

Abstract
Familial partial lipodystrophy, Dunnigan variety (FPLD) is a well-recognized autosomal dominant disorder due to heterozygous missense mutations in lamin A/C (LMNA) gene. Most of the FPLD patients harbor mutations in the C-terminal of the lamin A/C and do not develop cardiomyopathy. On the other hand, affected subjects from three FPLD pedigrees with heterozygous R28W, R60G and R62G LMNA mutations in the amino-terminal had associated cardiomyopathy presenting as premature onset of congestive heart failure, dilated cardiomyopathy and conduction system disturbances. We report three new FPLD pedigrees presenting with cardiomyopathy associated with heterozygous LMNA mutations in the amino-terminal region. Two of them had previously reported R60G and R62G mutations and one has a novel D192V mutation. Affected subjects belonging to the pedigree with heterozygous R62G mutation had atrial fibrillation and required pacemaker implantation. The affected subjects from the other pedigrees with R60G and D192V mutation developed severe cardiomyopathy requiring defibrillator implantation and cardiac transplantation before 30 years of age in some and premature death in the fourth decade in others. Thus, our report provides further evidence of association of a multisystem dystrophy syndrome in FPLD patients harboring amino-terminal mutations in LMNA. Increased understanding of the genotype-phenotype association might help devise clinical strategies aimed at preventing devastating manifestations of cardiomyopathy including heart failure, arrhythmias and sudden death. Furthermore, the underlying molecular mechanisms by which these amino-terminal mutations cause lipodystrophy as well as cardiomyopathy remain to be understood.
Keywords: Cardiomyopathy, lamin A/C, lipodystrophy
Introduction
Mutations in LMNA (lamin A/C) have been associated with a wide variety of disorders which include familial partial lipodystrophy, Dunnigan variety (FPLD), childhood-onset generalized lipodystrophy, Hutchinson Gilford Progeria syndrome, muscular dystrophies, dilated cardiomyopathy, mandibuloacral dysplasia and others (1-3). FPLD is a rare autosomal dominant disorder with an estimated prevalence of less than 1 in 10 million (4). Most of the mutations in patients with FPLD cluster in carboxy-terminal immunoglobulin domain of lamin A/C (4). Approximately 75% of patients with FPLD have one of the heterozygous missense mutations in a single allele affecting the codon encoding the arginine residue at position 482 (5). These affected patients develop partial lipodystrophy, insulin resistance and subsequent complications, but they rarely develop cardiomyopathy or muscular dystrophy (6).
Previously, we reported two heterozygous missense mutations in exon 1 of LMNA – p.R28W (c.82C>T), which affects the head domain and p.R62G (c.184C>G), which affects the alpha helical rod domain of the amino terminus in two pedigrees (7). The affected subjects from these pedigrees showed features of a multisystem dystrophy syndrome involving the adipose tissue and the cardiac muscle leading to overlapping features of FPLD and cardiomyopathy. Cardiac manifestations in these subjects included atrioventricular conduction defects and atrial fibrillation requiring pacemaker implantation, and dilated cardiomyopathy and heart failure requiring cardiac transplantation. Subsequently, van der Kooi et al. (8) reported a family with two affected females who presented at 44 and 52 years of age with lipodystrophy, cardiac conduction system disturbances and p.R60G (c.178C>G) missense mutation in exon 1 of LMNA.
We now report data from three additional pedigrees with an overlapping phenotype of typical FPLD and severe cardiomyopathy, all with amino-terminal mutations in LMNA.
Subjects and Methods
The protocol was approved by Institutional Review Board of the University of Texas Southwestern Medical Center and written informed consent was obtained from all participating subjects. We included clinical information obtained from history and physical examination, review of medical records, and responses to written questionnaire. Information about cardiac involvement was obtained by interviews and review of medical records. The pedigree information is shown in Figure 1.
Fig. 1.

Pedigree of families with FPL, Dunnigan and cardiomyopathy.
Panels A, B and C show the pedigrees. Circles denote females and squares represent males. Arrows indicate probands. Half-filled symbols indicate affected subjects based on clinical information and unfilled symbols indicate unaffected subjects. A diagonal line across a symbol indicates a deceased subject. The genotypes of the subjects for whom DNA was available are given under the symbols. Patients with heterozygous mutations are shown as RG or DV and unaffected subjects with RR and DD. Panel D Sequence electropherogram of exon 3 of LMNA showing the novel heterozygous mutation (Asp192Val) in the proband from F9800 pedigree. The arrow indicates the site of mutation. The numbers below the electropherogram represent amino acid numbers in lamin A protein.
Pedigree F7200
This Caucasian pedigree is of European descent from the United States. The proband was referred to us with a diagnosis of lipodystrophy. Two affected subjects from this pedigree (7200.7 and 7200.8) visited the General Clinical Research Center at Dallas and underwent complete evaluation.
F7200.1
The father of the proband, who had similar phenotypic features as the proband, was diagnosed with diabetes at age 40 and had a history of cardiomegaly. He underwent pacemaker implantation at 40 years of age for ‘irregular heart beat’ and died at age 46 due to myocardial infarction. Interestingly, his brother reportedly had features of lipodystrophy and died at 48 years of age due to unknown cardiac cause.
F7200.4
This 43-year-old male is the older brother of the proband. He has lipodystrophy and developed diabetes at the age of 26 years along with dyslipidemia. He developed shortness of breath while taking pioglitazone and cardiac evaluation including coronary angiography revealed severe coronary artery disease. He underwent four vessel coronary artery bypass grafting at the age of 39 years. He developed atrial fibrillation/flutter at 39 years of age for which he underwent DC cardioversion and is now maintained on medical treatment. He has 2 children who have lipodystrophy.
F7200.7
The proband is a 40-year-old male diagnosed with FPLD during his evaluation at our center. He developed diabetes, hyperlipidemia and hypertension at the age of 36 years, and atrial fibrillation/flutter at the age of 37. After two unsuccessful attempts at DC cardioversion, he underwent electrophysiologic ablation for atrial fibrillation at 38 years of age. Due to symptoms of angina, he underwent an angiogram at 37 years of age and it revealed distal left anterior descending artery stenosis. He underwent coronary angioplasty with stent placement. Three months later, he underwent repeat angioplasty and brachytherapy for stent occlusion. He also had liposuction of chin and appendectomy in the past. He is a non-smoker and consumes about 56-60 g of alcohol per week. Physical examination showed a relative lack of fat over the upper and lower extremities with prominent musculature and superficial veins and excess fat around the abdomen, neck and chin (Fig. 2). He had acanthosis nigricans in the axillary regions. He had a regular heart rate with a grade 2/6 systolic murmur. Echocardiography done at 38 years of age showed normal left ventricular size with an estimated ejection fraction of 55%, mild tricuspid regurgitation, mild to moderate mitral regurgitation, and sclerotic aortic valve without any aortic regurgitation. He is now 45 years of age and had recently required cardioversion for persistent atrial fibrillation and has developed complete heart block. He also underwent near total thyroidectomy for goiter.
Fig. 2.
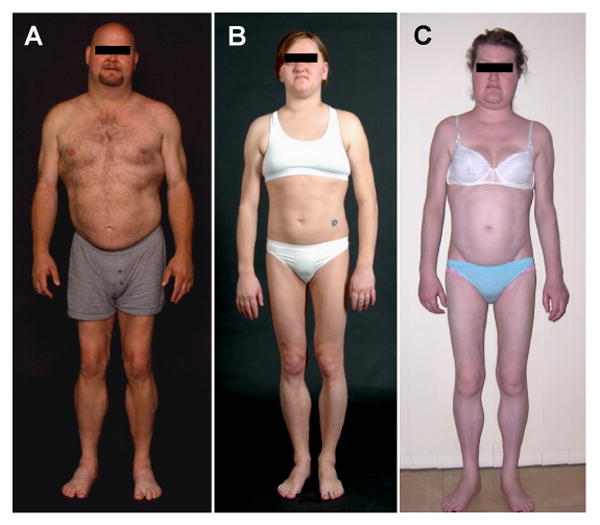
Body fat distribution pattern in Patients with FPLD and cardiomyopathy overlap syndrome.
(A) A 40-year-old male (FPL7200.7) with R62G LMNA mutation showing marked loss of subcutaneous fat from the upper and lower extremities and accumulation of sc fat in the face, chin and axillary region (B) A 26-year-old female (FPL9800.6) with D192V LMNA mutation showing marked loss of sc fat from the upper and lower extremities and accumulation of sc fat in the face and chin; (C) A 26-year-old female (FPL139.13) with R60G LMNA mutation showing marked loss of sc fat from the upper and lower extremities and accumulation of sc fat in the face and chin.
F7200.8
This 34-year-old sister of the proband developed diabetes at the age of 19, hypertension and dyslipidemia at the age of 20. She had a normal coronary angiogram at age 35 and a recent Thallium stress test was negative at age 40. She has not developed any arrhythmias. She takes rosiglitazone, metformin, fenofibrate, simvastatin, niacin, quinapril, iron and aspirin. She has a history of hirsutism, acanthosis nigricans and polycystic ovarian syndrome. She underwent liposuction of the chin and dorso-cervical hump at 25 years of age and cholecystectomy at 20 years of age. She showed typical clinical features of FPLD on physical examination.
Pedigree F9800
The subjects of this pedigree are from the United States and are of European ancestry. The proband was the only affected member of the family available for evaluation.
F9800.1
Paternal grandfather of the proband died suddenly at 25 years of age due to unknown cause.
F9800.4
Father of the proband was tall and muscular in appearance. He developed idiopathic dilated cardiomyopathy at the age of 25 and underwent cardiac transplantation at 28 years of age. He died 3 years later due to pneumonia.
F9800.5
Paternal aunt of the proband had features of lipodystrophy and developed idiopathic dilated cardiomyopathy and underwent cardiac transplantation at the age of 29 and died at the age of 34 due to pneumonia.
F9800.6
The proband is a 26-year-old woman who noticed loss of fat from the extremities and excess accumulation of fat in the face at the age of 19 (Fig. 2). She was noted to have hypertriglyceridemia, hyperinsulinemia and fatty liver at 25 years of age and was started on fenofibrate 160 mg once daily, metformin 750 mg twice daily, and pioglitazone 45 mg daily. Echocardiography at the age of 22 revealed mitral valve prolapse without regurgitation, mild tricuspid valve prolapse and a normal left ventricular function with an ejection fraction of 65%. Comparison of electrocardiograms at age 22 and 26, showed increased QRS duration from 86 to 116 ms, prolonged PR interval from 130 to 170 ms and a poor R wave progression. An oral glucose tolerance test revealed diabetes. Eight months later, she developed exertional dyspnea, cough, orthopnea, paroxysmal nocturnal dyspnea and fatigue. Chest radiograph showed right sided pleural effusion. A trans-thoracic echocardiogram revealed severe global hypokinesis with moderate to severe tricuspid regurgitation and a left ventricular ejection fraction of 30%. Electrocardiogram showed a new left bundle branch block. She responded well to initial medical therapy for congestive heart failure which included furosemide, carvedilol and lisinopril but is being considered for cardiac transplantation.
Pedigree F139
The subjects of this pedigree are from Australia and are of European ancestry. The proband was the only affected member of the family available for evaluation.
FPL 139.2
Paternal grandmother of the proband had FPLD features. She died at the age of 31 shortly after giving birth to her third child.
FPL 139.5
The father of the proband had phenotypic features, suggestive of FPLD. He was diagnosed with cardiomyopathy at the age of 29 and died at 31 years of age in the hospital due to congestive heart failure. He also had ulcerative colitis.
FPL 139.13
The proband is a 26-year-old woman who developed palpitations at age 18 when an echocardiogram showed moderate left ventricular systolic dysfunction with an ejection fraction of 44%, thickening of aortic valve leaflets with moderate aortic regurgitation, moderate mitral and tricuspid regurgitation with an elevated pulmonary artery pressure of 42 mm Hg. She was diagnosed with hypertension, fatty liver and pre-diabetes at age 21. She developed exertional dyspnea and orthopnea since 21 years of age and was diagnosed with congestive heart failure. She was medically managed on irbesartan/hydrochlorothiazide 300/12.5 mg once daily, carvedilol 25 mg twice daily, aldactone 25 mg once daily, metformin 1000 mg twice daily and furosemide 40 mg once daily. She underwent cholecystectomy due to gall stones and appendectomy at the age of 25. Due to her phenotypic features of fat loss over the extremities and trunk and increased submental and dorsocervical fat, she was diagnosed to have FPLD at the age of 26 (Fig. 2). Follow-up echocardiogram showed mild to moderate systolic dysfunction with an ejection fraction of 35% and mild to moderate aortic regurgitation. An electrocardiogram showed normal sinus rhythm with first degree atrioventricular block. Due to her family history of cardiomyopathy and early death, she underwent implantable cardioverter defibrillator placement.
Anthropometry
a) Anthropometric measurements
Height and body weight were measured by standard procedures. Skinfold thickness was measured with a Lange caliper (Cambridge Scientific Industries, Cambridge, Maryland) at five truncal sites (chest, mid-axillary, abdomen, subscapular, and suprailiac), six peripheral sites (biceps, triceps, forearm, hip, thigh and calf) on the right side of the body, and at the chin. The mean of three repeat measurements at each site was calculated.
b) Magnetic Resonance Imaging (MRI)
Whole body MRI studies were performed using a 1.5 Tesla imaging device (Philips Medical Systems, Best, Netherlands) and 5.2-2 software. The patients were evaluated using 10-mm thick T1 weighted imaging technique with TR (repetition time) of 580 milliseconds (ms) and a TE (echo time) of 8 ms and a 384 × 512 matrix combined with a 45-cm field of view. Adipose tissue distribution and thickness was assessed by visual inspection of the films.
c) Dual-energy x-ray absorptiometry (DEXA)
Whole body and regional fat in the head, trunk, upper and lower extremities were determined using a DEXA scan with a multiple detector fan-beam Hologic QDR-2000 densitometer (Hologic, Inc., Waltham, MA). The proportion of fat in individual regions as well as whole body was calculated as percentage of body mass.
d) Cardiac MRI
MR images of the heart were obtained using a 1.5 Tesla imaging device (Philips Medical Systems, Best, Netherlands) with the following imaging characteristics: slice thickness of 6 mm, TR of 4 ms, TE of 2 ms, 256 × 256 matrix combined with a 36-cm field of view, and a flip angle of 55 degrees.
Metabolic assessments and genetic analysis
a) Biochemical analyses
Plasma glucose was measured by the glucose oxidase method with a glucose analyzer (Beckman Coulter, Inc., Fullerton, CA). Serum insulin and leptin levels were determined by immunoassays using commercial kits (Linco Research, Inc., St. Charles, MO). Serum cholesterol, triglycerides, high density lipoprotein (HDL) cholesterol, and chemistry as well as blood hemoglobin A1C were analyzed as part of a systematic multichannel analysis (Synchron CX9 ALX Clinical System, Beckman, Fullerton, CA).
b) Oral glucose tolerance test (OGTT)
After an overnight fast, an OGTT using 75 g dextrose was performed in only those patients who did not have diabetes mellitus. Venous blood was collected for determination of serum glucose and insulin concentrations at 30 min, 15 min, and immediately before glucose administration, and at 30 min intervals thereafter, for 180 min.
c) Mutational analyses
The exons and exon-intron boundaries of the LMNA gene were sequenced according to Chen et al. (9)
Results
Three subjects, F7200.7, F7200.8 and F9800.6 underwent detailed anthopometric, metabolic and imaging studies at the GCRC. All 3 had physical features consistent with FPLD. With skinfold thickness over the extremities (triceps, biceps, forearm, thigh and calf) close to or below the 5th percentile of normal age and sex matched controls, while they were in the normal range or close to the 95th percentile over the chest, axillary and subscapular regions. Whole body MRI images also showed loss of subcutaneous fat from the extremities and hips, with normal to slightly increased fat in the face, trunk, submental, dorsocervical and intraabdominal regions. The total body fat percentage, as assessed by DEXA scan was in the normal range, but there was markedly less fat in the lower extremities compared to the trunk. Metabolic testing revealed diabetes mellitus and hypertriglyceridemia in F7200.7 and F7200.8, while F9800.6 had impaired glucose tolerance. Patient F139.13 had hepatic steatosis and impaired glucose tolerance.
Cardiac MRI in 3 subjects (F7200.7, F7200.8 and F9800.6) revealed no abnormality. The estimated ventricular volumes, mass and ejection fraction were in the normal range, and there was no evidence of cardiac fibrosis.
Mutational analysis showed that the proband from the F7200 family had a heterozygous missense mutation in exon 1 of LMNA, p.Arg62Gly (R62G; c.184C>G, CGC→GGC). The mutation segregated with the disease in affected members of the family. The proband from the F9800 family harbored a novel heterozygous missense mutation in exon 3 of LMNA, Asp192Val (D192V; c.575A>T, GAT→GTT) (Fig. 1D). Unaffected family members without lipodystrophy did not show the mutation. This patient also had homozygous variant p.H566H, c.1698C>T, a known single nucleotide polymorphism. The proband from the F139 family had a heterozygous missense mutation in exon 1 of LMNA, p.Arg60Gly (R60G; c.178C>G, CGC→GGC) and a heterozygous variant p.H566H.
Discussion
Laminopathies, which encompass a heterogeneous group of inherited disorders, can be caused by many different mutations in the LMNA gene. These mutations can give rise to distinct clinical syndromes or syndromes with overlapping phenotypes. For example, certain mutations in LMNA cause FPLD alone whereas some mutations cause an overlapping phenotype of FPLD and cardiomyopathy or FPLD, cardiomyopathy and skeletal muscle dystrophy (6, 7, 10).
The current report strengthens the previously reported association of exon 1 mutations (R60G and R62G) in LMNA with FPLD and cardiomyopathy phenotype (7). We also report a novel exon 3 mutation with similar phenotype of typical FPLD and cardiomyopathy leading to early cardiac deaths. In addition, we previously described R28W mutation in a family of 5 phenotypically affected members with FPLD out of whom 3 had overlapping features of cardiomyopathy and conduction system defects leading to sudden cardiac death, cardiac transplant and pacemaker implantation (7). Besides these, 2 pedigrees with heterozygous R527P exon 9 mutation have been reported with FPLD, muscular dystrophy and cardiac arrhythmias (8) and one with heterozygous C591F exon 11 mutation with FPLD and hypertrophic cardiomyopathy and aortic stenosis (11).
Interestingly, the patients with exon 1 and 3 mutations have similar pattern of body fat loss as seen in patients with typical FPLD harboring R482Q, R482W and R482L mutations in exon 8 of LMNA. The affected patients also had variable phenotypic manifestations of insulin resistance, diabetes or impaired glucose tolerance, hepatic steatosis, hypertriglyceridemia, acanthosis nigricans and polycystic ovarian syndrome as seen in patients with typical FPLD. Besides exon 8 mutations, mutations in exons 7 and 9 also cause typical FPLD without cardiomyopathy while mutations in exon 11 have been shown to cause both typical and atypical, mild FPLD without cardiomyopathy (5, 12).
Previously reported patients by us with FPLD and cardiomyopathy overlap syndrome by us did not show early onset cardiac disease or a severe cardiac phenotype (7). Out of the 8 affected subjects, one had cardiac transplantation at age 48 and 4 had pacemaker implantation between the ages of 37 to 68 due to conduction system defects. In contrast, the newly reported patients with the R60G and D192V mutations have severe cardiac manifestations and present in early 20s. LMNA mutations causing predominantly cardiomyopathy without associated FPLD have been described in almost all the exons of the gene (1, 13, 14). It is the most common gene defect found in patients with dilated cardiomyopathy and it is shown to be associated with poor prognosis (14, 15). Cardiomyopathy is rarely seen in affected subjects under the age of 20 and tends to occur mostly in the middle aged subjects (14). Some of the affected subjects in the F7200 pedigree also had evidence of coronary heart disease which could also contribute to arrhythmias and heart failure. Patients with FPLD are indeed at high risk for coronary atherosclerotic heart disease, but cardiomyopathy and heart failure are seldom reported in other FPLD patients with exon 8 mutation despite marked dyslipidemia. Further, patients in the other 2 pedigrees reported had no evidence of atherosclerotic heart disease, strongly suggesting the non-ischemic nature of LMNA-associated cardiomyopathy.
The early onset of fatal cardiomyopathy described in F9800 and F139 pedigrees brings up the question of which patients with FPLD need to be screened for cardiomyopathy and how frequently they need to be followed up. Mutations in exon 3 suggest that not only extreme amino-terminal mutations but also those affecting the α-helical rod domain can also cause the overlapping phenotype.
There are no management guidelines for patients with cardiomyopathy due to LMNA mutations. Pasotti et al. (14). studied the long-term outcome of dilated cardiomyopathies due to heterozygous LMNA mutations in 27 families with 60 phenotypically affected members. During a median follow-up of 57 months, there were about 15 heart transplants, 15 sudden cardiac deaths, 12 implantable cardioverter-defibrillator placements, 1 death from end stage heart failure in the affected members suggesting the aggressive nature of these mutations and poor prognosis. They proposed that patients LMNA-related dilated cardiomyopathies should undergo an implantable cardioverter defibrillator implantation earlier than that suggested by the current American College of Cardiology guidelines for patients with ventricular arrhythmias (16). They also recommended avoiding highly competitive sports in patients with clinically overt disease (14). This strategy may also prevent sudden deaths in patients with cardiomyopathy due to LMNA mutations (17).
But, the questions remain as to how to recognize the high risk subjects prior to the development of cardiomyopathy and risk stratify these subjects especially when they have combined features of lipodystrophy and cardiomyopathy. Definitely, prior family history of premature death or cardiac transplantation can alert the physicians to aggressively manage manifestations of cardiomyopathy in the affected subjects. Whether medical therapy including diuretics, beta-adrenergic blockers, angiotensin-converting enzyme inhibitors and other drugs for congestive heart failure can postpone the age at which cardiac transplantation is required, remains unclear. Whether such therapy impacts the natural progression of cardiomyopathy also remains unknown. Implantable cardioverter defibrillators and pacemakers can help in managing arrhythmias such as complete heart block, atrial fibrillation and ventricular arrhythmias. However, how proactively we should advise these interventions in patients who have not yet demonstrated any syncope or arrhythmias on Holter monitoring, is not clear. Whether subtle electrocardiographic parameters such as prolongation of the PR, QRS, or QT interval can predicate serious arrhythmias in patients with cardiomyopathy due to LMNA mutations, remains to be studied.
There are probably multiple mechanisms causing cellular toxicity in patients with LMNA mutations (2, 3, 18). We reported that accumulation of mutant farnesylated prelamin A may cause toxicity by affecting the nuclear pore complexes (19). However, the LMNA mutations in exon 1 and 3, which are far away from the carboxy-terminus are unlikely to affect the post-translational processing of prelamin A. On the other hand, interaction of mutant lamin A with other nuclear lamina proteins, transcription factors or chromatin may be responsible for cellular toxicity. Finally, the molecular mechanisms for pleiotropy associated with LMNA mutations remain to be elucidated to explain why the reason(s) behind some unique mutations causing FPLD while others leading to an overlapping phenotype.
Fig. 3.
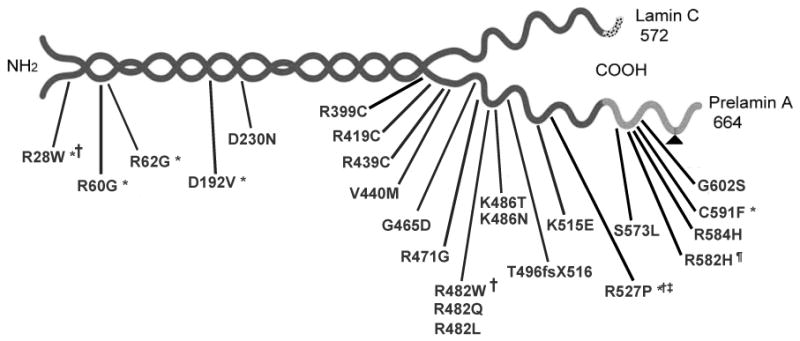
The structure of lamins A and C with site of mutations described in patients with FPLD.
Lamin A and C isoforms share the same 566 amino acids. Lamin C has 6 unique carboxy-terminal amino acids and the precursor of lamin A, prelamin A, has 98 unique carboxy-terminal amino acids. Post-translational processing of prelamin A including proteolysis of 18 C-terminal amino acids (indicated by a triangle) is catalyzed by zinc metalloproteinase. Most of the mutations in familial partial lipodystrophy are located in the tail region. Peculiar phenotypic features associated with various mutations are represented by symbols. For example, *, represents associated cardiomyopathy; †, associated Emery-Dreifuss muscular dystrophy; ‡, associated limb-girdle muscular dystrophy; #, mild myopathy; and § mild lipodystrophy.
Table.1. Cardiac manifestations in affected subjects from the three pedigrees with LMNA mutations and FPLD.
| Affected subject | LMNA genotype | Age at report / Sex | Cardiac manifestations (Age of onset in years) | Age of death | |||||
|---|---|---|---|---|---|---|---|---|---|
| Atrial Fibrillation | CHF | Conduction disturbances | ICD/Pacemaker implant | Heart transplant | CHD | ||||
| F7200.1 | NA | 46/M | + | Unknown | Unknown | + (40) | - | + | 46 |
| F7200.4 | NA | 39/M | + (39) | - | - | - | - | + | |
| F7200.7 | R62G | 40/M | + (36) | - | - | - | - | + | |
| F7200.8 | R62G | 36/F | - | - | - | - | - | - | |
| F9800.1 | NA | 25/M | Unknown | Unknown | Unknown | - | - | N/A | 25 |
| F9800.4 | NA | 25/M | - | + (25) | - | - | + (28) | - | 31 |
| F9800.5 | NA | 29/F | - | + (29) | - | - | + (29) | - | 34 |
| F9800.6 | D192V | 26/F | - | + (27) | - | - | - | - | |
| F139.2 | NA | 31/F | Unknown | Unknown | Unknown | - | - | N/A | 31 |
| F139.5 | NA | 29/M | - | + (29) | - | - | - | - | 31 |
| F139.13 | R60G | 26/F | - | + (21) | - | + (26) | - | - | |
Abbreviations: NA, not available; M, male; F, female; ICD, Implantable cardioverter defibrillator; CHF, congestive heart failure; CHD, coronary heart disease.
Acknowledgments
We are thankful to Drs. Amit Vora, David Rees and Bruce Bode for sharing information on their patients with us. This work was supported by the National Institutes of Health grant R01-DK54387, M01-RR00633, UL1-RR024982 and Southwest Medical Foundation. We thank Sarah Masood for illustrations and Crystal Kittisopikul for genotyping.
References
- 1.Jacob KN, Garg A. Laminopathies: multisystem dystrophy syndromes. Molecular genetics and metabolism. 2006;87:289–302. doi: 10.1016/j.ymgme.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 2.Rankin J, Ellard S. The laminopathies: a clinical review. Clin Genet. 2006;70:261–274. doi: 10.1111/j.1399-0004.2006.00677.x. [DOI] [PubMed] [Google Scholar]
- 3.Worman HJ, Fong LG, Muchir A, et al. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest. 2009;119:1825–1836. doi: 10.1172/JCI37679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garg A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350:1220–1234. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- 5.Haque WA, Oral EA, Dietz K, et al. Risk factors for diabetes in familial partial lipodystrophy, Dunnigan variety. Diabetes Care. 2003;26:1350–1355. doi: 10.2337/diacare.26.5.1350. [DOI] [PubMed] [Google Scholar]
- 6.Hegele RA. Familial partial lipodystrophy: a monogenic form of the insulin resistance syndrome. Molecular Genetics & Metabolism. 2000;71:539–544. doi: 10.1006/mgme.2000.3092. [DOI] [PubMed] [Google Scholar]
- 7.Garg A, Speckman RA, Bowcock AM. Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene. Am J Med. 2002;112:549–555. doi: 10.1016/s0002-9343(02)01070-7. [DOI] [PubMed] [Google Scholar]
- 8.van der Kooi AJ, Bonne G, Eymard B, et al. Lamin A/C mutations with lipodystrophy, cardiac abnormalities, and muscular dystrophy. Neurology. 2002;59:620–623. doi: 10.1212/wnl.59.4.620. [DOI] [PubMed] [Google Scholar]
- 9.Chen L, Lee L, Kudlow BA, et al. LMNA mutations in atypical Werner's syndrome. Lancet. 2003;362:440–445. doi: 10.1016/S0140-6736(03)14069-X. [DOI] [PubMed] [Google Scholar]
- 10.Hegele R. LMNA mutation position predicts organ system involvement in laminopathies. Clin Genet. 2005;68:31–34. doi: 10.1111/j.1399-0004.2005.00447.x. [DOI] [PubMed] [Google Scholar]
- 11.Araujo-Vilar D, Lado-Abeal J, Palos-Paz F, et al. A novel phenotypic expression associated with a new mutation in LMNA gene, characterized by partial lipodystrophy, insulin resistance, aortic stenosis and hypertrophic cardiomyopathy. Clin Endocrinol (Oxf) 2008;69:61–68. doi: 10.1111/j.1365-2265.2007.03146.x. [DOI] [PubMed] [Google Scholar]
- 12.Garg A, Vinaitheerthan M, Weatherall P, et al. Phenotypic heterogeneity in patients with familial partial lipodystrophy (Dunnigan variety) related to the site of mis-sense mutations in Lamin A/C (LMNA) gene. J Clin Endocrinol Metab. 2001;86:59–65. doi: 10.1210/jcem.86.1.7121. [DOI] [PubMed] [Google Scholar]
- 13.Pasotti M, Repetto A, Pisani A, et al. Diseases associated with lamin A/C gene defects: what the clinical cardiologist ought to know. Ital Heart J Suppl. 2004;5:98–111. [PubMed] [Google Scholar]
- 14.Pasotti M, Klersy C, Pilotto A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008;52:1250–1260. doi: 10.1016/j.jacc.2008.06.044. [DOI] [PubMed] [Google Scholar]
- 15.Mestroni L, Taylor MR. Lamin A/C gene and the heart: how genetics may impact clinical care. J Am Coll Cardiol. 2008;52:1261–1262. doi: 10.1016/j.jacc.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) J Am Coll Cardiol. 2006;48:e247–346. doi: 10.1016/j.jacc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 17.Meune C, Van Berlo JH, Anselme F, et al. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006;354:209–210. doi: 10.1056/NEJMc052632. [DOI] [PubMed] [Google Scholar]
- 18.Davies BS, Fong LG, Yang SH, et al. The posttranslational processing of prelamin A and disease. Annu Rev Genomics Hum Genet. 2009;10:153–174. doi: 10.1146/annurev-genom-082908-150150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan Y, Garg A, Agarwal AK. Mislocalization of prelamin A Tyr646Phe mutant to the nuclear pore complex in human embryonic kidney 293 cells. Biochem Biophys Res Commun. 2007;355:78–84. doi: 10.1016/j.bbrc.2007.01.116. [DOI] [PMC free article] [PubMed] [Google Scholar]