

Abstract
ADP is the endogenous agonist for both P2Y1 and P2Y12 receptors, which are important therapeutic targets. It was previously demonstrated that ADP and a synthetic agonist, 2-methylthioadenosine 5′-diphosphate (2MeSADP), can induce apoptosis by activating the human P2Y1 receptor heterologously expressed in astrocytoma cells. However, it was not known whether the P2Y12 receptor behaved similarly. We demonstrated here that, unlike with the Gq-coupled P2Y1 receptor, activation of the Gi-coupled P2Y12 receptor does not induce apoptosis. Furthermore, activation of the P2Y12 receptor by either ADP or 2MeSADP significantly attenuates the tumor necrosis factor α (TNFα)-induced apoptosis in 1321N1 human astrocytoma cells. This protective effect was blocked by the P2Y12 receptor antagonist 2-methylthioAMP and by inhibitors of phospholipase C (U73122) and protein kinase C (chelerythrin), but not by the P2Y1 receptor antagonist MRS2179. Toward a greater mechanistic understanding, we showed that hP2Y12 receptor activation by 10 nM 2MeSADP, activates Erk1/2, Akt, and JNK by phosphorylation. However, at a lower protective concentration of 100 pM 2MeSADP, activation of the hP2Y12 receptor involves only phosphorylated Erk1/2, but not Akt or JNK. This activation is hypothesized as the major mechanism for the protective effect induced by P2Y12 receptor activation. Apyrase did not affect the ability of TNFα to induce apoptosis in hP2Y12-1321N1 cells, suggesting that the endogenous nucleotides are not involved. These results may have important implications for understanding the signaling cascades that follow activation of P2Y1 and P2Y12 receptors and their opposing effects on cell death pathways.
Keywords: Apoptosis, Nucleotides, G protein-coupled receptors, Tumor necrosis factor, Phospholipase C, Protein kinase C
1. Introduction
P2Y1 and P2Y12 purinergic receptors are G protein-coupled receptors that are activated by endogenous ADP and are important drug targets [1,2]. Activation of both Gq-coupled P2Y1 and Gi-coupled P2Y12 receptors in platelets induces aggregation, although via different mechanisms [3–5]. The P2Y12 receptor is the site of action of several clinically used antithrombotic agents, i.e. clopidogrel (also known as Plavix) and ticlopidine [1,4–6], which must be metabolized in vivo prior to receptor inhibition. Recently, the wide distribution of P2Y12 mRNA in human, mouse, and rat brain tissues was reported [7,8], and the evidence indicates that this receptor is associated with astrocyte function. However, the role of the P2Y12 receptor in brain function is largely unclear.
Activation of the widely distributed P2Y1 receptor was demonstrated to induce apoptosis in 1321N1 astrocytoma cells heterologously expressing the receptor [9]. In the present study, we explore the intriguing possibility that the effects of ADP and its analogues on intracellular signaling pathways involving the Ras/extracellular signal-regulated protein kinase (Erk) and phosphatidylinositol 3-kinase (PI3-K) may depend on both of these purinergic receptors. Erk1/2 and PI3-K are associated with cell proliferation and differentiation [10,11]. The aims of this study were to determine whether the ADP-sensitive P2Y12 nucleotide receptor affects apoptotic pathways involving the regulation of Erk1/2 and PI3-K activity and to investigate the possible bridge between signaling pathways triggered by the P2Y1 and P2Y12 receptors. For this purpose, we used 1321N1 astrocytoma cells stably expressing the human (h) P2Y1 or P2Y12 receptor. This study demonstrated that the activation of the P2Y1 receptor induced apoptosis, but the P2Y12 receptor activation did not. Furthermore, it was demonstrated that 2-methylthioadenosine 5′-diphosphate (2Me-SADP) activates the P2Y12 receptor to antagonize tumor necrosis factor α (TNFα)-induced apoptosis and that this protection occurs principally with modulation of Erk1/2 phosphorylation, with possible involvement of pAkt and phosphorylated c-Jun N-terminal kinase (pJNK) signaling pathways.
2. Materials and methods
2.1. Materials
The 1321N1 astrocytoma cells stably transfected with the hP2Y1 or hP2Y12 receptor were generously provided by Prof. T.K. Harden (University of North Carolina, Chapel Hill, NC). Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were purchased from Life Technologies (Rockville, MD). Plastic collagen-coated cellware was purchased from Becton Dickinson (Bedford, MA). Horseradish peroxidase (HRP)-linked anti-rabbit IgG, HRP-linked anti-mouse IgG antibodies, p38, Akt1/2, caspase-3, Erk1 and Erk2, JNK, and α, β isoforms of protein kinase C (PKC) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The antibodies to the phosphorylated forms were also supplied by Santa Cruz Biotechnology. TNFα was purchased from Biosource International (Camarillo, CA). The rabbit polyclonal antibodies for P2Y1 and P2Y12 receptors were purchased from Alomone Labs, Ltd. (Jerusalem, Israel). APO-BrdU TUNEL Assay Kit was purchased from Molecular Probes (Invitrogen Detection Technologies, Carlsbad, CA). ATP Assay Kit was purchased from Perkin-Elmer (Boston, MA). Calcium Mobilization Assay Kit was purchased from Molecular Devices (Sunnyvale, CA). Phospholipase C (PLC) inhibitor 1-[6-((17b-3-Methoxyestra-1,3,5(10)-trien-17-yl)-amino)hexyl]-1Hpyrrole-2,5-dione (U73122), IP3 (inositol trisphosphate) receptor inhibitor 2-APB, hematoxylin solution, cycloheximide, pertussis toxin (PTX), and all other reagents were purchased from Sigma (St. Louis, MO).
2.2. Cell culture
Human 1321N1 astrocytoma cells stably transfected with the hP2Y12 receptor were grown at 37 °C in a humidified incubator with 5% CO2/95% air in DMEM/F-12 medium (1:1) supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine. The cells were passaged using trypsinization every 4–5 days.
2.3. Induction and detection of apoptosis
Cells were plated in six-well collagen-coated plates at an original seeding density of 200,000–500,000 cells per well and cultured to ~70% confluence for the experiments. Cells were used at this stage since fully confluent cultures are easily detached and generally express a reduced number of receptors per cell [12]. TNFα was used to induce apoptosis in the astrocytoma cells [13]. Medium containing 5 μg/ml cycloheximide was added to the cells grown to ~70% confluence. Cycloheximide, an inhibitor of protein synthesis, was included in all experiments concerning TNFα-induced apoptosis. The cells were treated with both 2MeSADP and TNFα for 4 h. Antagonists were added to the incubation medium 20 min prior to addition of 2MeSADP (1–10 nM) and TNFα (10–20 ng/ml). After 4 h, the medium was changed and was left for 16 h. The medium contained 5 ng/ml cycloheximide during the entire incubation. Cell death was observed 16 h later.
The cleavage of genomic DNA into small oligonucleosomal fragments is a late hallmark of cells that succumb to apoptosis. DNA fragmentation of apoptotic cells is detected by exploiting the fact that the DNA breaks expose a large number of 3′-hydroxyl ends. These fragments can be labeled in cells by the Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) method. Briefly, treated cells were fixed with 1% paraformaldehyde for 15 min at 4 °C and permeabilized with 70% ethanol at −20 °C for at least 12–18 h. After washing, the presence in apoptotic cells of ladder fragments was detected by labeling their termini with 5-bromo-2′-deoxyuridine 5′-triphosphate (BrdUTP) and incubating the cells in the DNA-labeling solution for 60 min at 37 °C. Subsequently, the reaction was blocked by the anti-BrdU antibody, and then propidium iodide (PI)/RNase A staining buffer was added for 30 min at room temperature. For microscopy applications, the cells were deposited onto slides.
2.4. Histochemical staining and cell viability
Cells were grown on poly-L-lysine-treated (100 μM/ml) or collagen-coated six-well plates until they reached ~70% confluence. For histochemical staining, the medium was removed from the wells, and the plates were rinsed with phosphate-buffered saline (PBS), fixed with methanol for 10 min, and washed three times with PBS for 5 min each. The cells were incubated for 3 min in 4 g/l hematoxylin solution (35.2 g/l aluminum sulfate, 0.4 g/l sodium iodate). After three additional washes for 5 min each in PBS, the plates were allowed to air dry before adding glycerol. The image was visualized with a Zeiss (Thornwood, NY) wide-field microscope.
For the cell viability assay, after treatment to induce apoptosis, cells detached by trypsin-EDTA were combined and centrifuged. The cells were washed with PBS and resuspended to 2–5 × 105 ml−1 in PBS. The cells were then treated with PI solution (final concentration: 2 μg/ml). After 10 min incubation at room temperature in the dark, the PI-positive cell fraction was analyzed by flow cytometry (BD FacsCalibur, Becton Dickinson).
2.5. ATP assay
Control and treated cells were resuspended in ice-cold lysis buffer, and the cells extracts were used to measure ATP content with a luciferin-luciferase bioluminescence kit (ATP Lite, Perkin-Elmer, USA) following the manufacturer’s protocol. The values are expressed as μmol/mg protein.
2.6. Western blotting
Cells were washed with PBS and solubilized in 2 mM EGTA, 25 mM β-glycerophosphate, 1% Triton X-100, 10% glycerol, 1 mM DTT, 1 mM Na3VO4, 5 mM NaF, 10 μg/μl leupeptin, 10 μg/μl aprotinin, 1 mM PMSF, and 20 mM HEPES.
Whole-cell lysates (50 μg) were separated on 10% SDS-PAGE, then transfered to a nitrocellulose membrane (Invitrogen), and incubated with primary antibodies at the appropriate dilution: antibodies to JNK1 (1:800), caspase-3 (1:800), p38 (1:500), Akt1/2 (1:1000), or PKC α and βI (1:800). For detection of the phosphorylated forms of the kinases, the nitrocellulose membrane was incubated with a 1:800 dilution of the antiphosphospecific antibodies.
For analysis of Erk activation, Western blots were generated as described above but developed with an affinity-purified mouse monoclonal antibody that specifically recognizes the dually Thr202/Tyr204-phosphorylated, active form of Erk (anti-phospho-Erk; 1:1000; Santa Cruz Biotechnology). The Western blots shown are representative of three separate experiments, and each panel is taken from a single immunoblot.
2.7. Calcium mobilization assay
Cells were grown overnight in 100 μl of medium in 96-well flat-bottom plates at 37 °C at 5% CO2 or until they reached ~60–80% confluency. The calcium assay kit (Molecular Devices) was used as directed with no washing of cells and with probenecid added to the loading dye at a final concentration of 2.5 mM to increase dye retention. Cells were loaded with 50 μl dye with probenecid in each well and incubated for 45 min at room temperature. The compound plate was prepared with dilutions of various compounds in Hank’s Buffer at pH 7.2. For antagonist studies, both agonist and antagonist were added to the sample plate. Samples were run in duplicate with a Flexstation I (Molecular Devices) at room temperature. Cell fluorescence (excitation = 485 nm; emission = 525 nm) was monitored following exposure to a compound. Increases in intracellular calcium are reported as the maximum fluorescence value after exposure minus the basal fluorescence value before exposure.
2.8. Statistical analysis
Pharmacological parameters were analyzed with the Graph-PAD Prism software (Version 4.0, GraphPAD Prism, San Diego, CA). Data were expressed as mean ± standard error. Statistical significance was calculated using the Student’s t-test. P values less than 0.05 (P < 0.05) were considered to be statistically significant.
3. Results
3.1. Detection of the hP2Y1 and hP2Y12 receptors by specific anti-rabbit antibodies
Nucleotide effects in 1321N1 astrocytoma cells stably expressing the hP2Y receptors have been studied extensively [14]. Expression of hP2Y receptors was confirmed using specific anti-rabbit antibodies (against hP2Y1 and hP2Y12). We were unsuccessful to demonstrate modulation of cAMP levels by 2MeSADP in either astrocytoma cells or CHO cells stably transfected with the hP2Y12 receptor (data not shown). However, Western blot analysis demonstrated the presence of the hP2Y12 receptor (Fig. 1A) but not the P2Y1 receptor, in astrocytoma cells stably transfected with the hP2Y12 receptor. The P2Y12 receptor was not expressed in control astrocytoma cells. As a positive control experiment, we also detected the P2Y12 receptor in rat C6 glioma cells, as reported [15]. Similar positive results were shown for detection of the hP2Y1 receptor in astrocytoma cells stably transfected with this subtype (data not shown).
Fig. 1.

(A) Western blot detection of the hP2Y12 receptor expressed in various cells. Band 1, control 1321N1 astrocytoma cells; 2, stably transfected hP2Y1-1321N1 cells; 3, stably transfected hP2Y12-expressing CHO cells; 4, stably transfected hP2Y12-1321N1 cells; 5, rat C6 glioma cells. For measurement of apoptotic effects, the control 1321N1 cells and hP2Y1 and hP2Y12-expressing 1321N1 cells were pretreated with 2MeSADP at the concentration indicated for 20 min and further treated with TNFα (20 ng/ml) for 4 h. The medium was then replaced with fresh medium following washing with Ca2+-free PBS. The medium always contained 5 μg/ml cycloheximide. In all experiments, cell death was observed on the following day (total of 16 h incubation). Groups labeled * are significantly different from control (P < 0.05). (B) Effects of 2MeSADP (100 nM) in control astrocytoma cells. Data shown are mean ± S.D. from two independent experiments in triplicate. (C) Effects of 2MeSADP (300 nM) detected by the PI method in hP2Y1-1321N1 cells. The PI-positive cells were analyzed with a FacsCalibur instrument. Data shown are mean ± S.D. from two independent experiments in duplicate. (D) Concentration-dependent protection by 2MeSADP (0.001–100 nM) in hP2Y12-1321N1 cells. Data shown are mean ± S.D. from three independent experiments in triplicate.
3.2. TNFα induced cell death in 1321N1 astrocytoma cells and effects of nucleotides
As shown in Fig. 1A, TNFα induced cell death in 1321N1 astrocytoma cells stably expressing the hP2Y12 receptor. There was no protection following TNFα treatment upon activation of control 1321N1 astrocytoma cells and cells stably expressing the P2Y1 receptor by the agonist 2MeSADP (100 nM) (Fig. 1B and C). It was found previously that 2MeSADP (300 nM) induced cell death in 1321N1 cells stably expressing P2Y1 receptors [9]; we made the same observations (Fig. 1C). It was demonstrated that activation of the P2Y12 receptor by 2MeSADP protects against TNFα induced cell death in a concentration dependent manner (Fig. 1D). Thus, 2MeSADP activates the P2Y12 receptor to protect against cell death and activates the P2Y1 receptor to induce cell death. However, administration of 2MeSADP alone in P2Y12-expressing cells did not influence the percentage of PI-positive cells indicative of the degree of cell death, although at a concentration of 100 pM it protected against TNFα-induced cell death by 40%.
The protective effect of hP2Y12 receptor activation was antagonized by a relatively nonselective antagonist of the P2Y12 receptor, 2MeSAMP [16], but not by a selective antagonist of the P2Y1 receptor, N6-methyl-2′-deoxyadenosine-3′,5′-bisphosphate (MRS2179; Fig. 2) at 30 nM. MRS2179 at a concentration of 100 nM also did not show any blocking effect (data not shown).
Fig. 2.
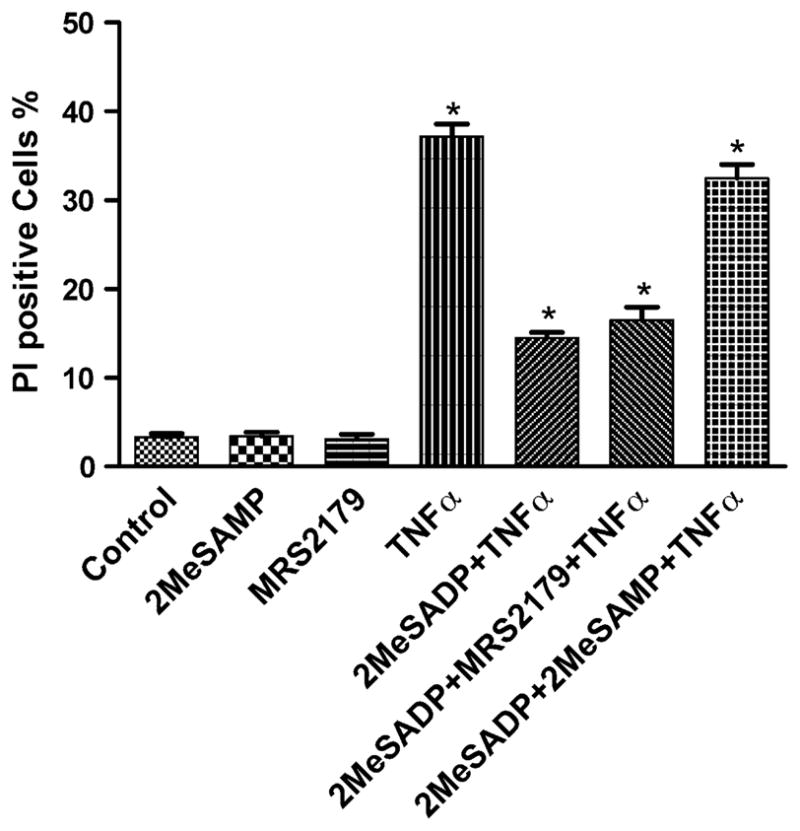
Effect of P2Y12 and P2Y1 receptor antagonists on the protection by 2MeSADP against TNFα-induced apoptosis. The P2Y12-expressing cells were pretreated with 2MeSAMP, an antagonist of the P2Y12 receptor (50 μM), and MRS2179, an antagonist of the P2Y1 receptor (100 nM), 20 min before treatment with 2MeSADP (10 nM) and TNFα 20 ng/ml for 4 h. The medium was then replaced with fresh medium following washing with Ca2+-free PBS. Cell death was observed on the following day (total of 16 h incubation). The medium always contained 5 μg/ml cycloheximide. The cell suspension was stained with a PI solution (final concentration: 2 μg/ml). The PI-positive cells were analyzed with a FacsCalibur instrument. Data shown are mean ± S.D. from three independent experiments in duplicate. Groups labeled * are significantly different from control (P < 0.05).
Basal constitutive release of nucleotides occurs from most, if not all, cell types [17–20], and this release is counterbalanced by ectonucleotidase-catalyzed degradation [19,21]. Baseline apoptosis in the presence of TNFα depends on extracellular calcium, and ATP is released from most eukaryotic cells into the extracellular environment [22]. Treatment with apyrase (2 U/ml) did not affect the ability of TNFα to induce apoptosis in hP2Y12-1321N1 cells (Fig. 3). Therefore, there was no indication of an effect of endogenous nucleotides on the outcome of the apoptosis experiments.
Fig. 3.
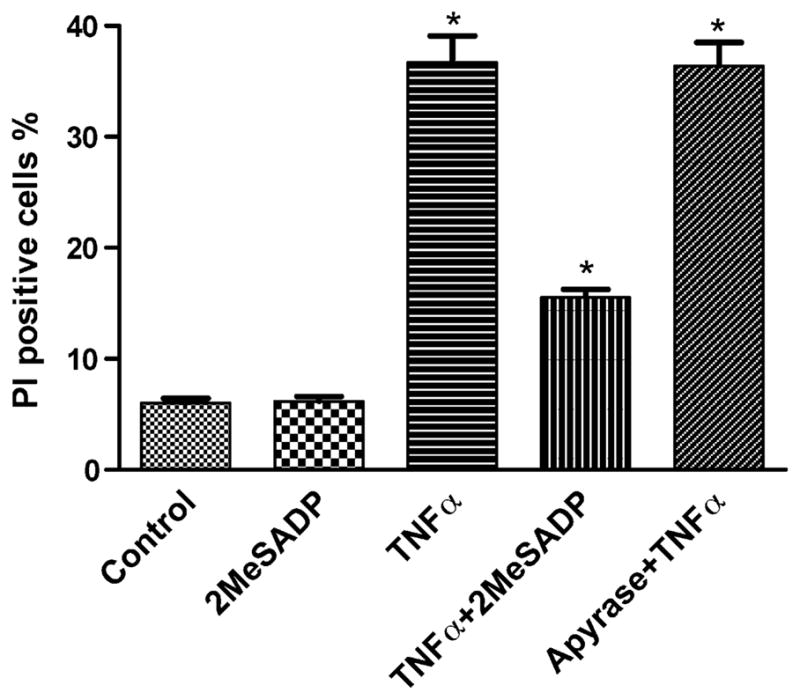
Effect of apyrase on TNFα-treated hP2Y12-1321N1 cells. Apyrase (2 U/ml) was added 20 min before treatment with 2MeSADP (10 nM) and TNFα 20 ng/ml for 4 h. The medium was then replaced with fresh medium following washing with Ca2+-free PBS. Cell death was observed on the following day (total of 16 h incubation). The medium always contained 5 μg/ml cycloheximide. The cell suspension was stained with a PI solution (final concentration: 2 μg/ml). The PI-positive cells were analyzed with a FacsCalibur instrument. Data shown are mean ± S.D. from three independent experiments in duplicate. Groups labeled * are significantly different from control (P < 0.05).
3.3. Measurement of ATP levels
Apoptosis requires energy in the form of intracellular ATP, indicating that programmed cell death, as opposed to necrosis, is an energy-dependent, active physiological and pathophysiological phenomenon. In this study, we measured ATP levels in homogenates of P2Y12 receptor-expressing astrocytoma cells after apoptotic treatment. It was shown that the level of ATP decreased upon TNFα-induced apoptosis in P2Y12 receptor-expressing astrocytoma cells (Fig. 4). Under conditions of protection against apoptosis by P2Y12 receptor activation, the level of intracellular ATP was partially restored, and this protective effect was blocked by the P2Y12 receptor antagonist 2MeSAMP.
Fig. 4.
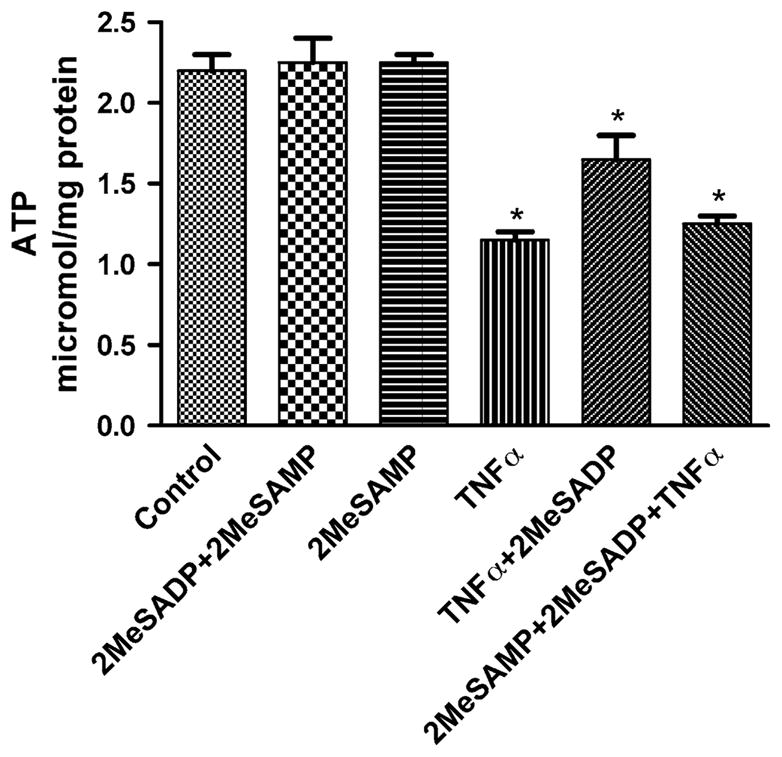
ATP levels in hP2Y12-1321N1 cells following treatment with TNFα to induce cell death. The cells were pretreated with 2MeSADP (10 nM) for 20 min and further treated with TNFα 20 ng/ml for 4 h. The medium was then replaced with fresh medium following washing with Ca2+-free PBS. Cell death was observed on the following day (total of 16 h incubation). The ATP levels were analyzed by the luciferin-luciferase bioluminescence kit (ATP Lite, Perkin-Elmer) following the manufacturer’s protocol. Data shown are mean ± S.D. from three independent experiments in duplicate. *Significantly different from control (P < 0.05).
3.4. Morphological analysis of apoptosis by the APO-BrdU TUNEL assay and by histochemical staining with hematoxylin
Histochemical staining and TUNEL assay results clearly showed that activation of the P2Y12 receptor by 2MeSADP protected the cells from TNFα-induced apoptosis (Fig. 5A–L). The cells that were treated with TNFα and 2MeSADP in the presence of cycloheximide and were fixed with methanol and stained with hematoxylin solution demonstrated that activation of the P2Y12 receptor by 2MeSADP protects the cells from TNFα-induced apoptosis. The DNA fragmentation assay also provides evidence of the protective effect of 2MeSADP. The corresponding picture illustrates apoptotic cells visualized by the TUNEL method (Fig. 5F–H) in comparison to control cells (Fig. 5B–D). The protective effect of 2MeSADP was demonstrated in Fig. 5J–L.
Fig. 5.
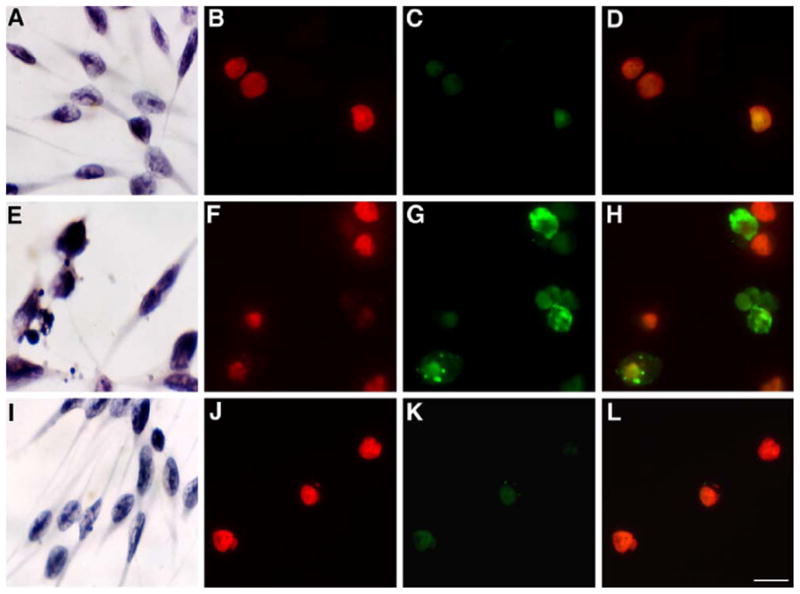
Histochemical staining by hematoxylin and TUNEL in hP2Y12-1321N1 astrocytoma cells. Morphological changes of hP2Y12 receptor-transfected 1321N1 astrocytoma cells 16 h after the treatments as indicated in Section 2. Cells were stained by hematoxylin and TUNEL. Light microscope pictures by hematoxylin staining in astrocytoma cells stably transfected with the hP2Y12 receptor. (A) Nontreated control cells, (E) TNFα-treated cells, and (I) TNFα + 2MeSADP-treated cells. Fluorescence micrographs of TUNEL-assayed cells. Nontreated control cells (B) stained with PI, (C) negative TUNEL-stained, and (D) negative stained TUNEL with PI. TNFα-treated cells: (F) cells stained by PI, (G) positive TUNEL-stained cells, and (H) positive TUNEL-stained with PI. TNFα + 2MeSADP-treated cells (protected cells), (J) PI-stained cells, (K) negative TUNEL-stained cells, and (L) negative TUNEL-stained cells with PI. Pictures were taken by Zeiss wide-field microscopy. Magnification of figures was 63× (TUNEL) or 20× (hematoxylin). Bars = 10 μm. Each image is representative of three experiments.
3.5. P2Y12 receptor-elicited activation of mitogen-activated protein kinases (MAPKs)
We observed that the level of phosphorylated Erk1/2 in cells increased within 5 min after the application of 10 nM 2MeSADP, and this effect persisted for 1 h (Fig. 6B). There was no change in the basal level of Erk1/2 (Fig. 6A).
Fig. 6.
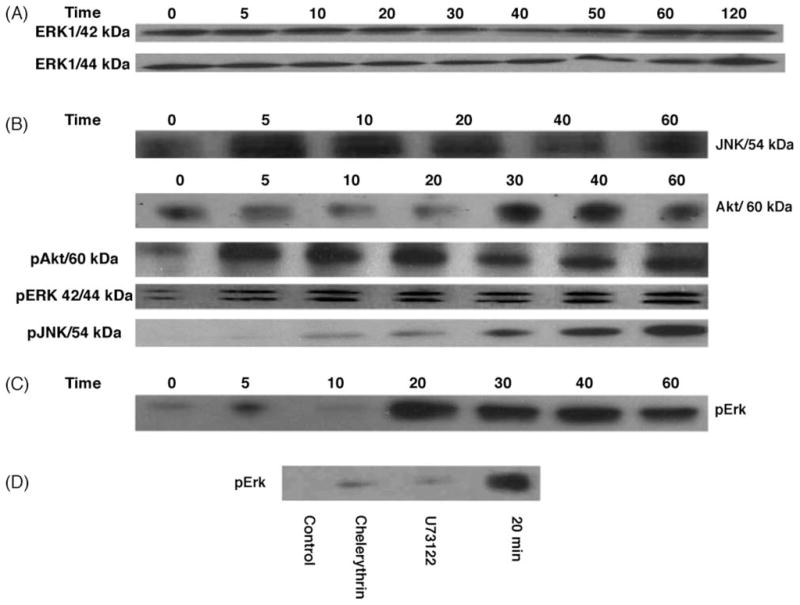
(A) Effect of 2MeSADP on the expression of Erk1/2, after incubation of hP2Y12-1321N1 cells with the agonist 2MeSADP (10 nM) for a period ranging from 5 to 120 min. (B) The effect of 2MeSADP (10 nM) on the phosphorylation of Erk1/2, JNK, and Akt. (C) The effect of 2MeSADP (100 pM) on the phosphorylation of Erk1/2. (D) Effects of U73122 and chelerythrin on 2MeSADP-induced (100 pM) Erk phosphorylation in hP2Y12-1321N1 cells, compared to 20 min control. The cells were pretreated for 30 min with 10 μM U73122 and 10 μM chelerythrin. 2MeSADP (100 pM) was added for 20 min, and then proteins were extracted and applied to immunoblotting as described in Section 2. A total of 40 μg of protein was applied to each lane. The Western blots are representative of two separate transfections, and each panel is taken from a single immunoblot. Following separation by 10% polyacrylamide gel electrophoresis and transfer onto nitrocellulose, detection was made with an anti-β-actin antibody to demonstrate consistency of protein loading (not shown).
2MeSADP also increased phosphorylation of both Akt and JNK (Fig. 6B) but did not induce expression or phosphorylation of p38 (data not shown). However, induction of phosphorylated JNK displayed different time characteristics. While induction of phosphorylated Akt increased within 5 min after application of 2MeSADP, phosphorylated JNK began to increase only 20 min after application of 2MeSADP.
Consistent with the inability of the P2Y12 receptor to induce apoptosis demonstrated by the PI method, additional Western blot results for caspase-3 also demonstrated that 2MeSADP did not induce this mediator of apoptosis in astrocytoma cells expressing the P2Y12 receptor (data not shown). However, 2MeSADP inhibited the activation of caspase-3 that was induced by TNFα in astrocytoma cells expressing the P2Y12 receptor (data not shown).
Consistent with the observation that hP2Y12 receptor activation by pM concentrations of 2MeSADP protected against TNFα-induced apoptosis, we observed changes in the level of phosphorylated Erk1/2 in P2Y12 receptor-expressing astrocytoma cells at a low agonist concentration. Phosphorylated Erk1/2 increased within 20 min following the application of 100 pM 2MeSADP, and this effect persisted for 1 h (Fig. 6C). However, following exposure to 100 pM 2MeSADP there was no detectable phosphorylation of Akt or JNK.
3.6. Probing the involvement of calcium, PKC, and IP3 pathways in the protective effects of 2MeSADP
We first used the calcium chelator BAPTA-AM (5–10 μM) to see if calcium-dependent PKC is responsible for the P2Y12 receptor-induced protective effect. Also, we used the PKC inhibitor chelerythrin (10 μM); a PLC inhibitor, U73122 (10 μM); and an IP3 receptor inhibitor, 2ABP (10 μM), to determine if this protection occurs through activation of PKC. Exposure to BAPTA-AM, U73122, 2ABP, and chelerythrin increased the PI-positive cell fraction in the 2MeSADP-treated group in comparison to control (Fig. 7). The effect of the PLC inhibitor U73122 was more pronounced than that of other pathway inhibitors. The addition of the PLC inhibitor U73122 (10 μM) and the PKC inhibitor chelerythrin (10 μM) 30 min prior to the activation of P2Y12 receptors inhibited the 2MeSADP-induced phosphorylation of Erk (Fig. 6D).
Fig. 7.
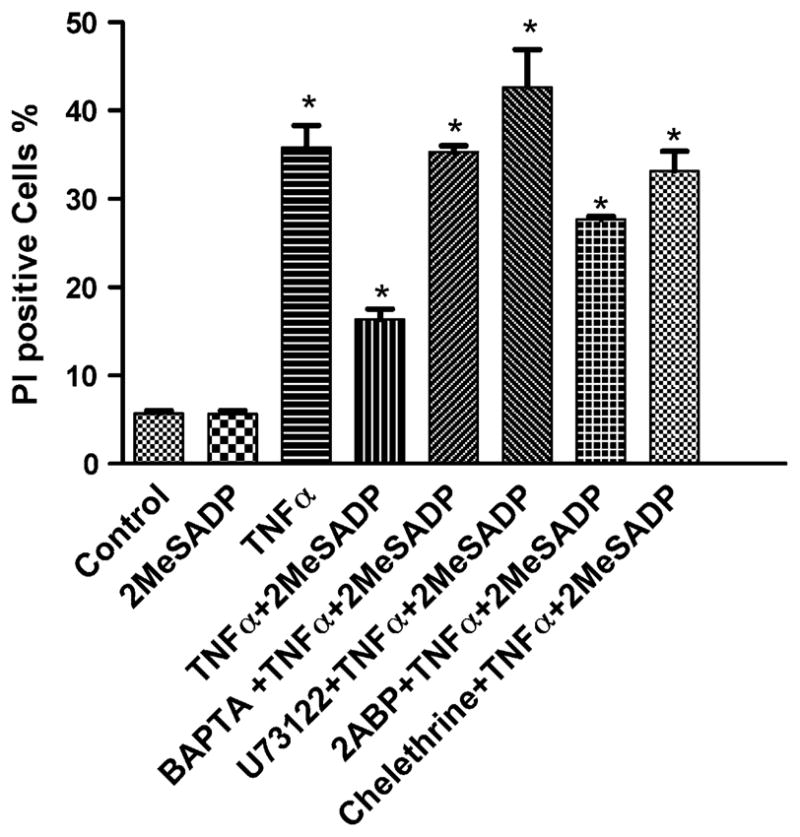
Effect of a calcium chelator, BAPTA-AM (10 μM); a PLC inhibitor, U73122 (10 μM), an IP3 receptor inhibitor, 2ABP (10 μM), and a PKC inhibitor, chelerythrin (10 μM), on protection by 2MeSADP against TNFα-induced cell death in hP2Y12-1321N1 cells. The cells were pretreated with various inhibitors for 10 min and treated further with TNFα 20 ng/ml for 4 h in the absence or presence of 10 nM 2MeSADP. The medium was then replaced with fresh medium following washing with PBS. Cell death was observed on the following day (total of 16 h incubation). The medium always contained 5 μg/ml cycloheximide. The PI-positive cells were analyzed with a FacsCalibur instrument. Data shown are mean ± S.D. from a representative result in triplicate, out of three independent experiments performed in duplicate. Groups labeled * are significantly different from control (P < 0.05).
3.7. P2Y12 receptor-elicited [Ca2+]i elevation
Exposure of 1321N1 cells expressing the P2Y12 receptor to ADP or 2MeSADP induced a concentration-dependent rise in [Ca2+]i levels, with EC50 values of 191 ± 66 and 23 ± 6 nM, respectively (Fig. 8A). This agonist effect was inhibited by PTX (Fig. 8B). The P2Y12 competitive antagonist 2MeSAMP (10 μM) right-shifted the concentration–response curve for 2MeSADP, indicative of action at the hP2Y12 receptor (Fig. 8C).
Fig. 8.

(A) Concentration–response curves for the increase in [Ca2+]i induced by the endogenous agonist, ADP, in hP2Y12-1321N1 cells. (B) Effects of PTX on the [Ca2+]i response to the agonist 2MeSADP in hP2Y12-1321N1 cells. The cells were pretreated with PTX (200 ng/ml) and in the absence and presence of 2MeSADP for 24 h. For all experiments, before measurement cells were washed with Ca2+-free PBS and incubated with a Ca2+ dye as provided in a kit. [Ca2+]i levels were measured with a Flexstation I instrument. (C) Effects of 10 μM 2MeSAMP on the [Ca2+]i response to the agonist 2MeSADP in hP2Y12-1321N1 cells. The cells were pretreated with 2MeSAMP for 25 min before the assay.
4. Discussion
In this study, we demonstrated that the activation of P2Y12 receptors by both the endogenous agonist, ADP, and a synthetic agonist, 2MeSADP, significantly protected astrocytoma cells from TNFα-induced cell death. This result complements an earlier report that activation of P2Y1 receptors induces cell death under the same conditions [9], which is also confirmed in the present study. Thus, it was demonstrated that ADP might simultaneously activate both P2Y1 and P2Y12 receptors, which might play a role in balancing cell growth and cell death.
Native astrocytes have been reported to express both P2Y1 and P2Y12 receptors [7,23]. In the present study, stably transfected astrocytoma cells that do not express these receptors endogenously were used. We did not create a stable cell line expressing both receptors in order to examine the interplay of the two subtypes.
Also, it should be noted that cells expressing transfected receptors are good models for receptor/ligand studies and cellular biochemical processing studies but may not represent cell signaling in the same manner as cells that naturally express the receptor. There also may be differences in internalization. Both receptors (P2Y1 and P2Y12) are internalized, however, with different kinetics and through different pathways. This resulted in a greater persistence of the P2Y12 receptor at the plasma membrane in the presence of nucleotide in the medium, while the P2Y1 receptor remained internalized [24–26].
The mechanism by which P2Y12 receptor activation protects cells was explored. Western blots showed that 2MeSADP induced activation of Erk1/2 but had no effect on the activity of p38 kinase. Activation of the hP2Y6 receptor expressed in astrocytoma cells also protected against TNFα-induced apoptosis through activation of Erk [27]. Also, both U73122 and chelerythrin reversed the protective effect of 2MeSADP, suggesting that both PLC and PKC are required for the antiapoptotic effect of P2Y12 receptors.
ADP is an agonist for both P2Y1 and P2Y12 receptors. Activation of the P2Y1 receptor is coupled to the Gq-PLC pathway, and P2Y12 receptor activation is negatively coupled to the adenylyl cyclase pathway via Giα [1,2,20] and positively coupled to PLC through Gβγ subunits. Consequently, different signaling pathways are activated in response to ADP in systems such as platelets, which express both P2Y1 and P2Y12 receptors, and astroglial cells. The present study demonstrated that 2MeSADP-induced calcium mobilization is abolished by treatment with PTX, suggesting that Gq is not involved. All three groups of PKC (conventional, novel, and atypical) are able to activate p42 MAPK as well as its immediate upstream activator, MEK-1 [28]. Thus, one signaling pathway generated by P2Y12 receptor activation is attributable to the activation of PLCβ leading to the formation of diacylglycerol and IP3 and the mobilization of [Ca2+]i, and most significantly the subsequent activation of PKCβ1, β2, and γ. It is known that activation of certain isoforms of PKC can activate Erks. In this study, the protective effect of 2MeSADP was blocked by BAPTA-AM, indicative of a Ca2+-dependent mechanism, however, elevation of [Ca2+]i levels required relatively high concentrations of ADP or 2MeSADP. The calcium mobilization was antagonized by a PLC inhibitor (U73122) and an IP3 receptor inhibitor (2-ABP). In addition to an apoptotic signal, TNFα can induce a survival signal through NF-κB, which inhibits caspase 3 [29]. We did not examine the effects of P2Y12 receptor signaling on NF-κB.
The anti-apoptotic effect of extracellular ADP has been reported [30], but it was not known previously if the P2Y12 receptor was involved. Both Gi-dependent and -independent mechanisms downstream of P2Y12 receptors have been reported [31]. Crosstalk between the downstream pathways of P2Y1 and P2Y12 receptors was demonstrated to be critical for various events, including platelet aggregation and cell proliferation [2,32,33].
Caspases are responsible for the deliberate disassembly of a cell into apoptotic bodies. Caspases are present as inactive proenzymes, most of which are activated by proteolytic cleavage. Caspase-9 can activate caspase-3 by proteolytic cleavage, which can in turn cleave vital cellular proteins, leading to apoptosis [34]. Activation of the P2Y12 receptor significantly suppresses the induction of caspases by TNFα. The present study demonstrated that activation of the P2Y12 receptor by 2MeSADP or ADP did not stimulate caspase-3.
We have shown that a chelator of intracellular calcium eliminated the protective effect of 2MeSADP, suggesting a possible role of calcium in the antiapoptotic pathway. Recently, several other examples of Ca2+ channel modulation via endogenous P2Y receptors were reported [35]. It was found that adenine nucleotides activate P2Y12 receptors to inhibit voltage-gated Ca2+ currents in PC12 cells via a voltage-dependent and PTX-sensitive mechanism [36,37]. P2Y12 receptors in rat sympathetic neurons were shown to mediate a voltage-dependent and PTX-sensitive inhibition of N-type calcium channels [38,39].
Although the activation mechanisms of JNK have been extensively investigated, the biological consequence of JNK activation in cell death is still controversial [29,40–43]. While the functions of the JNKs under physiological conditions are diverse and incompletely understood, there is increasing evidence that JNKs are potent effectors of apoptosis in both the brain and the mammalian inner ear following a variety of injuries. The activation of the inducible transcription factor c-Jun by N-terminal phosphorylation is a central event in JNK-mediated neural and inner ear hair cell death [44]. Our results demonstrated that P2Y12 receptor stimulation by 10 nM 2MeSADP activates phosphorylation of JNK. However, activation begins slowly, only after 20 min of incubation with 2MeSADP. It is possible that the transient activation of pJNK through P2Y12 receptor activation may regulate apoptotic events rather than induce cell death. However, a lower concentration of 2MeSADP (100 pM), which significantly protected against cell death, did not activate phosphorylation of JNK. Interestingly, the induction of phosphorylated Akt was detected after 5 min of incubation with 10 nM 2MeSADP, however, no phosphorylation was detected in the presence of 100 pM 2MeSADP. Thus, activation of JNK and Akt pathways may not contribute significantly to the antiapoptotic effect of low concentrations of 2MeSADP via the P2Y12 receptor.
Thus, ADP and 2MeSADP have dual effects on cell death and survival, inducing apoptosis via activation of the P2Y1 receptor and protecting cells against TNFα-induced apoptosis through the activation of the P2Y12 receptor. P2Y12 receptors protect cells against TNFα-induced apoptosis, at least in part, through the activation of the PLC-PKC-Erk pathway, and this protection might also involve activation of Akt and JNK. Future experiments will be needed to determine the effects of ADP treatment on normal astrocytes expressing both P2Y1 and P2Y12 receptors.
Acknowledgments
We thank Prof. T.K. Harden (University of North Carolina) and Prof. Ivar von Kügelgen (University of Bonn, Germany) for helpful discussions. We thank Wanda Williams (NIDDK) for technical assistance. This research was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases.
Abbreviations
- BrdUTP
5-bromo-2′-deoxyuridine 5′-triphosphate
- caspase-3
cytosolic aspartate-specific protease-3
- DMEM
Dulbecco’s modified Eagle’s medium
- Erk1/2
extracellular signal-regulated protein kinases 1 and 2
- FACS
fluorescence-activated cell sorting
- FBS
fetal bovine serum
- HRP
horseradish peroxidase
- IP3
inositol trisphosphate
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- 2MeSADP
2-methylthioadenosine 5′-diphosphate
- MRS2179
N6-methyl-2′-deoxyadenosine-3′,5′-bisphosphate
- PI
propidium iodide
- PI3-K
phosphatidylinositol 3-kinase
- PKC
protein kinase C
- PTX
pertussis toxin
- TNF
tumor necrosis factor
- TUNEL
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling
- U73122
1-[6-((17b-3-Methoxyestra-1,3,5(10)-trien-17-yl)-amino)hexyl]-1Hpyrrole-2,5-dione
References
- 1.Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409:202–7. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- 2.Hardy AR, Conley PB, Luo J, Benovic JL, Poole AW, Mundell SJ. P2Y1 and P2Y12 receptors for ADP desensitize by distinct kinase-dependent mechanisms. Hem Thromb Vasc Biol. 2005;105:3552–60. doi: 10.1182/blood-2004-07-2893. [DOI] [PubMed] [Google Scholar]
- 3.Hechler B, Vigne P, Leon C, Breittmayer JP, Gachet C, Frelin C. ATP derivatives are antagonists of the P2Y1 receptor: similarities to the platelet ADP receptor. Mol Pharmacol. 1998;53:727–33. [PubMed] [Google Scholar]
- 4.Leon C, Ravanat C, Freund M, Cazenave JP, Gachet C. Differential involvement of the P2Y1 and P2Y12 receptors in platelet procoagulant activity. Arterioscler Thromb Vasc Biol. 2003;23:1941–7. doi: 10.1161/01.ATV.0000092127.16125.E6. [DOI] [PubMed] [Google Scholar]
- 5.Dorsam RT, Kunapuli SP. Central role of the P2Y12 receptor in platelet activation. J Clin Invest. 2004;13:340–5. doi: 10.1172/JCI20986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding Z, Kim S, Dorsam RT, Jin J, Kunapuli SP. Inactivation of the human P2Y12 receptor by thiol reagents requires interaction with both extracellular cysteine residues, Cys17 and Cys270. Blood. 2003;101:3908–14. doi: 10.1182/blood-2002-10-3027. [DOI] [PubMed] [Google Scholar]
- 7.Gustafson EL, Yang S, Laverty M, Luo L, Vassileva ZM, Laz T, et al. Expression of P2Y12 and P2Y13 purinergic receptors in human and mouse brain. IUPHAR meeting; San Francisco, CA. 2003. [abstract 142.11] [Google Scholar]
- 8.Nasu-tada K, Koizumi S, Inoue K. Involvement of β1 integrin in microglial chemotaxis and profileration on fibronectin: different regulations by ADP through PKA. Glia. 2005;52:98–107. doi: 10.1002/glia.20224. [DOI] [PubMed] [Google Scholar]
- 9.Sellers LA, Simon J, Lundahl TS, Cousens DJ, Humphrey PP, Barnard EA. Adenosine nucleotides acting at the human P2Y1 receptor stimulate mitogen-activated protein kinases and induce apoptosis. J Biol Chem. 2001;276:16379–90. doi: 10.1074/jbc.M006617200. [DOI] [PubMed] [Google Scholar]
- 10.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–35. [PubMed] [Google Scholar]
- 11.Ptasznik A, Beattie GM, Mally MI, Cirulli V, Lopez A, Hayek A. Phosphatidylinositol 3-kinase is negative regular of cellular differentiation. J Cell Biol. 1997;137:1127–36. doi: 10.1083/jcb.137.5.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doss RC, Perkins JP, Harden TK. Recovery of beta-adrenergic receptors following long term exposure of astrocytoma cells to catecholamine. Role of protein synthesis. J Biol Chem. 1981;256:12281–6. [PubMed] [Google Scholar]
- 13.Kim SG, Soltysiak KA, Gao ZG, Chang TS, Chung E, Jacobson KA. Tumor necrosis factor α-induced apoptosis in astrocytes is prevented by the activation of P2Y6, but not P2Y4 nucleotide receptors. Biochem Pharmacol. 2003;65:923–31. doi: 10.1016/s0006-2952(02)01614-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filtz TM, Li Q, Boyer JL, Nicholas RA, Harden TK. Expression of a cloned P2Y purinergic receptor that couples to phospholipase C. Mol Pharmacol. 1994;46:8–14. [PubMed] [Google Scholar]
- 15.Czajkowski R, Banachewicz W, Ilnytska O, Drobot LB, Baranska J. Differential effects of P2Y1 and P2Y12 nucleotide receptors on ERK1/ERK2 and phosphatidylinositol 3-kinase signalling and cell proliferation in serum-deprived and nonstarved glioma C6 cells. Br J Pharmacol. 2004;141:497–507. doi: 10.1038/sj.bjp.0705639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bodor ET, Waldo GL, Hooks SB, Corbitt J, Boyer JL, Harden TK. Purification and functional reconstitution of the human P2Y12 receptor. Mol Pharmacol. 2003;64:1210–6. doi: 10.1124/mol.64.5.1210. [DOI] [PubMed] [Google Scholar]
- 17.Lazarowski ER, Watt WC, Stutts MJ, Boucher RC, Harden TK. Pharmacological selectivity of the cloned human P2U—purinergic receptor potent activation by diadenosine tetraphosphate. Br J Pharmacol. 1995;116:1619–27. doi: 10.1111/j.1476-5381.1995.tb16382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lazarowski ER, Homolya L, Boucher RC, Harden TK. Direct demonstration of mechanically induced release of cellular UTP and its implication for uridine nucleotide receptor activation. J Biol Chem. 1997;272:24348–54. doi: 10.1074/jbc.272.39.24348. [DOI] [PubMed] [Google Scholar]
- 19.Lazarowski ER, Boucher RC, Harden TK. Constitutive release of ATP and evidence for major contribution of ecto-nucleotide pyrophosphatase and nucleoside diphosphokinase to extracellular nucleotide concentrations. J Biol Chem. 2000;275:31061–8. doi: 10.1074/jbc.M003255200. [DOI] [PubMed] [Google Scholar]
- 20.Ostrom RS, Gregorian C, Insel PA. Cellular release of and response to ATP as key determinants of the set-point of signal transduction pathways. J Biol Chem. 2000;275:11735–9. doi: 10.1074/jbc.275.16.11735. [DOI] [PubMed] [Google Scholar]
- 21.Lazarowski ER, Harden TK. Quantitation of extracellular UTP using a sensitive enzymatic assay. Br J Pharmacol. 1999;127:1272–8. doi: 10.1038/sj.bjp.0702654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dubyak GR, El-Moatassim C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am J Physiol. 1993;265:C577–606. doi: 10.1152/ajpcell.1993.265.3.C577. [DOI] [PubMed] [Google Scholar]
- 23.Shinozaki Y, Koizumi S, Ishida S, Sawada J, Ohno Y, Inoue K. Cytoprotection against oxidative stress-induced damage of astrocytes by extracellular ATP via P2Y1 receptors. Glia. 2005;49:288–300. doi: 10.1002/glia.20118. [DOI] [PubMed] [Google Scholar]
- 24.Baurand A, Eckly A, Hechler B, Kauffenstein G, Galzi J-L, Cazenave J-P, et al. Differential regulation and relocation of the platelet P2Y receptors after activation: A way to avoid loss of hemostatic properties? Mol Pharmacol. 2005;67:721–33. doi: 10.1124/mol.104.004846. [DOI] [PubMed] [Google Scholar]
- 25.Mundell SJ, Jones ML, Hardy AR, Barton JF, Beaucourt SM, Conley PB, et al. Distinct roles for protein kinase C isoforms in regulating platelet purinergic receptor function. Mol Pharmacol. 2006;70:1132–42. doi: 10.1124/mol.106.023549. [DOI] [PubMed] [Google Scholar]
- 26.Dubyak GR. Knock-out mice reveal tissue-specific roles of P2Y receptor subtypes in different epithelia. Mol Pharmacol. 2003;63:773–6. doi: 10.1124/mol.63.4.773. [DOI] [PubMed] [Google Scholar]
- 27.Kim SG, Gao ZG, Soltysiak KA, Chang TS, Brodie C, Jacobson KA. P2Y6 nucleotide receptor activates PKC to protect 1321N1 astrocytoma cells against tumor necrosis factor-induced apoptosis. Cell Mol Neurobiol. 2003;23:410–8. doi: 10.1023/a:1023696806609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790–8. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karin M, Lin A. NF-κB at the crossroads of life and death. Nat Immunol. 2002;3:221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 30.Vitolo OV, Ciotti MT, Galli C, Borsello T, Calissano P. Adenosine and ADP prevent apoptosis in cultured rat cerebellar granule cells. Brain Res. 1998;809:297–301. doi: 10.1016/s0006-8993(98)00713-6. [DOI] [PubMed] [Google Scholar]
- 31.Soulet C, Sauzeau V, Plantavid M, Herbert JM, Pacaud P, Payrastre B, et al. Gi-dependent and -independent mechanisms downstream of the P2Y12 ADP-receptor. J Thromb Haemost. 2004;2:135–46. doi: 10.1111/j.1538-7836.2004.00556.x. [DOI] [PubMed] [Google Scholar]
- 32.Daniel JL, Dangelmaier C, Jin J, Kim YB, Kunapuli SP. Role of intracellular signaling events in ADP-induced platelet aggregation. Thromb Haemost. 1999;82:1322–6. [PubMed] [Google Scholar]
- 33.Hardy AR, Jones ML, Mundell SJ, Poole AW. Reciprocal cross-talk between P2Y1 and P2Y12 receptors at the level of calcium signaling in human platelets. Blood. 2004;104:1745–52. doi: 10.1182/blood-2004-02-0534. [DOI] [PubMed] [Google Scholar]
- 34.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–6. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 35.Vartian N, Boehm S. P2Y receptor-mediated inhibition of voltage-activated Ca2+ currents in PC12 cells. Eur J Neurosci. 2001;13:899–908. doi: 10.1046/j.1460-9568.2001.01461.x. [DOI] [PubMed] [Google Scholar]
- 36.Kulick MB, von Kügelgen I. P2Y-receptors mediating an inhibition of the evoked entry of calcium through N-type calcium channels ay neuronal processes. J Pharmacol Exp Ther. 2002;303:520–6. doi: 10.1124/jpet.102.037960. [DOI] [PubMed] [Google Scholar]
- 37.Kubista H, Lechner SG, Wolf AM, Boehm S. Attenuation of the P2Y receptor-mediated control of neuronal Ca2+ channels in PC12 cells by antithrombotic drugs. Br J Pharmacol. 2003;138:343–50. doi: 10.1038/sj.bjp.0705037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lechner SG, Boehm S. Regulation of neuronal ion channels via P2Y receptors. Purinergic Signal. 2004;1:31–41. doi: 10.1007/s11302-004-4746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simon J, Filippov AK, Goransson S, Wong YH, Frelin C, Michel AD, et al. Characterization and channel coupling of the P2Y12 nucleotide receptor of brain capillary endothelial cells. J Biol Chem. 2002;277:31390–400. doi: 10.1074/jbc.M110714200. [DOI] [PubMed] [Google Scholar]
- 40.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 41.Lin A. Activation of the JNK signaling pathway: breaking the brake on apoptosis. Bioessays. 2003;25:17–24. doi: 10.1002/bies.10204. [DOI] [PubMed] [Google Scholar]
- 42.Varfolomeev EE, Ashkenazi A. Tumor necrosis factor: an apoptosis JuNKie? Cell. 2004;116:491–7. doi: 10.1016/s0092-8674(04)00166-7. [DOI] [PubMed] [Google Scholar]
- 43.Nakano H, Nakajima A, Sakon-Komazawa S, Piao J-H, Xue X, Okumura K. Cell Death Differ. 2006;13:730–7. doi: 10.1038/sj.cdd.4401830. [DOI] [PubMed] [Google Scholar]
- 44.Bonny C, Borsello T, Zine A. Targeting the JNK pathway as atherapeutic protective strategy for nervous system diseases. Rev Neurosci. 2005;16:57–67. doi: 10.1515/revneuro.2005.16.1.57. [DOI] [PubMed] [Google Scholar]