusively an active process resembling processes observed in bone. The relative importance of true “ectopic osteogenesis”, however, is not entirely clear in humans or in mice with CAVS. In our opinion, there are several potential mechanisms whereby calcium nodules may initiate or expand in CAVS (Figure 5).
Figure 5.

Potential pathways contributing to calcified nodule formation in CAVS. A) recapitulation of classical skeletal osteogenesis, in which osteoblast and osteoclast cells respond to exogenous stressors (such as oxidative stress) in a manner similar to that found in bone-derived osteoblasts. B) formation of amorphous calcific nodules without a requirement for osteoblast-like cells, in which stressors initiate cellular aggregation, apoptosis or necrosis, and nodule formation. C) “pseudo-skeletal” ossification, in which cells expressing a subset of osteoblast or osteoclast genes are present in the aortic valve, but respond to exogenous stimuli in fundamentally different ways. For example, previous studies in vitro have shown that—unlike skeletal osteoblasts—cells from cardiovascular tissue typically increase their osteogenic potential in response to exogenous oxidative stress. Bone matrix, replete with marrow hematopoietic elements, has been identified in aortic valves of some patients with CAVS. It is not clear whether this requires processes identical to skeletal osteogenesis (panel A), or if similar structures can be formed by osteoblast-like cells (panel C).
First, valvular calcification may progress by a process that parallels bone. Approximately 15–20% of valve cusps from patients with CAVS have evidence of bone matrix, including osteoid cells, highly organized collagen scaffolds, multi-nucleated osteoclast-like cells, and marrow pockets151. To date, similar structures have not been described in murine CAVS.
A second possible mechanism of valvular calcium accumulation is accumulation of amorphous calcium deposits. Calcified nodules of this type typically have a crystalline ultrastructure, and lack live cells within the core of the calcified mass itself151, 152. Cellular necrosis and apoptosis are classical mechanisms of nodule formation and expansion of amorphous calcium 152–154. In vitro, TGFβ induces caspase-dependent apoptosis and formation of calcified nodules, and TGFβ is markedly increased in valves from humans with CAVS52, 55. Although mechanisms of TGFβ-induced calcification are highly substrate/matrix sensitive in vitro57, these data support the concept that formation of osteoid cells is not always a primary event in initiation or expansion of calcified nodules. It is important to note that, although accumulation of calcium may not occur via a process that resembles skeletal ossification, initiation (and perhaps progression) of calcium deposition via this mechanism may occur via: 1) tightly regulated “active” processes, such as caspase-dependent apoptosis, or 2) “passive” processes, via accumulation of calcium secondary to tissue necrosis. The relative contributions of both pathways to valve calcification remains poorly understood, and both may prove to be therapeutic targets for patients with CAVS.
A third mechanism may lead to valvular calcification in humans and mice. Active mineralization of valvular tissues may occur by cells that express a subset of osteogenic genes12, 13, 19, 155, but is regulated by processes that are fundamentally different from skeletal ossification. In vitro, calcifying cells of cardiovascular origin respond to several external stimuli in a manner that is fundamentally different from skeletal osteoblasts82. As mentioned above, cells expressing osteogenic markers frequently are found near calcified areas in both humans and mice12, 13, 19, 78, 151, although the functionality (and malleability of their function) has yet to be determined experimentally.
In summary, it is not clear which signaling cascades are responsible for initiation and progression of aortic valve calcification in vivo, or which mechanisms predominate in CAVS in humans or mice. These questions deserve attention, and will ultimately be addressed through careful histomorphometric studies which examine the cellularity, ultrastructure, composition, and molecular fingerprint of calcified nodules in human and murine CAVS.
Role of fibrosis in CAVS
The conventional wisdom is that calcification is the major determinant of stenosis in CAVS. Acquired fusion of valve cusps also may contribute to stenosis, but commissural fusion is not common in CAVS, in contrast to rheumatic and congential aortic stenosis. We suggest that fibrosis of the valve, as well as calcification, may contribute importantly to CAVS (see Figure 6).
Figure 6.

Progression and “regression” of CAVS in “Reversa” mice12, 13. Early stages of CAVS in mice involve myofibroblast activation and lipid insudation/foam cell formation, and are followed by the appearance of osteoblast-like cells, valvular calcification, and substantial increases in valvular fibrosis. Following reduction of blood lipids (“regression” in right panel), there are substantial reductions in valvular lipid content and calcium content, but valvular fibrosis remains increased. Despite reduction of valvular lipid and calcium content, aortic valve function does not improve with substantial lipid lowering.
There is extensive fibrosis of the aortic valve in humans30 and mice12 with CAVS. Extracellular matrix synthesis (ECM) can originate from activated myofibroblasts (i.e., alpha-smooth muscle actin positive cells in the valve). Myofibroblast activation occurs early in the development of aortic valve disease12, and myofibroblasts may actively secrete collagen156, hyaluronan157, 158, and other ECM components during development and progression of CAVS. ECM composition and stiffness may have a profound impact on the phenotype of valve interstitial cells, and ECM may contribute to differentiation of cells to an osteoblast-like phenotype57, 58, 159–161. If additional studies continue to implicate fibrosis in the pathogenesis of CAVS, it may be more accurate to use the term fibrocalcific aortic valve stenosis.
TGF-β and fibrosis
TGFβ is an antiinflammatory and profibrotic cytokine162, 163. TGFβ plays a critical role in fibrosis of the myocardium after injury, and also may “stabilize” atherosclerotic plaques in arteries, by its antiinflammatory and profibrotic effects. TGFβ signaling and myofibroblast activation are markedly increased during development of CAVS in mice12, 13 and in valves from patients with severe CAVS55, making it an attractive candidate as a primary driver of fibrosis in CAVS.
Twist1
Re-activation of developmental gene expression programs may occur in the stenotic valve. Specifically, increases in Twist1, which is essential for normal endocardial cushion development and remodeling, have been reported in the peri-calcific regions of stenotic aortic valves30. Overexpression of Twist1 in mice produces valvular hypercellularity and excessive cusp fibrosis, which suggests that Twist1 may contribute to valvular fibrosis and interstitial cell proliferation in advanced CAVS30.
Aging, valvular calcification, and valvular fibrosis: failure of multiple regulatory mechanisms?
Increasing age is one of the strongest predictors of cardiovascular calcification 164 and the development of aortic valve stenosis165. Several regulatory mechanisms may have a profound effect on lifespan, genomic stability, and age-related diseases. We will discuss a few areas of research in aging that hold promise for advancing our understanding of cardiovascular calcification during aging.
Progeric humans and mice
Patients with progeroid syndromes (e.g., Hutchinson-Gilford syndrome or Werner’s syndrome) have increased prevalence of severe aortic valve calcification and stenosis166–170. Non-progeroid humans with atherosclerotic lesions accumulate prelamin A (whose expression is increased in some forms of progeria) in areas close to senescent or calcifying smooth muscle cells171. Mice that overexpress progerin develop robust vascular calcification at a young age172. We speculate that characterizing changes in expression of these molecules in stenotic human valves, and the valvular phenotypes of progeric mouse models, will provide important insights mechanisms that contribute to CAVS.
Other mouse models of aging and progeria develop cardiovascular calcification. For example, klotho-deficient mice develop premature vascular calcification173. These mice, however, also develop calcification of the gut and other soft tissues, which are not typically associated with normal aging173, 174.
Post-transcriptional regulation of gene expression in CAVS
An emerging field of study is the role of micro-RNA in the regulation of mRNA stability and translation. One microRNA may target many, perhaps hundreds, of mRNAs175, predominantly by destabilization of target mRNAs resulting in their subsequent degradation176. MicroRNA expression is dramatically altered in numerous tissues with aging177. A recent report describing microRNAs in cardiac valves suggested that several microRNAs are decreased in stenotic bicuspid aortic valves, compared to insufficient valves, and may modulate mRNA levels of several pro-calcific genes178. Downregulation of micro-RNAs modulates development of fibrosis in myocardium179, but the role of micro-RNAs in the regulation of cardiac valve fibrosis is not known.
Epigenetic modifications
Changes in acetylation levels of transcription factors and histones are a critical determinant of availability and affinity of transcription factor binding sites, result from perturbations in the balance between acetyltransferase activity and deacetylase activity (class I–IV histone deacetylases), and are significantly altered by aging. Both class I deacetylases (such as histone deacetylase 3) and class III deacetylases (the sirtuins) influence several proteins involved in cardiovascular calcification: 1) HDAC3 suppresses Runx2 activity and prevents osteoblastic differentiation180, 2) reductions in Sirt1increase vascular inflammation and endothelial cell activation181, 3) reductions in Sirt1 and Sirt6 increase histone acetylation, promote genomic instability, and are permissive for increases in NFκB binding in the nucleus182, and 4) the histone acetyltransferase GCN5 increases TGF-β binding efficiency and overall genomic instability183. Mice deficient in acetyltransferase or deacetylase enzymes have been generated, and will be useful in determining the role of histone and protein acetylation in the progression of CAVS.
DNA methylation also contributes to regulation of both global and specific gene expression, and aberrations in DNA methylation profiles are present in atherosclerosis, stroke, and cancer184. Interestingly, recent work has shown that CpG island methylation at the α-smooth muscle actin promoter contributes to gene silencing in cultured smooth muscle cells, thereby facilitating re-differentiation of these cells to an osteoblast-like phenotype185. In contrast, hypermethylation of pluripotency-inducing transcription factors could potentially prevent cellular de-differentiation 184 and maintain resident valvular or vascular cells in a non-osteogenic lineage. While changes in DNA and histone methylation in stenotic aortic valves have not been examined at this time, we speculate that use of genetically altered mice and methyltransferase inhibitors will lend important insights into the role of epigenetic modifications in CAVS.
Histological and molecular changes with treatment of valve disease: is regression of CAVS possible?
At present, surgical (i.e., valve replacement) or emerging interventional (i.e., percutaneous valve implantation) approaches are the only treatments for CAVS. When hypercholesterolemia is the primary driver of valve calcification, lipid lowering therapy may be a useful intervention in hypercholesterolemic patients with relatively early stages of CAVS186. Based on data from three large clinical trials (SEAS187, SALTIRE188, and ASTRONOMER189), however, lipid lowering is not likely to be beneficial for patients with severe CAVS. To determine molecular, histological, and functional changes with lipid lowering in early- and late-stage CAVS, we used a mouse model of CAVS in which lipids could be altered with a “genetic switch”, thereby avoiding confounding/pleiotropic effects of pharmacological interventions190–192.
Histological changes following lipid lowering
Reduction of blood lipids in mice reduces valvular lipid content and inflammatory cell infiltrate in both early and late stages of hypercholesterolemia-induced CAVS 12, 13 (see Figure 6). Lipid lowering therapy also reduces BMP12 and Wnt/β-catenin signaling13 and, remarkably, reduces valvular calcium12, 13. This finding is markedly different from observations in advanced atherosclerotic lesions, where activity of osteoblast-like cells is markedly reduced by statins23, but calcium deposits are resistant to reduction and/or resorption193, 194. These contrasting effects in valves and arteries are somewhat surprising because osteoclast-like cells are present in both calcifying aortic valves79, 195–197 and in arteries198, 199. Further work to examine differences in calcified plaque composition/ultrastructure, and differences in osteoblast-/osteoclast-like function in arteries and valves, will be important to understanding the susceptibility of calcified deposits to resorption in different cardiovascular tissues.
In contrast to valvular lipid and calcium, valvular fibrosis is remarkably refractory to lipid lowering (Figure 6). Fibrosis, extending into the spongiosa of the valve, persists even after 6 months of lipid lowering12. This finding is concordant with observations in atherosclerotic lesions during the first few months of reduction of blood lipids in hyperlipidemic monkeys, as decreases in lipid content of lesions are not accompanied by reduction of fibrosis200.
Although phospho-smad2/3 levels (indicative of TGF-β signaling) and myofibroblast activation are reduced following lipid lowering in early stages of valve disease12, both remain elevated if lipid lowering is initiated in advanced valve disease13. We speculate that persistent myofibroblast activation may be due to epigenetic modifications resulting in sustained expression of TGF-β and smad2201, or cell-matrix interactions that result in sustained activation of TGF-β1 signaling202. Thus, interventions that target both pro-calcific and pro-fibrotic signaling may be required to slow progression of valvular dysfunction in end-stage CAVS. Similarly, improving valvular function/reversing histopathological changes in advanced CAVS may require interventions that effectively reduce valvular calcium content, connective tissue content, and their respective signaling cascades.
Functional changes after lipid lowering
When initiated in early stages of valve disease (i.e., before reduction in aortic valve orifice area), lipid lowering in mice halts progression to severe aortic valve stenosis12 (see Figure 6), although it is not clear whether lipid lowering confers similar benefits to patients with mild/moderate valve disease203, especially those with hyperlipidemia186. Similar to observations from large clinical trials in humans187–189, however, lipid lowering in advanced stages of aortic valve disease does not improve aortic valve function in hypercholesterolemic mice13. Taken in the context of the histological changes described above, it is clear that reducing valvular calcium per se is not sufficient to improve valvular function with lipid lowering. Thus, we speculate that reduction of valvular fibrosis may also be critical to improving valvular function.
Interventions that may inhibit development and/or progression of CAVS in humans and mice
Based on previous studies, and discussion of endogenous mechanisms that may modulate CAVS (see above), we speculate that several interventions might potentially inhibit development and/or progression of calcification, fibrosis, and stenosis of the aortic valve (Figure 7).
Figure 7.

Potential targets and treatments to slow the progression of aortic valve stenosis. Risk factors (including hypercholesterolemia, hypertension, metabolic syndrome, and smoking) can be treated. Possible signaling cascades and treatments (in red), although supported by some experimental evidence, are speculative. See text for rationale. Abbreviations: ACE-I: angiotensin converting enzyme inhibitor; AT1R-B, angiotension II receptor type I blocker; n-Ab, neutralizing antibody against TGFβ; miRNA, micro-RNA; ROS, reactive oxygen species; NO, nitric oxide; OPG, osteoprotegerin; PPARg, peroxisome proliferator-activated receptor gamma.
First, lipid lowering therapy could slow progression of aortic valve disease under some conditions. Specifically, lipid lowering stops progression—but does not induce regression—of aortic valve stenosis in hypercholesterolemic mice12, 13. Thus, lipid lowering therapy may be a useful intervention in hypercholesterolemic patients in early stages of CAVS.
Second, we propose that altering oxidative stress and/or nitric oxide bioavailability may be useful in slowing progression of CAVS. High doses of antioxidants (e.g., vitamin E) rarely confer long-term therapeutic benefit 204, and chronic treatment with tempol unexpectedly increased apoptosis and calcification in a rabbit model of CAVS79. Nevertheless, targeting antioxidants to different subcellular compartments may be more beneficial than those which affect redox state throughout the cell205. Reducing oxidative stress is also likely to increase nitric oxide bioavailability8, 68, which may be augmented by combining antioxidant therapy with treatments that reduce endogenous inhibitors of nitric oxide synthases (i.e., asymmetric dimethylarginine, which is increased in patients with CAVS87, 88).
Third, inhibition of the renin-angiotensin system may slow the development and/or progression of CAVS. Although retrospective studies suggest that ACE inhibitors do not slow the progression of CAVS100, 101, it is possible that AT1r inhibitors may be more effective at slowing the progression of CAVS100 by allowing angiotensin II to exert beneficial effects via both the AT2 receptor206 and through its cleavage product angiotensin-1-7207. Indeed, overexpression of AT2r reduces atherosclerosis in mice208, and administration of exogenous angiotensin 1–7 suppresses fibrosis in models of vascular injury 209.
Fourth, inhibition of the RANK/RANKL pathway may prevent and/or slow progression of CAVS. Based on previous findings from models of intimal plaque calcification, it is likely that osteoprotegerin will slow progression of CAVS when initiated during early stages of disease111. Because RANKL may be a key mediator of monocyte recruitment and osteoclastogenesis in vascular calcification, however, it is difficult to predict whether osteoprotegerin will induce regression of calcification in advanced vascular or valvular lesions.
Fifth, a PPARγ ligand may inhibit development of CAVS. Because PPARγ ligands prevent cells from differentiating to an osteoblast-like cell lineage, suppress inflammation, and increase antioxidant protein levels, it is possible that PPAR may slow the development of CAVS. However, the time at which treatment is initiated is likely to be of great importance. Thiazolidinediones appear to inhibit development of early atherosclerotic lesions210, but have little or no beneficial effect on advanced lesions in ldlr−/− mice, perhaps because they promote cell death in advanced lesions211.
Sixth, we speculate that manipulation of microRNA levels may be useful in prevention of fibrosis and calcification of the aortic valve, and inhibit development of CAVS. For example, antisense oligonucleotide-mediated (antimiR) knockdown and overexpression techniques are under development for prevention and/or treatment of cardiac fibrosis175, 179, 212. MicroRNAs that alter pro-calcific and pro-fibrotic signaling are dysregulated in CAVS178. Thus, altering expression of microRNAs may be effective in attenuating progression of valve disease, especially because microRNAs can modulate the functions of multiple genes in a signaling cascade (and may affect multiple signaling cascades), thereby conferring higher therapeutic efficacy.
Finally, pharmacological manipulation of epigenetic modifications and post-translational modifications of transcription factors may be useful in preventing calcification and fibrosis of the aortic valve. Acetylation levels of histones and proteins have a profound impact on genomic stability and transcription factor binding affinity, and the beneficial effects of acetyltransferase inhibitors and deacetylase activators on inflammation and atherosclerosis make them promising candidates for use in CAVS213. Small molecule methyltransferase inhibitors or demethylase activators may also further our understanding of the role of epigenetic modifications in CAVS, facilitate favorable changes in gene expression184, and perhaps slow progression of CAVS.
Integration of findings from cardiovascular calcification and skeletal ossification
There is strong support for the concept that CAVS is an active process mediated by ectopic osteoblastogenesis, and that these osteoblast-like cells express markers similar to those found in skeletal osteoblasts12, 13, 214. Patients with CAVS generally are over the age of 65, and at risk of developing osteoporosis. Thus, therapies that are designed to slow the progression of CAVS via inhibition of osteoblastogenesis may not be useful, because they may augment osteoporosis. An example of this side effect is PPARγ agonists, which may slow progression of CAVS by diverting cells to an adipocyte-like lineage, but increase the prevalence of bone fractures in patients with type II diabetes via a similar mechanism215–217. An ideal pharmacological intervention would simultaneously suppress ectopic osteogenesis and improve skeletal osteogenesis. Two potential therapies may accomplish this challenging goal: antioxidants or suppression of RANKL signaling.
First, increasing oxidative stress promotes osteogenic differentiation of vascular smooth muscle cells, but attenuates mineralization of bone-derived osteoblasts in vitro. Antioxidants such as N-acetylcysteine attenuate bone loss in genetically altered mice218, but, direct evidence for a role of oxidative stress in initiation or progression of CAVS in vivo is limited. Antioxidant specificity 79, 219, and perhaps subcellular compartmentalization/ targeting205, may be required for successful therapeutic interventions.
Second, modulation of the OPG/RANKL-axis attenuates calcification of atherosclerotic plaque, but preserves bone mineral density220, 221. Furthermore, estrogen appears to tonically inhibit aortic calcification through the suppression of RANKL signaling222. Thus, suppression of RANKL signaling with monoclonal antibodies or other strategies may be an efficacious treatment to preserve bone mineral density and attenuate progression of valvular calcification in men and post-menopausal women.
Conclusions
Major advances have been made towards our understanding of mechanisms that contribute to aortic valve stenosis. Much of this insight has been gleaned from analysis of human tissue, because animal models that consistently develop hemodynamically significant CAVS have been available only for the past 5 years. These murine models are dependent upon hypercholesterolemia or background strain of the mice for the development of CAVS. The context dependence of cell signaling will always be an underlying issue with these models, which makes the development of additional models of CAVS important. These mouse models will provide an opportunity to examine mechanisms that lead to CAVS, and to test the efficacy of pharmacological interventions. We suggest that interventions outlined in this review hold promise for slowing the progression of CAVS and delaying the need for valve replacement surgery.
Supplementary Material
Table 1.
Echocardiographic and hemodynamic changes in animal models of aortic valve sclerosis and stenosis.
| Species/Strain | DietREF | Histopathological changes in aortic valve | Hemodynamically significant stenosis? |
|---|---|---|---|
| Mice | |||
| C57BL/6 | HF16 | Lipid deposition Modest calcification |
No |
| ApoE−/− | Chow15 | Lipid deposition Calcification Monocyte/Inflammatory cell infiltration |
<2% |
| HF/HC19, 33 | Lipid deposition Fibrosis19, 33 Calcification19, 33 Monocyte/Inflammatory cell infiltration19, 33 |
<2% | |
| Ldlr−/− | HF/HC16, 223 | Lipid deposition Calcification Monocyte/Inflammatory cell infiltration |
No |
| Ldlr−/−/apoB100/100 | Chow14 | Lipid deposition Calcification Monocyte/Inflammatory cell infiltration Myofibroblast activation |
Yes, ~30% of mice |
| HF/HC12, 13 | Lipid deposition Calcification Fibrosis Monocyte/Inflammatory cell infiltration Myofibroblast activation |
Yes, >50% of mice | |
| EGFRWa2/Wa2 | Chow11 | Fibrosis Calcification Inflammatory cell infiltration |
Yes, but background strain dependent |
| eNOS−/− | Chow224 | Bicuspid aortic valves in ~40% of mice | Not known |
| Notch1+/− | HF/HC125, 126 | Calcification | No |
| Periostin−/− | Chow225 | Calcification Fibrosis |
Not known |
| HF/HC226 | Reduced valve thickening and fibrosis | No | |
| MGP−/− | Chow122 | Calcification | Not known |
| Chm1−/− | Chow227 | Neoangiogenesis Lipid deposition Calcification |
Not known |
| Rabbits | |||
| New Zealand | HF/HC49, 79, 89, 209, 228–238 | Lipid deposition | <10% |
| White | Calcification Inflammatory cell infiltration |
Mostly moderate sclerosis | |
| Chow + | Fibrosis | <10% | |
| HTN229 | Inflammation | ||
| Watanabe | HF/HC49 | Lipid deposition Fibrosis Calcification Inflammatory cell infiltration |
No |
| Pigs | |||
| Yorkshire Landrace |
HF/HC83, 239 | Lipid deposition | No |
Acknowledgments
Sources of Funding
Original studies by the authors were supported by National Institutes of Health grants HL092235, HL62984, NS24621, RR026293, and by a Carver Research Program of Excellence.
The authors would like to thank Elise Oehler and Kathy Zimmerman for assistance with acquisition and processing of echocardiographic images.
NON-STANDARD ABBREVIATIONS
- CAVS
calcific aortic valve stenosis
- RT-PCR
reverse transcriptase-polymerase chain reaction
- BMP
bone morphogenetic protein
- NOS
nitric oxide synthase
- RANKL
receptor activator of nuclear factor κB ligand
- OPG
osteoprotegerin
- PPARγ
peroxisome proliferator-activated receptor
- RAS
renin-angiotensin system
- TGF-β
transforming growth factor-β ROS, reactive oxygen species
- VSMC
vascular smooth muscle cell
- RAGE
receptor for advanced glycosylation end products
- MGP
matrix gamma-carboxyglutamic (Gla) protein
- MMP
matrix metalloproteinase
- TNF
tumor necrosis factor
- HDAC
histone deacetylase
- AT1r/AT2R
angiotensin receptor 1/2
- CpG
linear base sequence of cytosine and guanine in DNA
Footnotes
Conflict of Interest
Two of the authors (DDH and RMW) have received osteoprotegerin, and a research grant, from Amgen, Inc.
References
- 1.Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: A population-based study. Lancet. 2006;368:1005–1011. doi: 10.1016/S0140-6736(06)69208-8. [DOI] [PubMed] [Google Scholar]
- 2.Bonow RO, Carabello BA, Chatterjee K, de Leon AC, Jr, Faxon DP, Freed MD, Gaasch WH, Lytle BW, Nishimura RA, O’Gara PT, O’Rourke RA, Otto CM, Shah PM, Shanewise JS. 2008 focused update incorporated into the acc/aha 2006 guidelines for the management of patients with valvular heart disease: A report of the american college of cardiology/american heart association task force on practice guidelines (writing committee to revise the 1998 guidelines for the management of patients with valvular heart disease): Endorsed by the society of cardiovascular anesthesiologists, society for cardiovascular angiography and interventions, and society of thoracic surgeons. Circulation. 2008;118:e523–661. doi: 10.1161/CIRCULATIONAHA.108.190748. [DOI] [PubMed] [Google Scholar]
- 3.Beckmann E, Grau JB, Sainger R, Poggio P, Ferrari G. Insights into the use of biomarkers in calcific aortic valve disease. J Heart Valve Dis. 2010;19:441–452. [PMC free article] [PubMed] [Google Scholar]
- 4.Messika-Zeitoun D, Bielak LF, Peyser PA, Sheedy PF, Turner ST, Nkomo VT, Breen JF, Maalouf J, Scott C, Tajik AJ, Enriquez-Sarano M. Aortic valve calcification: Determinants and progression in the population. Arterioscler Thromb Vasc Biol. 2007;27:642–648. doi: 10.1161/01.ATV.0000255952.47980.c2. [DOI] [PubMed] [Google Scholar]
- 5.Freeman RV, Otto CM. Spectrum of calcific aortic valve disease: Pathogenesis, disease progression, and treatment strategies. Circulation. 2005;111:3316–3326. doi: 10.1161/CIRCULATIONAHA.104.486738. [DOI] [PubMed] [Google Scholar]
- 6.Qian J, Chen Z, Ge J, Ma J, Chang S, Fan B, Liu X, Ge L. Relationship between aortic valve calcification and the severity of coronary atherosclerotic disease. J Heart Valve Dis. 2010;19:466–470. [PubMed] [Google Scholar]
- 7.Mazzone A, Venneri L, Berti S. Aortic valve stenosis and coronary artery disease: Pathophysiological and clinical links. J Cardiovasc Med (Hagerstown) 2007;8:983–989. doi: 10.2459/JCM.0b013e32802e6c3d. [DOI] [PubMed] [Google Scholar]
- 8.Heistad DD. Oxidative stress and vascular disease: 2005 duff lecture. Arterioscler Thromb Vasc Biol. 2006;26:689–695. doi: 10.1161/01.ATV.0000203525.62147.28. [DOI] [PubMed] [Google Scholar]
- 9.Shao JS, Cheng SL, Sadhu J, Towler DA. Inflammation and the osteogenic regulation of vascular calcification: A review and perspective. Hypertension. 2010;55:579–592. doi: 10.1161/HYPERTENSIONAHA.109.134205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. 2010;7:528–536. doi: 10.1038/nrcardio.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrick CJ, Roberts RB, Rojas M, Rajamannan NM, Suitt CB, O’Brien KD, Smyth SS, Threadgill DW. Reduced egfr causes abnormal valvular differentiation leading to calcific aortic stenosis and left ventricular hypertrophy in c57bl/6j but not 129s1/svimj mice. Am J Physiol Heart Circ Physiol. 2009;297:H65–75. doi: 10.1152/ajpheart.00866.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller JD, Weiss RM, Serrano KM, Brooks RM, 2nd, Berry CJ, Zimmerman K, Young SG, Heistad DD. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation. 2009;119:2693–2701. doi: 10.1161/CIRCULATIONAHA.108.834614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller JD, Weiss RM, Serrano KM, Castaneda LE, Brooks RM, Zimmerman K, Heistad DD. Evidence for active regulation of pro-osteogenic signaling in advanced aortic valve disease. Arterioscler Thromb Vasc Biol. 2010;30:2482–2486. doi: 10.1161/ATVBAHA.110.211029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation. 2006;114:2065–2069. doi: 10.1161/CIRCULATIONAHA.106.634139. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka K, Sata M, Fukuda D, Suematsu Y, Motomura N, Takamoto S, Hirata Y, Nagai R. Age-associated aortic stenosis in apolipoprotein e-deficient mice. J Am Coll Cardiol. 2005;46:134–141. doi: 10.1016/j.jacc.2005.03.058. [DOI] [PubMed] [Google Scholar]
- 16.Drolet MC, Roussel E, Deshaies Y, Couet J, Arsenault M. A high fat/high carbohydrate diet induces aortic valve disease in c57bl/6j mice. J Am Coll Cardiol. 2006;47:850–855. doi: 10.1016/j.jacc.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 17.Hinton RB, Jr, Alfieri CM, Witt SA, Glascock BJ, Khoury PR, Benson DW, Yutzey KE. Mouse heart valve structure and function: Echocardiographic and morphometric analyses from the fetus through the aged adult. Am J Physiol Heart Circ Physiol. 2008;294:H2480–2488. doi: 10.1152/ajpheart.91431.2007. [DOI] [PubMed] [Google Scholar]
- 18.Kupfahl C, Honold M, Meinhardt G, Vogelsberg H, Wagner A, Mahrholdt H, Sechtem U. Evaluation of aortic stenosis by cardiovascular magnetic resonance imaging: Comparison with established routine clinical techniques. Heart. 2004;90:893–901. doi: 10.1136/hrt.2003.022376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115:377–386. doi: 10.1161/CIRCULATIONAHA.106.654913. [DOI] [PubMed] [Google Scholar]
- 20.Berry CJ, Miller JD, McGroary K, Thedens DR, Young SG, Heistad DD, Weiss RM. Biventricular adaptation to volume overload in mice with aortic regurgitation. J Cardiovasc Magn Reson. 2009;11:27. doi: 10.1186/1532-429X-11-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berry CJ, Thedens DR, Light-McGroary K, Miller JD, Kutschke W, Zimmerman KA, Weiss RM. Effects of deep sedation or general anesthesia on cardiac function in mice undergoing cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2009;11:16. doi: 10.1186/1532-429X-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanada K, Vermeij M, Garinis GA, de Waard MC, Kunen MG, Myers L, Maas A, Duncker DJ, Meijers C, Dietz HC, Kanaar R, Essers J. Perturbations of vascular homeostasis and aortic valve abnormalities in fibulin-4 deficient mice. Circ Res. 2007;100:738–746. doi: 10.1161/01.RES.0000260181.19449.95. [DOI] [PubMed] [Google Scholar]
- 23.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850. doi: 10.1161/CIRCULATIONAHA.107.732867. [DOI] [PubMed] [Google Scholar]
- 24.Mehrabian M, Demer LL, Lusis AJ. Differential accumulation of intimal monocyte-macrophages relative to lipoproteins and lipofuscin corresponds to hemodynamic forces on cardiac valves in mice. Arterioscler Thromb. 1991;11:947–957. doi: 10.1161/01.atv.11.4.947. [DOI] [PubMed] [Google Scholar]
- 25.Ovchinnikova O, Gylfe A, Bailey L, Nordstrom A, Rudling M, Jung C, Bergstrom S, Waldenstrom A, Hansson GK, Nordstrom P. Osteoprotegerin promotes fibrous cap formation in atherosclerotic lesions of apoe-deficient mice--brief report. Arterioscler Thromb Vasc Biol. 2009;29:1478–1480. doi: 10.1161/ATVBAHA.109.188185. [DOI] [PubMed] [Google Scholar]
- 26.Ovchinnikova O, Robertson AK, Wagsater D, Folco EJ, Hyry M, Myllyharju J, Eriksson P, Libby P, Hansson GK. T-cell activation leads to reduced collagen maturation in atherosclerotic plaques of apoe(−/−) mice. Am J Pathol. 2009;174:693–700. doi: 10.2353/ajpath.2009.080561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whittaker P, Kloner RA, Boughner DR, Pickering JG. Quantitative assessment of myocardial collagen with picrosirius red staining and circularly polarized light. Basic Res Cardiol. 1994;89:397–410. doi: 10.1007/BF00788278. [DOI] [PubMed] [Google Scholar]
- 28.Burke AP, Kolodgie FD, Virmani R. Fetuin-a, valve calcification, and diabetes: What do we understand? Circulation. 2007;115:2464–2467. doi: 10.1161/CIRCULATIONAHA.107.698324. [DOI] [PubMed] [Google Scholar]
- 29.Matsumoto Y, Adams V, Jacob S, Mangner N, Schuler G, Linke A. Regular exercise training prevents aortic valve disease in low-density lipoprotein-receptor-deficient mice. Circulation. 2010;121:759–767. doi: 10.1161/CIRCULATIONAHA.109.892224. [DOI] [PubMed] [Google Scholar]
- 30.Chakraborty S, Wirrig EE, Hinton RB, Merrill WH, Spicer DB, Yutzey KE. Twist1 promotes heart valve cell proliferation and extracellular matrix gene expression during development in vivo and is expressed in human diseased aortic valves. Dev Biol. 2010;347:167–179. doi: 10.1016/j.ydbio.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alfieri CM, Cheek J, Chakraborty S, Yutzey KE. Wnt signaling in heart valve development and osteogenic gene induction. Dev Biol. 2010;338:127–135. doi: 10.1016/j.ydbio.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller AA, Drummond GR, De Silva TM, Mast AE, Hickey H, Williams JP, Broughton BR, Sobey CG. Nadph oxidase activity is higher in cerebral versus systemic arteries of four animal species: Role of nox2. Am J Physiol Heart Circ Physiol. 2009;296:H220–225. doi: 10.1152/ajpheart.00987.2008. [DOI] [PubMed] [Google Scholar]
- 33.Aikawa E, Aikawa M, Libby P, Figueiredo JL, Rusanescu G, Iwamoto Y, Fukuda D, Kohler RH, Shi GP, Jaffer FA, Weissleder R. Arterial and aortic valve calcification abolished by elastolytic cathepsin s deficiency in chronic renal disease. Circulation. 2009;119:1785–1794. doi: 10.1161/CIRCULATIONAHA.108.827972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hjortnaes J, Butcher J, Figueiredo JL, Riccio M, Kohler RH, Kozloff KM, Weissleder R, Aikawa E. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: A role for inflammation. Eur Heart J. 2010;31:1975–1984. doi: 10.1093/eurheartj/ehq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyce R, Dorph-Petersen KA, Lyck L, Gundersen H. Design-based stereology: Introduction to basic concepts and practical approaches for estimation of cell number. Toxicol Pathol. 2010 doi: 10.1177/0192623310385140. [DOI] [PubMed] [Google Scholar]
- 36.Thomsen JS, Laib A, Koller B, Prohaska S, Mosekilde L, Gowin W. Stereological measures of trabecular bone structure: Comparison of 3d micro computed tomography with 2d histological sections in human proximal tibial bone biopsies. J Microsc. 2005;218:171–179. doi: 10.1111/j.1365-2818.2005.01469.x. [DOI] [PubMed] [Google Scholar]
- 37.van Elburg HJ, Kuypers LC, Decraemer WF, Dirckx JJ. Improved correction of axial geometrical distortion in index-mismatched fluorescent confocal microscopic images using high-aperture objective lenses. J Microsc. 2007;228:45–54. doi: 10.1111/j.1365-2818.2007.01822.x. [DOI] [PubMed] [Google Scholar]
- 38.Kuypers LC, Decraemer WF, Dirckx JJ, Timmermans JP. A procedure to determine the correct thickness of an object with confocal microscopy in case of refractive index mismatch. J Microsc. 2005;218:68–78. doi: 10.1111/j.1365-2818.2005.01457.x. [DOI] [PubMed] [Google Scholar]
- 39.Briley-Saebo KC, Cho YS, Shaw PX, Ryu SK, Mani V, Dickson S, Izadmehr E, Green S, Fayad ZA, Tsimikas S. Targeted iron oxide particles for in vivo magnetic resonance detection of atherosclerotic lesions with antibodies directed to oxidation-specific epitopes. J Am Coll Cardiol. 2011;57:337–347. doi: 10.1016/j.jacc.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang K, Francis SA, Aikawa E, Figueiredo JL, Kohler RH, McCarthy JR, Weissleder R, Plutzky J, Jaffer FA. Pioglitazone suppresses inflammation in vivo in murine carotid atherosclerosis: Novel detection by dual-target fluorescence molecular imaging. Arterioscler Thromb Vasc Biol. 2010;30:1933–1939. doi: 10.1161/ATVBAHA.110.206342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jarrett BR, Correa C, Ma KL, Louie AY. In vivo mapping of vascular inflammation using multimodal imaging. PLoS One. 2010;5:e13254. doi: 10.1371/journal.pone.0013254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamilton AM, Rogers KA, Belisle AJ, Ronald JA, Rutt BK, Weissleder R, Boughner DR. Early identification of aortic valve sclerosis using iron oxide enhanced mri. J Magn Reson Imaging. 2010;31:110–116. doi: 10.1002/jmri.22008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wirrig EE, Hinton RB, Yutzey KE. Differential expression of cartilage and bone-related proteins in pediatric and adult diseased aortic valves. J Mol Cell Cardiol. 2010 doi: 10.1016/j.yjmcc.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial vcam-1 and icam-1 in a bmp-4- and tgf-beta1-dependent pathway. Arterioscler Thromb Vasc Biol. 2009;29:254–260. doi: 10.1161/ATVBAHA.108.176347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ni CW, Qiu H, Rezvan A, Kwon K, Nam D, Son DJ, Visvader JE, Jo H. Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood. 2010;116:e66–73. doi: 10.1182/blood-2010-04-278192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Csiszar A, Labinskyy N, Jo H, Ballabh P, Ungvari Z. Differential proinflammatory and prooxidant effects of bone morphogenetic protein-4 in coronary and pulmonary arterial endothelial cells. Am J Physiol Heart Circ Physiol. 2008;295:H569–577. doi: 10.1152/ajpheart.00180.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galvin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz JN, Fairchild-Huntress V, Dixon KL, Dunmore JH, Gimbrone MA, Jr, Falb D, Huszar D. A role for smad6 in development and homeostasis of the cardiovascular system. Nat Genet. 2000;24:171–174. doi: 10.1038/72835. [DOI] [PubMed] [Google Scholar]
- 48.Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, Spelsberg TC, McCarthy PM, Rahimtoola SH, Rajamannan NM. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol. 2006;47:1707–1712. doi: 10.1016/j.jacc.2006.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajamannan NM, Subramaniam M, Caira F, Stock SR, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the lrp5 receptor pathway. Circulation. 2005;112:I229–234. doi: 10.1161/01.CIRCULATIONAHA.104.524306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benton JA, Kern HB, Anseth KS. Substrate properties influence calcification in valvular interstitial cell culture. J Heart Valve Dis. 2008;17:689–699. [PMC free article] [PubMed] [Google Scholar]
- 51.Kennedy JA, Hua X, Mishra K, Murphy GA, Rosenkranz AC, Horowitz JD. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur J Pharmacol. 2009;602:28–35. doi: 10.1016/j.ejphar.2008.11.029. [DOI] [PubMed] [Google Scholar]
- 52.Clark-Greuel JN, Connolly JM, Sorichillo E, Narula NR, Rapoport HS, Mohler ER, 3rd, Gorman JH, 3rd, Gorman RC, Levy RJ. Transforming growth factor-beta1 mechanisms in aortic valve calcification: Increased alkaline phosphatase and related events. Ann Thorac Surg. 2007;83:946–953. doi: 10.1016/j.athoracsur.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 53.Cushing MC, Liao JT, Anseth KS. Activation of valvular interstitial cells is mediated by transforming growth factor-beta1 interactions with matrix molecules. Matrix Biol. 2005;24:428–437. doi: 10.1016/j.matbio.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 54.Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor-beta: Implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. 2004;95:253–260. doi: 10.1161/01.RES.0000136520.07995.aa. [DOI] [PubMed] [Google Scholar]
- 55.Jian B, Narula N, Li QY, Mohler ER, 3rd, Levy RJ. Progression of aortic valve stenosis: Tgf-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–465. doi: 10.1016/s0003-4975(02)04312-6. discussion 465–456. [DOI] [PubMed] [Google Scholar]
- 56.Mohler ER, 3rd, Chawla MK, Chang AW, Vyavahare N, Levy RJ, Graham L, Gannon FH. Identification and characterization of calcifying valve cells from human and canine aortic valves. J Heart Valve Dis. 1999;8:254–260. [PubMed] [Google Scholar]
- 57.Chen JH, Chen WL, Sider KL, Yip CY, Simmons CA. {beta}-catenin mediates mechanically regulated, transforming growth factor-{beta}1-induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler Thromb Vasc Biol. 2010 doi: 10.1161/ATVBAHA.110.220061. [DOI] [PubMed] [Google Scholar]
- 58.Yip CY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29:936–942. doi: 10.1161/ATVBAHA.108.182394. [DOI] [PubMed] [Google Scholar]
- 59.Balooch G, Balooch M, Nalla RK, Schilling S, Filvaroff EH, Marshall GW, Marshall SJ, Ritchie RO, Derynck R, Alliston T. Tgf-beta regulates the mechanical properties and composition of bone matrix. Proc Natl Acad Sci U S A. 2005;102:18813–18818. doi: 10.1073/pnas.0507417102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rajamannan NM. Calcific aortic stenosis: Lessons learned from experimental and clinical studies. Arterioscler Thromb Vasc Biol. 2009;29:162–168. doi: 10.1161/ATVBAHA.107.156752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wylie-Sears J, Aikawa E, Levine RA, Yang JH, Bischoff J. Mitral valve endothelial cells with osteogenic differentiation potential. Arterioscler Thromb Vasc Biol. 2010 doi: 10.1161/ATVBAHA.110.216184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paranya G, Vineberg S, Dvorin E, Kaushal S, Roth SJ, Rabkin E, Schoen FJ, Bischoff J. Aortic valve endothelial cells undergo transforming growth factor-beta-mediated and non-transforming growth factor-beta-mediated transdifferentiation in vitro. Am J Pathol. 2001;159:1335–1343. doi: 10.1016/s0002-9440(10)62520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eghbali-Fatourechi GZ, Modder UI, Charatcharoenwitthaya N, Sanyal A, Undale AH, Clowes JA, Tarara JE, Khosla S. Characterization of circulating osteoblast lineage cells in humans. Bone. 2007;40:1370–1377. doi: 10.1016/j.bone.2006.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khosla S, Eghbali-Fatourechi GZ. Circulating cells with osteogenic potential. Ann N Y Acad Sci. 2006;1068:489–497. doi: 10.1196/annals.1346.022. [DOI] [PubMed] [Google Scholar]
- 65.Eghbali-Fatourechi GZ, Lamsam J, Fraser D, Nagel D, Riggs BL, Khosla S. Circulating osteoblast-lineage cells in humans. N Engl J Med. 2005;352:1959–1966. doi: 10.1056/NEJMoa044264. [DOI] [PubMed] [Google Scholar]
- 66.Olmsted-Davis EA, Gugala Z, Camargo F, Gannon FH, Jackson K, Kienstra KA, Shine HD, Lindsey RW, Hirschi KK, Goodell MA, Brenner MK, Davis AR. Primitive adult hematopoietic stem cells can function as osteoblast precursors. Proc Natl Acad Sci U S A. 2003;100:15877–15882. doi: 10.1073/pnas.2632959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Visconti RP, Ebihara Y, LaRue AC, Fleming PA, McQuinn TC, Masuya M, Minamiguchi H, Markwald RR, Ogawa M, Drake CJ. An in vivo analysis of hematopoietic stem cell potential: Hematopoietic origin of cardiac valve interstitial cells. Circ Res. 2006;98:690–696. doi: 10.1161/01.RES.0000207384.81818.d4. [DOI] [PubMed] [Google Scholar]
- 68.Heistad DD, Wakisaka Y, Miller J, Chu Y, Pena-Silva R. Novel aspects of oxidative stress in cardiovascular diseases. Circ J. 2009;73:201–207. doi: 10.1253/circj.cj-08-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rivera J, Sobey CG, Walduck AK, Drummond GR. Nox isoforms in vascular pathophysiology: Insights from transgenic and knockout mouse models. Redox Rep. 2010;15:50–63. doi: 10.1179/174329210X12650506623401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gielis JF, Lin JY, Wingler K, Van Schil PE, Schmidt HH, Moens AL. Pathogenetic role of enos-uncoupling in cardiopulmonary disorders. Free Radic Biol Med. 2010 doi: 10.1016/j.freeradbiomed.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 71.Touyz RM, Briones AM. Reactive oxygen species and vascular biology: Implications in human hypertension. Hypertens Res. 2010 doi: 10.1038/hr.2010.201. [DOI] [PubMed] [Google Scholar]
- 72.Harrison DG, Gongora MC. Oxidative stress and hypertension. Med Clin North Am. 2009;93:621–635. doi: 10.1016/j.mcna.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 73.Lassegue B, Griendling KK. Nadph oxidases: Functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leopold JA, Loscalzo J. Oxidative risk for atherothrombotic cardiovascular disease. Free Radic Biol Med. 2009;47:1673–1706. doi: 10.1016/j.freeradbiomed.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Essex DW. Redox control of platelet function. Antioxid Redox Signal. 2009;11:1191–1225. doi: 10.1089/ars.2008.2322. [DOI] [PubMed] [Google Scholar]
- 76.Forstermann U. Oxidative stress in vascular disease: Causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med. 2008;5:338–349. doi: 10.1038/ncpcardio1211. [DOI] [PubMed] [Google Scholar]
- 77.Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482–2494. doi: 10.1056/NEJMra071014. [DOI] [PubMed] [Google Scholar]
- 78.Miller JD, Chu Y, Brooks RM, Richenbacher WE, Pena-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. 2008;52:843–850. doi: 10.1016/j.jacc.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liberman M, Bassi E, Martinatti MK, Lario FC, Wosniak J, Jr, Pomerantzeff PM, Laurindo FR. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol. 2008;28:463–470. doi: 10.1161/ATVBAHA.107.156745. [DOI] [PubMed] [Google Scholar]
- 80.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. Nad(p)h oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 81.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor runx2 by akt signaling. J Biol Chem. 2008;283:15319–15327. doi: 10.1074/jbc.M800021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mody N, Parhami F, Sarafian TA, Demer LL. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic Biol Med. 2001;31:509–519. doi: 10.1016/s0891-5849(01)00610-4. [DOI] [PubMed] [Google Scholar]
- 83.Guerraty MA, Grant GR, Karanian JW, Chiesa OA, Pritchard WF, Davies PF. Hypercholesterolemia induces side-specific phenotypic changes and peroxisome proliferator-activated receptor-gamma pathway activation in swine aortic valve endothelium. Arterioscler Thromb Vasc Biol. 2010;30:225–231. doi: 10.1161/ATVBAHA.109.198549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Soini Y, Salo T, Satta J. Angiogenesis is involved in the pathogenesis of nonrheumatic aortic valve stenosis. Hum Pathol. 2003;34:756–763. doi: 10.1016/s0046-8177(03)00245-4. [DOI] [PubMed] [Google Scholar]
- 85.Charest A, Pepin A, Shetty R, Cote C, Voisine P, Dagenais F, Pibarot P, Mathieu P. Distribution of sparc during neovascularisation of degenerative aortic stenosis. Heart. 2006;92:1844–1849. doi: 10.1136/hrt.2005.086595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ozaki M, Kawashima S, Yamashita T, Hirase T, Namiki M, Inoue N, Hirata K, Yasui H, Sakurai H, Yoshida Y, Masada M, Yokoyama M. Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoe-deficient mice. J Clin Invest. 2002;110:331–340. doi: 10.1172/JCI15215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cagirci G, Cay S, Canga A, Karakurt O, Yazihan N, Kilic H, Topaloglu S, Aras D, Demir AD, Akdemir R. Association between plasma asymmetrical dimethylarginine activity and severity of aortic valve stenosis. J Cardiovasc Med (Hagerstown) 2010 doi: 10.2459/JCM.0b013e32833cdcea. [DOI] [PubMed] [Google Scholar]
- 88.Ngo DT, Heresztyn T, Mishra K, Marwick TH, Horowitz JD. Aortic stenosis is associated with elevated plasma levels of asymmetric dimethylarginine (adma) Nitric Oxide. 2007;16:197–201. doi: 10.1016/j.niox.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 89.Rajamannan NM, Subramaniam M, Stock SR, Stone NJ, Springett M, Ignatiev KI, McConnell JP, Singh RJ, Bonow RO, Spelsberg TC. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart. 2005;91:806–810. doi: 10.1136/hrt.2003.029785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schmidt TS, McNeill E, Douglas G, Crabtree MJ, Hale AB, Khoo J, O’Neill CA, Cheng A, Channon KM, Alp NJ. Tetrahydrobiopterin supplementation reduces atherosclerosis and vascular inflammation in apolipoprotein e-knockout mice. Clin Sci (Lond) 2010;119:131–142. doi: 10.1042/CS20090559. [DOI] [PubMed] [Google Scholar]
- 91.Jensky NE, Criqui MH, Wright MC, Wassel CL, Brody SA, Allison MA. Blood pressure and vascular calcification. Hypertension. 2010;55:990–997. doi: 10.1161/HYPERTENSIONAHA.109.147520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kitazono T, Padgett RC, Armstrong ML, Tompkins PK, Heistad DD. Evidence that angiotensin ii is present in human monocytes. Circulation. 1995;91:1129–1134. doi: 10.1161/01.cir.91.4.1129. [DOI] [PubMed] [Google Scholar]
- 93.Potter DD, Sobey CG, Tompkins PK, Rossen JD, Heistad DD. Evidence that macrophages in atherosclerotic lesions contain angiotensin ii. Circulation. 1998;98:800–807. doi: 10.1161/01.cir.98.8.800. [DOI] [PubMed] [Google Scholar]
- 94.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–853. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- 95.Helske S, Lindstedt KA, Laine M, Mayranpaa M, Werkkala K, Lommi J, Turto H, Kupari M, Kovanen PT. Induction of local angiotensin ii-producing systems in stenotic aortic valves. J Am Coll Cardiol. 2004;44:1859–1866. doi: 10.1016/j.jacc.2004.07.054. [DOI] [PubMed] [Google Scholar]
- 96.Didion SP, Kinzenbaw DA, Schrader LI, Chu Y, Faraci FM. Endogenous interleukin-10 inhibits angiotensin ii-induced vascular dysfunction. Hypertension. 2009;54:619–624. doi: 10.1161/HYPERTENSIONAHA.109.137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. Il-6 deficiency protects against angiotensin ii induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol. 2007;27:2576–2581. doi: 10.1161/ATVBAHA.107.153080. [DOI] [PubMed] [Google Scholar]
- 98.Marchesi C, Paradis P, Schiffrin EL. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367–374. doi: 10.1016/j.tips.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 99.Arishiro K, Hoshiga M, Negoro N, Jin D, Takai S, Miyazaki M, Ishihara T, Hanafusa T. Angiotensin receptor-1 blocker inhibits atherosclerotic changes and endothelial disruption of the aortic valve in hypercholesterolemic rabbits. J Am Coll Cardiol. 2007;49:1482–1489. doi: 10.1016/j.jacc.2006.11.043. [DOI] [PubMed] [Google Scholar]
- 100.Yamamoto K, Yamamoto H, Yoshida K, Kisanuki A, Hirano Y, Ohte N, Akasaka T, Takeuchi M, Nakatani S, Ohtani T, Sozu T, Masuyama T. Prognostic factors for progression of early- and late-stage calcific aortic valve disease in japanese: The japanese aortic stenosis study (jass) retrospective analysis. Hypertens Res. 2010;33:269–274. doi: 10.1038/hr.2009.225. [DOI] [PubMed] [Google Scholar]
- 101.Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U, Schemper M, Binder T, Maurer G, Baumgartner H. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation. 2004;110:1291–1295. doi: 10.1161/01.CIR.0000140723.15274.53. [DOI] [PubMed] [Google Scholar]
- 102.O’Brien KD, Probstfield JL, Caulfield MT, Nasir K, Takasu J, Shavelle DM, Wu AH, Zhao XQ, Budoff MJ. Angiotensin-converting enzyme inhibitors and change in aortic valve calcium. Arch Intern Med. 2005;165:858–862. doi: 10.1001/archinte.165.8.858. [DOI] [PubMed] [Google Scholar]
- 103.D’Amelio P, Isaia G, Isaia GC. The osteoprotegerin/rank/rankl system: A bone key to vascular disease. J Endocrinol Invest. 2009;32:6–9. [PubMed] [Google Scholar]
- 104.Boyce BF, Xing L. Biology of rank, rankl, and osteoprotegerin. Arthritis Res Ther. 2007;9 (Suppl 1):S1. doi: 10.1186/ar2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Byon CH, Sun Y, Chen J, Yuan K, Mao X, Heath JM, Anderson PG, Tintut Y, Demer LL, Wang D, Chen Y. Runx2-upregulated receptor activator of nuclear factor {kappa}b ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler Thromb Vasc Biol. 2011 doi: 10.1161/ATVBAHA.110.222547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sasaki H, Yamamoto H, Tominaga K, Masuda K, Kawai T, Teshima-Kondo S, Matsuno K, Yabe-Nishimura C, Rokutan K. Receptor activator of nuclear factor-kappab ligand-induced mouse osteoclast differentiation is associated with switching between nadph oxidase homologues. Free Radic Biol Med. 2009;47:189–199. doi: 10.1016/j.freeradbiomed.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 107.Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, Kim N, Lee SY. A crucial role for reactive oxygen species in rankl-induced osteoclast differentiation. Blood. 2005;106:852–859. doi: 10.1182/blood-2004-09-3662. [DOI] [PubMed] [Google Scholar]
- 108.Panizo S, Cardus A, Encinas M, Parisi E, Valcheva P, Lopez-Ongil S, Coll B, Fernandez E, Valdivielso JM. Rankl increases vascular smooth muscle cell calcification through a rank-bmp4-dependent pathway. Circ Res. 2009;104:1041–1048. doi: 10.1161/CIRCRESAHA.108.189001. [DOI] [PubMed] [Google Scholar]
- 109.Orita Y, Yamamoto H, Kohno N, Sugihara M, Honda H, Kawamata S, Mito S, Soe NN, Yoshizumi M. Role of osteoprotegerin in arterial calcification: Development of new animal model. Arterioscler Thromb Vasc Biol. 2007;27:2058–2064. doi: 10.1161/ATVBAHA.107.147868. [DOI] [PubMed] [Google Scholar]
- 110.Bennett BJ, Scatena M, Kirk EA, Rattazzi M, Varon RM, Averill M, Schwartz SM, Giachelli CM, Rosenfeld ME. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older apoe−/− mice. Arterioscler Thromb Vasc Biol. 2006;26:2117–2124. doi: 10.1161/01.ATV.0000236428.91125.e6. [DOI] [PubMed] [Google Scholar]
- 111.Morony S, Tintut Y, Zhang Z, Cattley RC, Van G, Dwyer D, Stolina M, Kostenuik PJ, Demer LL. Osteoprotegerin inhibits vascular calcification without affecting atherosclerosis in ldlr(−/−) mice. Circulation. 2008;117:411–420. doi: 10.1161/CIRCULATIONAHA.107.707380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kaden JJ, Bickelhaupt S, Grobholz R, Haase KK, Sarikoc A, Kilic R, Brueckmann M, Lang S, Zahn I, Vahl C, Hagl S, Dempfle CE, Borggrefe M. Receptor activator of nuclear factor kappab ligand and osteoprotegerin regulate aortic valve calcification. J Mol Cell Cardiol. 2004;36:57–66. doi: 10.1016/j.yjmcc.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 113.Steinmetz M, Skowasch D, Wernert N, Welsch U, Preusse CJ, Welz A, Nickenig G, Bauriedel G. Differential profile of the opg/rankl/rank-system in degenerative aortic native and bioprosthetic valves. J Heart Valve Dis. 2008;17:187–193. [PubMed] [Google Scholar]
- 114.Duan SZ, Usher MG, Mortensen RM. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ Res. 2008;102:283–294. doi: 10.1161/CIRCRESAHA.107.164384. [DOI] [PubMed] [Google Scholar]
- 115.Hamblin M, Chang L, Fan Y, Zhang J, Chen YE. Ppars and the cardiovascular system. Antioxid Redox Signal. 2009;11:1415–1452. doi: 10.1089/ars.2008.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kawaguchi H, Akune T, Yamaguchi M, Ohba S, Ogata N, Chung UI, Kubota N, Terauchi Y, Kadowaki T, Nakamura K. Distinct effects of ppargamma insufficiency on bone marrow cells, osteoblasts, and osteoclastic cells. J Bone Miner Metab. 2005;23:275–279. doi: 10.1007/s00774-005-0599-2. [DOI] [PubMed] [Google Scholar]
- 117.Yamashita A, Takada T, Nemoto K, Yamamoto G, Torii R. Transient suppression of ppargamma directed es cells into an osteoblastic lineage. FEBS Lett. 2006;580:4121–4125. doi: 10.1016/j.febslet.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 118.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 119.Keen HL, Ryan MJ, Beyer A, Mathur S, Scheetz TE, Gackle BD, Faraci FM, Casavant TL, Sigmund CD. Gene expression profiling of potential ppargamma target genes in mouse aorta. Physiol Genomics. 2004;18:33–42. doi: 10.1152/physiolgenomics.00027.2004. [DOI] [PubMed] [Google Scholar]
- 120.Yao Y, Watson AD, Ji S, Bostrom KI. Heat shock protein 70 enhances vascular bone morphogenetic protein-4 signaling by binding matrix gla protein. Circ Res. 2009;105:575–584. doi: 10.1161/CIRCRESAHA.109.202333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen-Dechent W, Weissberg PL, Shanahan CM. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: A potential mechanism for accelerated vascular calcification in esrd. J Am Soc Nephrol. 2004;15:2857–2867. doi: 10.1097/01.ASN.0000141960.01035.28. [DOI] [PubMed] [Google Scholar]
- 122.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix gla protein. Nature. 1997;386:78–81. doi: 10.1038/386078a0. [DOI] [PubMed] [Google Scholar]
- 123.Yao Y, Bennett BJ, Wang X, Rosenfeld ME, Giachelli C, Lusis AJ, Bostrom KI. Inhibition of bone morphogenetic proteins protects against atherosclerosis and vascular calcification. Circ Res. 2010;107:485–494. doi: 10.1161/CIRCRESAHA.110.219071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in notch1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 125.Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol. 2009;47:828–834. doi: 10.1016/j.yjmcc.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Luna-Zurita L, Prados B, Grego-Bessa J, Luxan G, del Monte G, Benguria A, Adams RH, Perez-Pomares JM, de la Pompa JL. Integration of a notch-dependent mesenchymal gene program and bmp2-driven cell invasiveness regulates murine cardiac valve formation. J Clin Invest. 2010;120:3493–3507. doi: 10.1172/JCI42666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ann EJ, Kim HY, Choi YH, Kim MY, Mo JS, Jung J, Yoon JH, Kim SM, Moon JS, Seo MS, Hong JA, Jang WG, Shore P, Komori T, Koh JT, Park HS. Inhibition of notch1 signaling by runx2 during osteoblast differentiation. J Bone Miner Res. 2010 doi: 10.1002/jbmr.227. [DOI] [PubMed] [Google Scholar]
- 128.Shimizu T, Tanaka T, Iso T, Doi H, Sato H, Kawai-Kowase K, Arai M, Kurabayashi M. Notch signaling induces osteogenic differentiation and mineralization of vascular smooth muscle cells: Role of msx2 gene induction via notch-rbp-jk signaling. Arterioscler Thromb Vasc Biol. 2009;29:1104–1111. doi: 10.1161/ATVBAHA.109.187856. [DOI] [PubMed] [Google Scholar]
- 129.Kaden JJ, Dempfle CE, Grobholz R, Fischer CS, Vocke DC, Kilic R, Sarikoc A, Pinol R, Hagl S, Lang S, Brueckmann M, Borggrefe M. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc Pathol. 2005;14:80–87. doi: 10.1016/j.carpath.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 130.Kaden JJ, Dempfle CE, Grobholz R, Tran HT, Kilic R, Sarikoc A, Brueckmann M, Vahl C, Hagl S, Haase KK, Borggrefe M. Interleukin-1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis. 2003;170:205–211. doi: 10.1016/s0021-9150(03)00284-3. [DOI] [PubMed] [Google Scholar]
- 131.Jian B, Jones PL, Li Q, Mohler ER, 3rd, Schoen FJ, Levy RJ. Matrix metalloproteinase-2 is associated with tenascin-c in calcific aortic stenosis. Am J Pathol. 2001;159:321–327. doi: 10.1016/S0002-9440(10)61698-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kaden JJ, Vocke DC, Fischer CS, Grobholz R, Brueckmann M, Vahl CF, Hagl S, Haase KK, Dempfle CE, Borggrefe M. Expression and activity of matrix metalloproteinase-2 in calcific aortic stenosis. Z Kardiol. 2004;93:124–130. doi: 10.1007/s00392-004-1021-0. [DOI] [PubMed] [Google Scholar]
- 133.Edep ME, Shirani J, Wolf P, Brown DL. Matrix metalloproteinase expression in nonrheumatic aortic stenosis. Cardiovasc Pathol. 2000;9:281–286. doi: 10.1016/s1054-8807(00)00043-0. [DOI] [PubMed] [Google Scholar]
- 134.Fondard O, Detaint D, Iung B, Choqueux C, Adle-Biassette H, Jarraya M, Hvass U, Couetil JP, Henin D, Michel JB, Vahanian A, Jacob MP. Extracellular matrix remodelling in human aortic valve disease: The role of matrix metalloproteinases and their tissue inhibitors. Eur Heart J. 2005;26:1333–1341. doi: 10.1093/eurheartj/ehi248. [DOI] [PubMed] [Google Scholar]
- 135.Helske S, Syvaranta S, Lindstedt KA, Lappalainen J, Oorni K, Mayranpaa MI, Lommi J, Turto H, Werkkala K, Kupari M, Kovanen PT. Increased expression of elastolytic cathepsins s, k, and v and their inhibitor cystatin c in stenotic aortic valves. Arterioscler Thromb Vasc Biol. 2006;26:1791–1798. doi: 10.1161/01.ATV.0000228824.01604.63. [DOI] [PubMed] [Google Scholar]
- 136.Helske S, Syvaranta S, Kupari M, Lappalainen J, Laine M, Lommi J, Turto H, Mayranpaa M, Werkkala K, Kovanen PT, Lindstedt KA. Possible role for mast cell-derived cathepsin g in the adverse remodelling of stenotic aortic valves. Eur Heart J. 2006;27:1495–1504. doi: 10.1093/eurheartj/ehi706. [DOI] [PubMed] [Google Scholar]
- 137.Pacifici R, Carano A, Santoro SA, Rifas L, Jeffrey JJ, Malone JD, McCracken R, Avioli LV. Bone matrix constituents stimulate interleukin-1 release from human blood mononuclear cells. J Clin Invest. 1991;87:221–228. doi: 10.1172/JCI114975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Qin X, Corriere MA, Matrisian LM, Guzman RJ. Matrix metalloproteinase inhibition attenuates aortic calcification. Arterioscler Thromb Vasc Biol. 2006;26:1510–1516. doi: 10.1161/01.ATV.0000225807.76419.a7. [DOI] [PubMed] [Google Scholar]
- 139.Isoda K, Matsuki T, Kondo H, Iwakura Y, Ohsuzu F. Deficiency of interleukin-1 receptor antagonist induces aortic valve disease in balb/c mice. Arterioscler Thromb Vasc Biol. 2010;30:708–715. doi: 10.1161/ATVBAHA.109.201749. [DOI] [PubMed] [Google Scholar]
- 140.Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, Cheng SL, Towler DA. Aortic msx2-wnt calcification cascade is regulated by tnf-alpha-dependent signals in diabetic ldlr−/− mice. Arterioscler Thromb Vasc Biol. 2007;27:2589–2596. doi: 10.1161/ATVBAHA.107.153668. [DOI] [PubMed] [Google Scholar]
- 141.Hamid T, Gu Y, Ortines RV, Bhattacharya C, Wang G, Xuan YT, Prabhu SD. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: Role of nuclear factor-kappab and inflammatory activation. Circulation. 2009;119:1386–1397. doi: 10.1161/CIRCULATIONAHA.108.802918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cecil DL, Terkeltaub RA. Arterial calcification is driven by rage in enpp1−/− mice. J Vasc Res. 2010;48:227–235. doi: 10.1159/000318805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ren X, Shao H, Wei Q, Sun Z, Liu N. Advanced glycation end-products enhance calcification in vascular smooth muscle cells. J Int Med Res. 2009;37:847–854. doi: 10.1177/147323000903700329. [DOI] [PubMed] [Google Scholar]
- 144.Tanikawa T, Okada Y, Tanikawa R, Tanaka Y. Advanced glycation end products induce calcification of vascular smooth muscle cells through rage/p38 mapk. J Vasc Res. 2009;46:572–580. doi: 10.1159/000226225. [DOI] [PubMed] [Google Scholar]
- 145.Towler DA. Vascular calcification: It’s all the rage! Arterioscler Thromb Vasc Biol. 2011;31:237–239. doi: 10.1161/ATVBAHA.110.220038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hofmann Bowman MA, Gawdzik J, Bukhari U, Husain AN, Toth PT, Kim G, Earley J, McNally EM. S100a12 in vascular smooth muscle accelerates vascular calcification in apolipoprotein e-null mice by activating an osteogenic gene regulatory program. Arterioscler Thromb Vasc Biol. 2011;31:337–344. doi: 10.1161/ATVBAHA.110.217745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gawdzik J, Mathew L, Kim G, Puri TS, Hofmann Bowman MA. Vascular remodeling and arterial calcification are directly mediated by s100a12 (en-rage) in chronic kidney disease. Am J Nephrol. 2011;33:250–259. doi: 10.1159/000324693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Katz R, Budoff MJ, Takasu J, Shavelle DM, Bertoni A, Blumenthal RS, Ouyang P, Wong ND, O’Brien KD. Relationship of metabolic syndrome with incident aortic valve calcium and aortic valve calcium progression: The multi-ethnic study of atherosclerosis (mesa) Diabetes. 2009;58:813–819. doi: 10.2337/db08-1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Geroldi D, Falcone C, Emanuele E. Soluble receptor for advanced glycation end products: From disease marker to potential therapeutic target. Curr Med Chem. 2006;13:1971–1978. doi: 10.2174/092986706777585013. [DOI] [PubMed] [Google Scholar]
- 150.Basta G, Corciu AI, Vianello A, Del Turco S, Foffa I, Navarra T, Chiappino D, Berti S, Mazzone A. Circulating soluble receptor for advanced glycation end-product levels are decreased in patients with calcific aortic valve stenosis. Atherosclerosis. 2010;210:614–618. doi: 10.1016/j.atherosclerosis.2009.12.029. [DOI] [PubMed] [Google Scholar]
- 151.Mohler ER, 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 152.Vattikuti R, Towler DA. Osteogenic regulation of vascular calcification: An early perspective. Am J Physiol Endocrinol Metab. 2004;286:E686–696. doi: 10.1152/ajpendo.00552.2003. [DOI] [PubMed] [Google Scholar]
- 153.Kalantari F, Miao D, Emadali A, Tzimas GN, Goltzman D, Vali H, Chevet E, Auguste P. Cellular and molecular mechanisms of abnormal calcification following ischemia-reperfusion injury in human liver transplantation. Mod Pathol. 2007;20:357–366. doi: 10.1038/modpathol.3800747. [DOI] [PubMed] [Google Scholar]
- 154.Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A. 2004;101:4477–4482. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rajamannan NM, Nealis TB, Subramaniam M, Pandya S, Stock SR, Ignatiev CI, Sebo TJ, Rosengart TK, Edwards WD, McCarthy PM, Bonow RO, Spelsberg TC. Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast-like bone formation. Circulation. 2005;111:3296–3301. doi: 10.1161/CIRCULATIONAHA.104.473165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yperman J, De Visscher G, Holvoet P, Flameng W. Molecular and functional characterization of ovine cardiac valve-derived interstitial cells in primary isolates and cultures. Tissue Eng. 2004;10:1368–1375. doi: 10.1089/ten.2004.10.1368. [DOI] [PubMed] [Google Scholar]
- 157.Stephens EH, Saltarrelli JG, Baggett LS, Nandi I, Kuo JJ, Davis AR, Olmsted-Davis EA, Reardon MJ, Morrisett JD, Grande-Allen KJ. Differential proteoglycan and hyaluronan distribution in calcified aortic valves. Cardiovasc Pathol. 2010 doi: 10.1016/j.carpath.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gupta V, Werdenberg JA, Blevins TL, Grande-Allen KJ. Synthesis of glycosaminoglycans in differently loaded regions of collagen gels seeded with valvular interstitial cells. Tissue Eng. 2007;13:41–49. doi: 10.1089/ten.2006.0091. [DOI] [PubMed] [Google Scholar]
- 159.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 160.Monzack EL, Gu X, Masters KS. Efficacy of simvastatin treatment of valvular interstitial cells varies with the extracellular environment. Arterioscler Thromb Vasc Biol. 2009;29:246–253. doi: 10.1161/ATVBAHA.108.179218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rodriguez KJ, Masters KS. Regulation of valvular interstitial cell calcification by components of the extracellular matrix. J Biomed Mater Res A. 2009;90:1043–1053. doi: 10.1002/jbm.a.32187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lutgens E, Gijbels M, Smook M, Heeringa P, Gotwals P, Koteliansky VE, Daemen MJ. Transforming growth factor-beta mediates balance between inflammation and fibrosis during plaque progression. Arterioscler Thromb Vasc Biol. 2002;22:975–982. doi: 10.1161/01.atv.0000019729.39500.2f. [DOI] [PubMed] [Google Scholar]
- 163.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*) Annu Rev Immunol. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Demer LL, Tintut Y. Mechanisms linking osteoporosis with cardiovascular calcification. Curr Osteoporos Rep. 2009;7:42–46. doi: 10.1007/s11914-009-0008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, Kitzman DW, Otto CM. Clinical factors associated with calcific aortic valve disease. Cardiovascular health study. J Am Coll Cardiol. 1997;29:630–634. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- 166.Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG, Gordon LB. Cardiovascular pathology in hutchinson-gilford progeria: Correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010;30:2301–2309. doi: 10.1161/ATVBAHA.110.209460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nair K, Ramachandran P, Krishnamoorthy KM, Dora S, Achuthan TJ. Hutchinson-gilford progeria syndrome with severe calcific aortic valve stenosis and calcific mitral valve. J Heart Valve Dis. 2004;13:866–869. [PubMed] [Google Scholar]
- 168.Makous N, Friedman S, Yakovac W, Maris EP. Cardiovascular manifestations in progeria. Report of clinical and pathologic findings in a patient with severe arteriosclerotic heart disease and aortic stenosis. Am Heart J. 1962;64:334–346. doi: 10.1016/0002-8703(62)90148-5. [DOI] [PubMed] [Google Scholar]
- 169.Sogawa M, Kasuya S, Yamamoto K, Koshika M, Oguma F, Hayashi J. Aortic valve replacement for aortic stenosis with a small aortic annulus in a patient having werner’s syndrome and liver cirrhosis. Ann Thorac Cardiovasc Surg. 2001;7:378–380. [PubMed] [Google Scholar]
- 170.Grubitzsch H, Beholz S, Wollert HG, Eckel L. Severe heart valve calcification in a young patient with werner syndrome. Cardiovasc Pathol. 2000;9:53–54. doi: 10.1016/s1054-8807(99)00036-8. [DOI] [PubMed] [Google Scholar]
- 171.Ragnauth CD, Warren DT, Liu Y, McNair R, Tajsic T, Figg N, Shroff R, Skepper J, Shanahan CM. Prelamin a acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation. 2010;121:2200–2210. doi: 10.1161/CIRCULATIONAHA.109.902056. [DOI] [PubMed] [Google Scholar]
- 172.Capell BC, Olive M, Erdos MR, Cao K, Faddah DA, Tavarez UL, Conneely KN, Qu X, San H, Ganesh SK, Chen X, Avallone H, Kolodgie FD, Virmani R, Nabel EG, Collins FS. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc Natl Acad Sci U S A. 2008;105:15902–15907. doi: 10.1073/pnas.0807840105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Masuda H, Chikuda H, Suga T, Kawaguchi H, Kuro-o M. Regulation of multiple ageing-like phenotypes by inducible klotho gene expression in klotho mutant mice. Mech Ageing Dev. 2005;126:1274–1283. doi: 10.1016/j.mad.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 174.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 175.Small EM, Frost RJ, Olson EN. Micrornas add a new dimension to cardiovascular disease. Circulation. 2010;121:1022–1032. doi: 10.1161/CIRCULATIONAHA.109.889048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian micrornas predominantly act to decrease target mrna levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chen LH, Chiou GY, Chen YW, Li HY, Chiou SH. Microrna and aging: A novel modulator in regulating the aging network. Ageing Res Rev. 2010;9 (Suppl 1):S59–66. doi: 10.1016/j.arr.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 178.Nigam V, Sievers HH, Jensen BC, Sier HA, Simpson PC, Srivastava D, Mohamed SA. Altered micrornas in bicuspid aortic valve: A comparison between stenotic and insufficient valves. J Heart Valve Dis. 2010;19:459–465. [PMC free article] [PubMed] [Google Scholar]
- 179.van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of micrornas after myocardial infarction reveals a role of mir-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Schroeder TM, Kahler RA, Li X, Westendorf JJ. Histone deacetylase 3 interacts with runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. J Biol Chem. 2004;279:41998–42007. doi: 10.1074/jbc.M403702200. [DOI] [PubMed] [Google Scholar]
- 181.Stein S, Schafer N, Breitenstein A, Besler C, Winnik S, Lohmann C, Heinrich K, Brokopp CE, Handschin C, Landmesser U, Tanner FC, Luscher TF, Matter CM. Sirt1 reduces endothelial activation without affecting vascular function in apoe−/− mice. Aging (Albany NY) 2010;2:353–360. doi: 10.18632/aging.100162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Schwer B, Schumacher B, Lombard DB, Xiao C, Kurtev MV, Gao J, Schneider JI, Chai H, Bronson RT, Tsai LH, Deng CX, Alt FW. Neural sirtuin 6 (sirt6) ablation attenuates somatic growth and causes obesity. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1016306107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Burgess RJ, Zhang Z. Roles for gcn5 in promoting nucleosome assembly and maintaining genome integrity. Cell Cycle. 2010;9:2979–2985. doi: 10.4161/cc.9.15.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.De Carvalho DD, You JS, Jones PA. DNA methylation and cellular reprogramming. Trends Cell Biol. 2010;20:609–617. doi: 10.1016/j.tcb.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Montes de Oca A, Madueno JA, Martinez-Moreno JM, Guerrero F, Munoz-Castaneda J, Rodriguez-Ortiz ME, Mendoza FJ, Almaden Y, Lopez I, Rodriguez M, Aguilera-Tejero E. High-phosphate-induced calcification is related to sm22alpha promoter methylation in vascular smooth muscle cells. J Bone Miner Res. 2010;25:1996–2005. doi: 10.1002/jbmr.93. [DOI] [PubMed] [Google Scholar]
- 186.Moura LM, Ramos SF, Zamorano JL, Barros IM, Azevedo LF, Rocha-Goncalves F, Rajamannan NM. Rosuvastatin affecting aortic valve endothelium to slow the progression of aortic stenosis. J Am Coll Cardiol. 2007;49:554–561. doi: 10.1016/j.jacc.2006.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Barwolf C, Holme I, Kesaniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, Willenheimer R. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 188.Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–2397. doi: 10.1056/NEJMoa043876. [DOI] [PubMed] [Google Scholar]
- 189.Chan KL, Teo K, Dumesnil JG, Ni A, Tam J. Effect of lipid lowering with rosuvastatin on progression of aortic stenosis: Results of the aortic stenosis progression observation: Measuring effects of rosuvastatin (astronomer) trial. Circulation. 2010;121:306–314. doi: 10.1161/CIRCULATIONAHA.109.900027. [DOI] [PubMed] [Google Scholar]
- 190.Rikitake Y, Liao JK. Rho gtpases, statins, and nitric oxide. Circ Res. 2005;97:1232–1235. doi: 10.1161/01.RES.0000196564.18314.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhou Q, Liao JK. Pleiotropic effects of statins. - basic research and clinical perspectives. Circ J. 2010;74:818–826. doi: 10.1253/circj.cj-10-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Makris GC, Lavida A, Nicolaides AN, Geroulakos G. The effect of statins on carotid plaque morphology: A ldl-associated action or one more pleiotropic effect of statins? Atherosclerosis. 2010;213:8–20. doi: 10.1016/j.atherosclerosis.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 193.Stary HC. The development of calcium deposits in atherosclerotic lesions and their persistence after lipid regression. Am J Cardiol. 2001;88:16E–19E. doi: 10.1016/s0002-9149(01)01713-1. [DOI] [PubMed] [Google Scholar]
- 194.Stary HC. Natural history of calcium deposits in atherosclerosis progression and regression. Z Kardiol. 2000;89 (Suppl 2):28–35. doi: 10.1007/s003920070097. [DOI] [PubMed] [Google Scholar]
- 195.Cappelli S, Epistolato MC, Vianello A, Mazzone A, Glauber M, Franzini M, Ottaviano V, Pompella A, Paolicchi A, Tanganelli P. Aortic valve disease and gamma-glutamyltransferase: Accumulation in tissue and relationships with calcific degeneration. Atherosclerosis. 2010;213:385–391. doi: 10.1016/j.atherosclerosis.2010.08.063. [DOI] [PubMed] [Google Scholar]
- 196.Shuvy M, Ben Ya’acov A, Zolotarov L, Lotan C, Ilan Y. Beta glycosphingolipids suppress rank expression and inhibit natural killer t cell and cd8+ accumulation in alleviating aortic valve calcification. Int J Immunopathol Pharmacol. 2009;22:911–918. doi: 10.1177/039463200902200406. [DOI] [PubMed] [Google Scholar]
- 197.Satta J, Melkko J, Pollanen R, Tuukkanen J, Paakko P, Ohtonen P, Mennander A, Soini Y. Progression of human aortic valve stenosis is associated with tenascin-c expression. J Am Coll Cardiol. 2002;39:96–101. doi: 10.1016/s0735-1097(01)01705-3. [DOI] [PubMed] [Google Scholar]
- 198.Doherty TM, Asotra K, Fitzpatrick LA, Qiao JH, Wilkin DJ, Detrano RC, Dunstan CR, Shah PK, Rajavashisth TB. Calcification in atherosclerosis: Bone biology and chronic inflammation at the arterial crossroads. Proc Natl Acad Sci U S A. 2003;100:11201–11206. doi: 10.1073/pnas.1932554100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Tyson KL, Reynolds JL, McNair R, Zhang Q, Weissberg PL, Shanahan CM. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler Thromb Vasc Biol. 2003;23:489–494. doi: 10.1161/01.ATV.0000059406.92165.31. [DOI] [PubMed] [Google Scholar]
- 200.Benzuly KH, Padgett RC, Kaul S, Piegors DJ, Armstrong ML, Heistad DD. Functional improvement precedes structural regression of atherosclerosis. Circulation. 1994;89:1810–1818. doi: 10.1161/01.cir.89.4.1810. [DOI] [PubMed] [Google Scholar]
- 201.Gomez D, Coyet A, Ollivier V, Jeunemaitre X, Jondeau G, Michel JB, Vranckx R. Epigenetic control of vascular smooth muscle cellsin marfan and non-marfan thoracic aorticaneurysms. Cardiovasc Res. 2010 doi: 10.1093/cvr/cvq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Webber J, Meran S, Steadman R, Phillips A. Hyaluronan orchestrates transforming growth factor-beta1-dependent maintenance of myofibroblast phenotype. J Biol Chem. 2009;284:9083–9092. doi: 10.1074/jbc.M806989200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gerdts E, Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gohlke-Barwolf C, Holme I, Kesaniemi YA, Malbecq W, Nienaber C, Ray S, Skjaerpe T, Wachtell K, Willenheimer R. Impact of baseline severity of aortic valve stenosis on effect of intensive lipid lowering therapy (from the seas study) Am J Cardiol. 2010;106:1634–1639. doi: 10.1016/j.amjcard.2010.07.042. [DOI] [PubMed] [Google Scholar]
- 204.Steinhubl SR. Why have antioxidants failed in clinical trials? Am J Cardiol. 2008;101:14D–19D. doi: 10.1016/j.amjcard.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 205.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Carey RM. Cardiovascular and renal regulation by the angiotensin type 2 receptor: The at2 receptor comes of age. Hypertension. 2005;45:840–844. doi: 10.1161/01.HYP.0000159192.93968.8f. [DOI] [PubMed] [Google Scholar]
- 207.Ferrario CM. New physiological concepts of the renin-angiotensin system from the investigation of precursors and products of angiotensin i metabolism. Hypertension. 2010;55:445–452. doi: 10.1161/HYPERTENSIONAHA.109.145839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hu C, Dandapat A, Chen J, Liu Y, Hermonat PL, Carey RM, Mehta JL. Over-expression of angiotensin ii type 2 receptor (agtr2) reduces atherogenesis and modulates lox-1, endothelial nitric oxide synthase and heme-oxygenase-1 expression. Atherosclerosis. 2008;199:288–294. doi: 10.1016/j.atherosclerosis.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 209.Zeng W, Chen W, Leng X, He JG, Ma H. Chronic angiotensin-(1–7) administration improves vascular remodeling after angioplasty through the regulation of the tgf-beta/smad signaling pathway in rabbits. Biochem Biophys Res Commun. 2009;389:138–144. doi: 10.1016/j.bbrc.2009.08.112. [DOI] [PubMed] [Google Scholar]
- 210.Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW, Valledor AF, Davis RA, Willson TM, Witztum JL, Palinski W, Glass CK. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by pparalpha, beta/delta, and gamma. J Clin Invest. 2004;114:1564–1576. doi: 10.1172/JCI18730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Thorp E, Kuriakose G, Shah YM, Gonzalez FJ, Tabas I. Pioglitazone increases macrophage apoptosis and plaque necrosis in advanced atherosclerotic lesions of nondiabetic low-density lipoprotein receptor-null mice. Circulation. 2007;116:2182–2190. doi: 10.1161/CIRCULATIONAHA.107.698852. [DOI] [PubMed] [Google Scholar]
- 212.van Rooij E. The art of microrna research. Circ Res. 2011;108:219–234. doi: 10.1161/CIRCRESAHA.110.227496. [DOI] [PubMed] [Google Scholar]
- 213.Quiles JL, Mesa MD, Ramirez-Tortosa CL, Aguilera CM, Battino M, Gil A, Ramirez-Tortosa MC. Curcuma longa extract supplementation reduces oxidative stress and attenuates aortic fatty streak development in rabbits. Arterioscler Thromb Vasc Biol. 2002;22:1225–1231. doi: 10.1161/01.atv.0000020676.11586.f2. [DOI] [PubMed] [Google Scholar]
- 214.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, Spelsberg T. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107:2181–2184. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Habib ZA, Havstad SL, Wells K, Divine G, Pladevall M, Williams LK. Thiazolidinedione use and the longitudinal risk of fractures in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2010;95:592–600. doi: 10.1210/jc.2009-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bilik D, McEwen LN, Brown MB, Pomeroy NE, Kim C, Asao K, Crosson JC, Duru OK, Ferrara A, Hsiao VC, Karter AJ, Lee PG, Marrero DG, Selby JV, Subramanian U, Herman WH. Thiazolidinediones and fractures: Evidence from translating research into action for diabetes. J Clin Endocrinol Metab. 2010;95:4560–4565. doi: 10.1210/jc.2009-2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kahn SE, Zinman B, Lachin JM, Haffner SM, Herman WH, Holman RR, Kravitz BG, Yu D, Heise MA, Aftring RP, Viberti G. Rosiglitazone-associated fractures in type 2 diabetes: An analysis from a diabetes outcome progression trial (adopt) Diabetes Care. 2008;31:845–851. doi: 10.2337/dc07-2270. [DOI] [PubMed] [Google Scholar]
- 218.Ambrogini E, Almeida M, Martin-Millan M, Paik JH, Depinho RA, Han L, Goellner J, Weinstein RS, Jilka RL, O’Brien CA, Manolagas SC. Foxo-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 2010;11:136–146. doi: 10.1016/j.cmet.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Salmon AB, Richardson A, Perez VI. Update on the oxidative stress theory of aging: Does oxidative stress play a role in aging or healthy aging? Free Radic Biol Med. 2010;48:642–655. doi: 10.1016/j.freeradbiomed.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 221.Leibbrandt A, Penninger JM. Rank(l) as a key target for controlling bone loss. Adv Exp Med Biol. 2009;647:130–145. doi: 10.1007/978-0-387-89520-8_9. [DOI] [PubMed] [Google Scholar]
- 222.Osako MK, Nakagami H, Koibuchi N, Shimizu H, Nakagami F, Koriyama H, Shimamura M, Miyake T, Rakugi H, Morishita R. Estrogen inhibits vascular calcification via vascular rankl system: Common mechanism of osteoporosis and vascular calcification. Circ Res. 2010;107:466–475. doi: 10.1161/CIRCRESAHA.110.216846. [DOI] [PubMed] [Google Scholar]
- 223.Shao JS, Cheng SL, Charlton-Kachigian N, Loewy AP, Towler DA. Teriparatide (human parathyroid hormone (1–34)) inhibits osteogenic vascular calcification in diabetic low density lipoprotein receptor-deficient mice. J Biol Chem. 2003;278:50195–50202. doi: 10.1074/jbc.M308825200. [DOI] [PubMed] [Google Scholar]
- 224.Lee TC, Zhao YD, Courtman DW, Stewart DJ. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation. 2000;101:2345–2348. doi: 10.1161/01.cir.101.20.2345. [DOI] [PubMed] [Google Scholar]
- 225.Tkatchenko TV, Moreno-Rodriguez RA, Conway SJ, Molkentin JD, Markwald RR, Tkatchenko AV. Lack of periostin leads to suppression of notch1 signaling and calcific aortic valve disease. Physiol Genomics. 2009;39:160–168. doi: 10.1152/physiolgenomics.00078.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hakuno D, Kimura N, Yoshioka M, Mukai M, Kimura T, Okada Y, Yozu R, Shukunami C, Hiraki Y, Kudo A, Ogawa S, Fukuda K. Periostin advances atherosclerotic and rheumatic cardiac valve degeneration by inducing angiogenesis and mmp production in humans and rodents. J Clin Invest. 2010;120:2292–2306. doi: 10.1172/JCI40973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Yoshioka M, Yuasa S, Matsumura K, Kimura K, Shiomi T, Kimura N, Shukunami C, Okada Y, Mukai M, Shin H, Yozu R, Sata M, Ogawa S, Hiraki Y, Fukuda K. Chondromodulin-i maintains cardiac valvular function by preventing angiogenesis. Nat Med. 2006;12:1151–1159. doi: 10.1038/nm1476. [DOI] [PubMed] [Google Scholar]
- 228.Cimini M, Boughner DR, Ronald JA, Aldington L, Rogers KA. Development of aortic valve sclerosis in a rabbit model of atherosclerosis: An immunohistochemical and histological study. J Heart Valve Dis. 2005;14:365–375. [PubMed] [Google Scholar]
- 229.Cuniberti LA, Stutzbach PG, Guevara E, Yannarelli GG, Laguens RP, Favaloro RR. Development of mild aortic valve stenosis in a rabbit model of hypertension. J Am Coll Cardiol. 2006;47:2303–2309. doi: 10.1016/j.jacc.2005.12.070. [DOI] [PubMed] [Google Scholar]
- 230.Drolet MC, Couet J, Arsenault M. Development of aortic valve sclerosis or stenosis in rabbits: Role of cholesterol and calcium. J Heart Valve Dis. 2008;17:381–387. [PubMed] [Google Scholar]
- 231.Gkizas S, Koumoundourou D, Sirinian X, Rokidi S, Mavrilas D, Koutsoukos P, Papalois A, Apostolakis E, Alexopoulos D, Papadaki H. Aldosterone receptor blockade inhibits degenerative processes in the early stage of calcific aortic stenosis. Eur J Pharmacol. 2010;642:107–112. doi: 10.1016/j.ejphar.2010.05.048. [DOI] [PubMed] [Google Scholar]
- 232.Hekimian G, Passefort S, Louedec L, Houard X, Jacob MP, Vahanian A, Michel JB, Messika-Zeitoun D. High-cholesterol + vitamin d2 regimen: A questionable in-vivo experimental model of aortic valve stenosis. J Heart Valve Dis. 2009;18:152–158. [PubMed] [Google Scholar]
- 233.Marechaux S, Corseaux D, Vincentelli A, Richardson M, Ung A, Susen S, Zawadzki C, Beregi JP, Ezekowitz MD, Jude B, Le Tourneau T. Identification of tissue factor in experimental aortic valve sclerosis. Cardiovasc Pathol. 2009;18:67–76. doi: 10.1016/j.carpath.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 234.Ngo DT, Stafford I, Kelly DJ, Sverdlov AL, Wuttke RD, Weedon H, Nightingale AK, Rosenkranz AC, Smith MD, Chirkov YY, Kennedy JA, Horowitz JD. Vitamin d(2) supplementation induces the development of aortic stenosis in rabbits: Interactions with endothelial function and thioredoxin-interacting protein. Eur J Pharmacol. 2008;590:290–296. doi: 10.1016/j.ejphar.2008.05.051. [DOI] [PubMed] [Google Scholar]
- 235.Ngo DT, Stafford I, Sverdlov AL, Qi W, Wuttke RD, Zhang Y, Kelly DJ, Weedon H, Smith MD, Kennedy JA, Horowitz JD. Ramipril retards development of aortic valve stenosis in a rabbit model: Mechanistic considerations. Br J Pharmacol. 2011;162:722–732. doi: 10.1111/j.1476-5381.2010.01084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Rajamannan NM, Sangiorgi G, Springett M, Arnold K, Mohacsi T, Spagnoli LG, Edwards WD, Tajik AJ, Schwartz RS. Experimental hypercholesterolemia induces apoptosis in the aortic valve. J Heart Valve Dis. 2001;10:371–374. [PubMed] [Google Scholar]
- 237.Rajamannan NM, Subramaniam M, Springett M, Sebo TC, Niekrasz M, McConnell JP, Singh RJ, Stone NJ, Bonow RO, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced cellular proliferation and bone matrix production in the rabbit aortic valve. Circulation. 2002;105:2660–2665. doi: 10.1161/01.cir.0000017435.87463.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Speidl WS, Cimmino G, Ibanez B, Elmariah S, Hutter R, Garcia MJ, Fuster V, Goldman ME, Badimon JJ. Recombinant apolipoprotein a-i milano rapidly reverses aortic valve stenosis and decreases leaflet inflammation in an experimental rabbit model. Eur Heart J. 2010;31:2049–2057. doi: 10.1093/eurheartj/ehq064. [DOI] [PubMed] [Google Scholar]
- 239.Simmons CA, Grant GR, Manduchi E, Davies PF. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ Res. 2005;96:792–799. doi: 10.1161/01.RES.0000161998.92009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.