

Abstract
Various dipyrroles possess important motifs for construction of pyrrole-containing pigments. A series of 1,2-dipyrrolylethynes (4a–d) has been efficiently synthesized using an improved one-pot double Sonagashira coupling from trimethylsilylethyne and various 2-iodopyrroles. The resulting 1,2-dipyrrolylethynes were further transformed into novel indolyl-, ethenyl- and carboranyl-dipyrroles (5–7) using the Larock indole synthesis, stereoselective catalytic hydrogenation, or B10H14. Indolyl-dipyrroles were found to selectively bind fluoride ions using one pyrrolic and the indolyl NHs, whereas the carboranyl- and ethenyl-dipyrroles are potentially valuable precursors for the synthesis of porphyrin isomers and expanded pigments.
Keywords: carborane, dipyrrole, indole, Larock, Sonagashira
INTRODUCTION
In recent years, the chemistry of pyrrole-containing pigments has received increasing attention in coordination chemistry, medicinal chemistry, and material science [1]. Various kinds of dipyrroles have been synthesized and some have been used as building blocks in the construction of various functional molecules [2]. Among these, dipyrrolylethynes such as A in Fig. 1, can be found only sparsely in the literature (five examples according to a SciFinder search). Compound A was first synthesized by Vogel and coworkers as a key intermediate during their synthesis of 22 π-electron expanded (or “stretched”) porphycene-(2.2.2.2) via B (Fig. 1) [3, 4]. Later, Lightner and coworkers adapted this methodology for the sysnthesis of alkynyl-homobilirubins [5]. The method for synthesis of dipyrrolylethynes involves a three-step 60% overall yield sequence (coupling-desilylation-coupling), using trimethylsilylethyne as the source of the alkyne bridge. At least two column separations are required for purification before the target compound is isolated.
Fig. 1.

Structures of dipyrrolylethyne (a) and expanded porphycene precursor (b) synthesized by Vogel and coworkers [3]
We are interested in new approaches to compound A due to its synthetic utility as a conjugated pyrrole-containing precursor for cycloadditive macrocyclization. Furthermore, the rich chemistry of the alkyne functionality provides the opportunity to synthesize other novel dipyrrole motifs. Inspired by Grieco’s recently developed one-pot synthesis of biarylalkynes [6], we have developed and now report a one-pot synthesis of dipyrrolylethynes. Our ethynyl products were then further transformed into various related dipyrroles (5–7) using the Larock indole synthesis, stereoselective catalytic hydrogenation, or reaction with decaborane. In particular, the new indolyldipyrroles were found to have high fluorescence quantum yields and to selectively bind fluoride ions in a 2:1 (host:F−) complex. Pyrrole and indole-based fluoride probes have been incorporated within conjugated systems for colorimetric and/or fluorescence detection of fluoride ions, because of their strong hydrogen bonding [F−····H-N] interactions, as well as their high stability over a wide range of pH values [7–9]. On the other hand, the carboranyl- and ethenyl-dipyrroles are valuable precursors for the synthesis of porphyrin isomers and other polypyrrolic materials [10, 11].
RESULTS AND DISCUSSION
Our overall synthesis of dipyrrole (4a) from readily available 2-iodopyrrole (1a) [12] is shown in Scheme 1. First, a three-step sequence (coupling-desilylation-coupling) using trimethylsilylethyne as the source of the 2-atom alkynyl bridge was used, as previously described [13]. Classical Sonagashira coupling between 1a and trimethylsilylethyne gave pyrrole 2 in 89% yield. Desilylation using tetrabutylammonium fluoride (TBAF) provided the terminal ethynylpyrrole (3) in 95% yield. The final coupling between compounds 1a and 3 afforded dipyrrolylethyne (4a) in 73% yield. Alternatively, a 1-step procedure for the formation of 4a using a double Sonagashira coupling reaction between 2-iodopyrrole 1a and trimethylsilyllethyne was investigated. Although Vogel [3] and later Kim [4] both reported very low yields for the direct formation of the dipyrrolylethyne using iodopyrrole and ethyne, using Grieco’s method [6] we were able to develop a one-pot reaction between 2-iodopyrrole 1a and trimethylsilylethyne (Scheme 1) which gave 4a in 62% yield. In this synthetic approach, the coupling reaction was performed using trimethylsilylethyne instead of ethyne, with DBU as the base. The reaction was followed using TLC since the target dipyrrolylethyne displays characteristic bright blue fluorescence under ultraviolet (366 nm) light. The workup of the reaction was very simple and required no chromatographic purification; after extraction with ethyl acetate it only required methanol washing to obtain analytically pure product. The 1-step methodology proved to be reproducible and routinely gave 55–65% yields of the target dipyrrolylethyne products, on a 10 mmol scale. Use of another solvent, such as acetonitrile in place of benzene, also gave similar results. The one-pot reaction was extended to the synthesis of dipyrrolylethynes (4b) and (4c) in 52 and 54% yields, respectively (Table 1). However, when N-tosyl-2-bromopyrrole (1d) was used, a poor yield of dipyrrolylethyne 4d was obtained (8%) using these conditions.
Scheme 1.

Synthesis of dipyrrolylethyne 4a using two different routes
Table 1.
Dipyrrolylethyne (4a–d) yields from one-pot coupling of 2-iodopyrroles (1), or 2-bromopyrrole
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | X | Yield of 4, % |
| a | Me | Et | CO2Bn | H | I | 62 |
| b | Me | Me | CO2Bn | H | I | 52 |
| c | Me | Me | CO2Et | H | I | 54 |
| d | H | H | H | Ts | Br | 8 |
We were able to grow suitable crystals for X-ray analysis of all dipyrrolylethynes 4a–d, by slow evaporation of dichloromethane solutions. The X-ray structures of 4a and 4c are shown in Figs 2 and 3, respectively. In compound 4a, the two pyrrole rings are twisted about the triple-bond line with a dihedral angle of 33.26(3)°, and the C–C triple bond distance is 1.205(2) Å. The molecule lies on a twofold axis in the crystal. In compound 4c, the molecule does not lie on a symmetry element in the crystal, and the pyrroles are twisted considerably more than in 4a, forming a dihedral angle of 69.25(6)°. The triple-bond distance is 1.203(3) Å. The difference in conformation between 4a and 4c appears to be related to hydrogen bonding. In both, each pyrrole and adjacent ester C=O form an intermolecular cyclic hydrogen bond dimer. However, in 4a, these 10-membered rings lie on inversion centers, thus the pyrroles are strictly parallel. In 4c, they deviate sharply from parallelism, forming a dihedral angle of 64.59(6)°. We have previously reported the molecular structures of 4b and 4d [14].
Fig. 2.
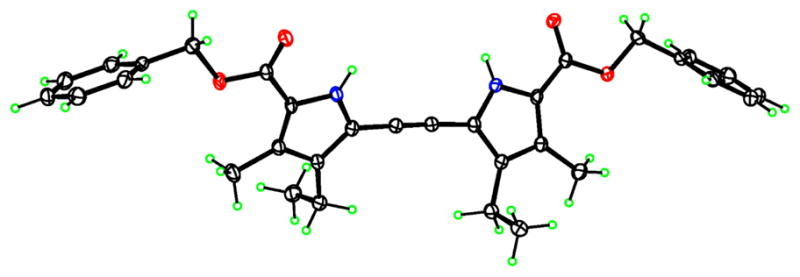
Molecular structure of dipyrrolylethyne 4a, with 50% ellipsoids
Fig. 3.

Molecular structure of dipyrrolylethyne 4c, with 50% ellipsoids
Novel indolyldipyrroles 5a and 5c were prepared in 55–60% yields via the Larock indole synthesis [15] from the corresponding dipyrrolylethynes 4a,c, as shown in Scheme 2. On the other hand, with several dipyrrolylethynes available we explored the Lindlar reduction of the alkyne bond to generate novel 1,2-dipyrrolylethene derivatives. Using Lindlar’s catalyst [16] in the absence of quinoline, dipyrrolylethene (6) was synthesized from 4c in 31% yield. Stereoselective semi-hydrogenation employing HSiEt3 as the reductant [17] also yielded the cis-dipyrrolylalkene 6. This transformation opens up the possibility for synthesis of porphycenes by way of Ullmann type self-coupling of derivatives of 6, and also of expanded 22 π-electron stretched porphycenes.
Scheme 2.

Synthesis of dipyrroles 5 and 6
The X-ray structure of 5c is shown in Fig. 4. One pyrrole at the α-position of the indole moiety is rotated to present its NH group approximately coplanar with that of the indole, forming an N–C–C–N torsion angle of 30.7(3)°. Thus, both of these two NH groups are able to donate their hydrogen bonds to the same acceptor, forming two convergent intermolecular hydrogen bonds to an ester carbonyl O atom.
Fig. 4.
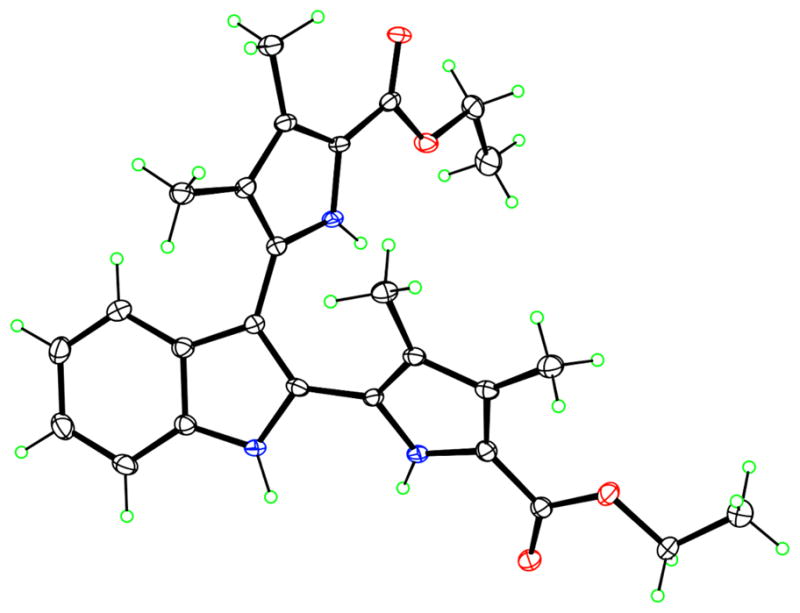
Molecular structure of bipyrrole 5c, with 50% ellipsoids
The photophysical properties of indolyl-dipyrroles 5a and 5c were investigated, compared with those of the dipyrrolylethyne precursors, and the results obtained are summarized in Table 2. Interestingly, dipyrroles 5a and 5c show strong absorption bands centered around 300 nm (blue shifted from those of the corresponding dipyrrolylethynes 4), and strong fluorescence emission bands at ~440 nm with surprisingly high quantum yields (0.91 for 5a and 0.88 for 5c) [18]. Fluorescence titration experiments using dipyrroles 5 and TBAF, revealed dramatic fluorescence quenching upon addition of TBAF, as seen in Fig. 5. Addition of other anions, such as bromide, chloride, and hydrogenosulfate did not cause such pronounced fluorescence quenching, probably due to their lower affinity for binding to the pyrrole and indole NH and their larger size. The Job’s plot shown in Fig. 6a indicates that compound 2a chelates with TABF in a 2:1 stoichiometry, with an association constant (K) of 8.32 × 104 M−1 based on the Hill plot shown in Fig. 6b. Although compound 5c also chelated with bromide and hydrogeno-sulfate in a 2:1 molar ratio with binding constants 5.89 × 104 M−1 and 7.94 × 103 M−1, respectively, the Hill coefficients are greater than one, suggesting that these two systems are positively cooperative. The crystal structure obtained of dipyrrole 5c in the presence of 10 equivalents excess of fluoride shows the formation of a dimer complex consisting of two molecules of 5c coordinating one fluoride ion that occupies the center of the molecule, as shown in Fig. 7. Dipyrrole 5c forms a 2:1 crystalline complex with TBAF in which the fluoride ion is chelated by four hydrogen-bonded pyrrole N–H groups. Two of the N–F distances are quite short, 2.566(4) and 2.574(4) Å, and those hydrogen bonds are fairly linear, with N–H–F angles 168 and 169°. The other two are longer, 2.855(4), 2.938(4) Å and less linear, in bifurcated hydrogen bonds with ester carbonyl acceptors. The TBA cation perches on top of this hydrogen-bonded aggregate, with N–F distance 3.894(5) Å.
Table 2.
Photophysical properties of dipyrroles 4 and 5 in CH2Cl2 at room temperature
| Compound | λmax, nm | λema, nm | Φb | Stokes’ shift, nm |
|---|---|---|---|---|
| 4c | 339 | 414 | 0.01 | 75 |
| 4a | 343 | 400 | 0.01 | 57 |
| 5c | 292 | 439 | 0.88 | 147 |
| 5a | 295 | 440 | 0.91 | 145 |
Excitation at 290 nm.
Fluorescence quantum yield calculated using quinine sulfate as the standard (0.56 in 0.1 M H2SO4 aqueous solution).
Fig. 5.

Fluorescence titration of 5c (a, 5 × 10−6 M in CH2Cl2, λex = 290 nm) and 5a (b, 5 × 10−6 M in CH2Cl2, λex = 290 nm) with TBAF (0–100 eq)
Fig. 6.

Job’s plot (a) and Hill plot (b) of 5c:F− complex with a 2:1 stoichiometry, (F0 – F) = change in fluorescence, [5c] and r[5c] = moles and mole fraction for 5c, excitation at 290 nm
Fig. 7.

Molecular structure of the 2:1 molar-ratio 5c-fluoride complex
Inspired by electropolymerization results previously reported [10] for carboranyl-containing pyrroles and thiophenes, in particular for the ortho-carboranyl systems which produced the most conjugated and conductive polymers, we turned our attention to the synthesis of ortho-carboranyldipyrrole 7, as shown in Scheme 3. The ortho-carborane cluster was obtained in 29% yield by reaction of dipyrrolylethyne 4a with decaborane and diethylsulfide in toluene [18]. Interestingly carboranyl-dipyrrole 7 showed the highest fluorescence quantum yield (0.98) of all dipyrroles synthesized.
Scheme 3.

Synthesis of carboranyldipyrrole 7
The X-ray structure of 7 is shown in Fig. 8. In the solid state, the two pyrroles are cofacial, presenting their NH groups on the same side of the carborane cage. The pyrrole rings are not parallel, forming a dihedral angle of 63.05(5)°. As in 4a and 4c, the pyrroles and their adjacent ester carbonyls form intermolecular hydrogen bonds within 10-membered rings. In this case, two molecules of 7 form a hydrogen-bonded dimer via four hydrogen bonds in two 10-membered rings, as shown in Fig. 8 (right panel). Carboranyl-dipyrrole 7 is a very valuable starting material to built carborane-containing macro-cycles and polymers.
Fig. 8.

Molecular structure of bipyrrole 7 (50% ellipsoids, left) and its hydrogen bonded dimer (right)
EXPERIMENTAL
General
Reagents were purchased as reagent-grade and used without further purification unless otherwise stated. Solvents were used as received from commercial suppliers unless noted otherwise. THF was freshly distilled from sodium benzophenone ketyl. All reactions were performed using oven-dried or flame-dried glassware unless otherwise stated, and were monitored by TLC using 0.25 mm silica gel plates with UV indicator (60F-254). Carborane was detected by immersion into PdCl2 in aqueous HCl solution (1 g PdCl2 in 80 mL water and 20 mL conc. HCl) and heated to develop a black spot on TLC. 1H and 13C NMR were obtained on a 250 MHz or 300 MHz spectrometer at room temperature. Chemical shifts (δ) are given in ppm relative to CDCl3 7.26 (1H) and 77 ppm (13C) or to internal TMS. High-resolution mass spectra were obtained by using quadrupole time-of-flight mass spectrometry with a nano LC system. MALDI-TOF mass spectra were obtained on an Applied Biosystems QSTAR XL instrument, using positive mode with dithranol as matrix unless otherwise indicated. Compounds 1a–c were prepared according to the literature [12]. UV-visible absorption spectra and fluorescence emission spectra were recorded on commercial spectrophotometers. The slit width was 2.5 nm for excitation and 5.0 nm for emission. Relative quantum efficiencies of fluorescence of dipyrroles were obtained by comparing the areas under the corrected emission spectrum of the test sample with that of quinine sulfate as the standard (0.56 in 0.1 M H2SO4 aqueous solutions). Non-degassed, spectroscopic grade solvents and a 10 mm quartz cuvette were used. Dilute solutions (0.01 < A < 0.05) were used to minimize reabsorption effects. Quantum yields were determined using the following equation [19]:
| (1) |
where ΦS is the reported quantum yield of the standard, I is the integrated emission spectra, A is the absorbance at the excitation wavelength and η is the refractive index of the solvent being used (η = 1 when the same solvent was used for both the test sample and the standard). An X subscript refers to the test sample, and an S subscript is for the standard.
Synthesis
Benzyl 5-ethynyl-4-ethyl-3-methylpyrrole-2-carboxy- late (3)
To a solution of benzyl 4-ethyl-5-iodo-3-methylpyrrole- 2-carboxylate (1a) (3.69 g, 10 mmol) in diethylamine (100 mL) were added under argon (trimethylsilyl)acetylene (1.5 g, 15 mmol), dichlorobis(triphenylphosphine)palladium(II) (125 mg, 0.175 mmol), and copper(I) iodide (65 mg, 0.35 mmol). The homogeneous mixture was stirred at 50 °C for 2 h until TLC showed that no starting material remained. After evaporation of the solvent under vacuum, the residue was subjected to chromatography on a short silica gel column with hexane/dichloromethane/ethyl acetate (5:1:1) as eluent, to give 2 (3.02 g, 89%). To a solution of 2 (2.71 g, 8.0 mmol) in THF (50 mL) was added Bu4NF (1.0 M THF solution, 8 mL). After being stirred for 1 h, the resulting mixture was concentrated under reduced pressure. The crude residue was subjected to chromatography on a short silica gel column with dichloromethane as eluent to give 3 as a white solid (2.03 g, 95% yield). 1H NMR (CDCl3, 250 MHz): δ, ppm 9.19 (1H, s), 7.31–7.36 (5H, m), 5.28 (2H, s), 3.29 (1H, s), 2.47–2.50 (2H, m), 2.25 (3H, s), 1.00–1.11 (3H, m). 13C NMR (CDCl3, 75 MHz): δ, ppm 161.2, 136.6, 133.7, 129.0, 128.9, 128.6, 126.5, 119.8, 114.1, 82.6, 75.5, 66.4, 18.4, 15.3, 10.8. MS (EI): m/z 267.15 [M]+.
1,2-bis[2-(5-benzyloxycarbonyl-4-ethyl-3-methyl-pyrrolyl)]ethyne (4a)
Method A: To a solution of benzyl 4-ethyl-5-iodo-3-methylpyrrole-2-carboxylate (1a) (1.85 g, 5 mmol) and benzyl 5-ethynyl-4-ethyl-3-methylpyrrole-2 -carboxylate (3) (1.34 g, 5 mmol) in diethylamine (50 mL) were added dichlorobis(triphenylphosphine)palladium(II) (59 mg, 0.08 mmol) and copper(I) iodide (30 mg, 0.16 mmol). The homogeneous mixture was stirred at 50 °C for 3 h. After evaporation of the solvent in vacuo, the residue was subjected to chromatography on a short silica gel column with hexane/dichloromethane/ethyl acetate (5:1:1) as eluent to give 4a (1.75 g, 73%). Method B: A 250 mL Schlenk tube with a teflon-coated magnetic stir bar was charged with PdCl2(PPh3)2 (210 mg, 6 mol%), CuI (188 mg, 10 mol%) and benzyl 4-ethyl-5-iodo-3-methylpyrrole-2-carboxylate (1a) (3.69 g, 10 mmol). The system was pumped with a vacuum pump and refilled with nitrogen gas three times. Then dry benzene 40 mL, sparged with dry argon was added by syringe. Argon-sparged DBU (9.0 mL, 6 equiv.) in 5 mL dry benzene was then added via syringe and ice-chilled trimethylsilylethyne (0.71 mL, 0.50 equiv.) in 5 mL dry benzene was added by syringe, followed immediately by distilled water (73 μL, 40 mol%). The reaction flask was covered with aluminum foil and left stirring at a high rate of speed for 2 d until no starting material was left according to TLC; the reaction mixture was partitioned between ethyl acetate and brine (200 mL each). The organic layer was washed with 10% HCl (3 × 200 mL), saturated aqueous NaCl (200 mL), dried over anhydrous NaSO4, gravity-filtered and the solvent then removed in vacuo. The crude product was washed with methanol and dried under vacuum, giving 1.57 g (62% yield) of the title compound. 1H NMR (CDCl3, 250 MHz): δ, ppm 8.86 (2H, s), 7.38–7.41 (10H, m), 5.31 (4H, s), 2.53–2.56 (4H, m), 2.30 (6H, s), 1.11–1.14 (6H, m). 13C NMR (CDCl3, 75 MHz): δ, ppm 162.2, 137.7, 134.6, 133.6, 130.2, 129.8, 127.9, 121.2, 115.8, 86.6, 67.6, 19.8, 16.5, 11.9. HRMS (ESI-TOF): m/z 509.2444 [M + H]+, calcd. for C32H33N2O4 509.2435.
1,2-bis[2-(5-benzyloxycarbonyl-3,4-dimethylpyrrolyl)]ethyne (4b)
This compound was obtained in 54% yield using the above method B and recrystallized from dichloromethane to afford colorless crystals. 1H NMR (250 MHz, CDCl3): δ, ppm 11.99 (2H, s), 7.47–7.32 (10H, m), 5.29 (4H, s), 2.2 (6H, s), 2.0 (6H, s). MS (EI): m/z 480.129 [M]+.
1,2-bis[2-(5-ethoxycarbonyl-3,4-dimethylpyrrolyl)]ethyne (4c)
This compound was obtained in 52% yield using the above method B and recrystallized from dichloromethane to afford colorless crystals. 1H NMR (250 MHz, CDCl3): δ, ppm 8.80 (2H, s), 4.34 (4H. q, J = 7.01 Hz), 2.35 (6H, s), 2.10 (6H, s), 1.37 (6H, t, J = 7.05 Hz). 13C NMR (63 MHz, CDCl3): δ, ppm 10.3, 10.9, 14.9, 60.6, 86.0, 114.8, 120.3, 126.9, 160.6. HRMS (MALDI-TOF): m/z 357.1807 [M + H]+, calcd. for C20H25N2O4 357.1808.
1,2-bis(N-tosylpyrrolyl)ethyne (4d)
To a 50 mL round bottom flask was added 2-bromo-1-tosyl-pyrrole (0.3 g, 1 mmol) followed by Pd(PPh)2Cl2 (0.042 g, 0.06 mmol) and CuI (0.038 g, 0.2 mmol). The flask was sealed and placed in a dry ice bath under N2. Trimethylsilylethyne (0.072 mL, 0.5 mmol), DBU (0.9 mL, 6 mmol) and water (0.0072 mL, 40 molar equiv.) were dissolved in benzene (5 mL) and added to the reaction flask. After freezing the mixture in a dry ice bath, the flask was evacuated and nitrogen gas was added. The resulting reaction mixture was allowed to warm slowly to room temperature and was stirred until complete disappearance of the starting material, as monitored by TLC. The mixture was worked up by adding ethyl acetate (100 mL), and washing the organic layer three times with brine. The organic phase was dried over anhydrous Na2CO3 and concentrated under reduced pressure. The crude mixture was purified by flash column chromatography using hexane/ethyl acetate (5:1) for elution. The bispyrrolylethyne (4d) was obtained in 8.4% yield (0.02 g) and recrystallized from dichloromethane to afford colorless crystals. mp 185–186 °C. 1H NMR (250 MHz, CDCl3,): δ, ppm 7.9 (4H, br), 7.4 (4H, br), 7.3 (2H, br, 2H), 6.71 (2H, br), 6.34 (2H, br), 2.42 (6H, s). HRMS (MALDI-TOF): m/z 465.0939 [M + H]+, calcd. for C24H21O4N2S2 465.0943.
2,3-bis[2-(5-ethoxycarbonyl-3-ethyl-4-methylpyrrolyl)]-indole (5c)
1,2-bis[2-(5-ethoxycarbonyl-3,4- dimethylpyrrolyl)]ethyne (4c) (0.36 g, 1 mmol) was added to a 100 mL reaction tube together with 2-iodoaniline (0.44 g, 2 mmol), Pd(OAc)2 (0.02 g, 0.1 mmol), LiCl (0.04 g, 1 mmol), and K2CO3 (1.38 g, 10 mmol) and dissolved in 25 mL of DMF. The mixture was placed in a dry ice bath, evacuated, purged with nitrogen gas and then allowed to warm to room temperature and heated at 80 °C for 2 d. The reaction was monitored by TLC. Ethyl acetate (150 mL) was added to the reaction mixture and the organic layer was washed with brine (3 × 100 mL). The organic phase was dried over anhydrous NaHCO3 and concentrated under reduced pressure. The crude mixture was purified by flash column chromatography on silica gel using hexane/ethyl acetate (7:1) as eluant. The product (5c) was obtained (0.25 g) in 55% yield and recrys-tallized from dichloromethane to give colorless crystals. mp 45–47 °C. 1H NMR (250 MHz, CDCl3): δ, ppm 8.86 (1H, s), 8.83 (1H, s), 8.47 (1H, s), 7.53 (1H, d, J = 7.45 Hz), 7.45 (1H, d, J = 7.91 Hz), 7.24 (2H, m), 4.33 (2H, q, J = 7.12 Hz), 4.23 (2H, q, J = 6.98 Hz), 2.37 (3H, s), 2.30 (3H, s), 2.11 (3H, s), 1.79 (3H, s), 1.39 (3H, t, J = 7.19 Hz), 1.32 (3H, t, J = 6.96 Hz). 13C NMR (63 MHz, CDCl3): δ, ppm 9.9, 10.4, 10.7, 11.2, 14.7, 14.9, 60.3, 60.4, 105.9, 111.4, 119.5, 119.6, 120.0, 120.2, 121.3, 123.6, 124.6, 126.6, 128.1, 128.2, 128.8, 136.1, 161.6, 161.2. HRMS (ESI-TOF): m/z 448.2221 [M + H]+, calcd. for C26H30N3O4 448.2231.
2,3-bis[2-(5-benzyloxycarbonyl-3,4-dimethylpyrrolyl)]-indole (5a)
Prepared using above method from 4a in 54% yield and recrystallized from dichloromethane to give colorless crystals. mp 53–54 °C. 1H NMR (250 MHz, CDCl3): δ, ppm 8.82 (1H, s), 8.80 (1H, s), 8.30 (1H, s), 7.34 (14H, m), 5.28 (2H, s), 5.18 (2H, s), 2.60 (2H, q, J = 7.56 Hz), 2.31 (3H, s), 2.34 (3H, s), 2.23 (2H, q, J = 7.58 Hz), 1.11 (3H, t, J = 7.53 Hz), 0.82 (3H, t, J = 7.58 Hz). 13C NMR (63 MHz, CDCl3): δ, ppm 10.6, 11.2, 15.2, 15.7, 18.1, 66.0, 106.1, 111.4, 119.5, 119.8, 123.6, 126.3, 127.3, 128.4, 128.5, 128.8, 128.9, 129.2, 136.0, 136.6, 161.7. HRMS (ESI-TOF): m/z 600.2889 [M + H]+, calcd. for C38H38N3O4 600.2863.
Cis-1,2-[2-(5-ethoxycarbonyl-4-ethyl-3-methylpyrrolyl)]ethene (6)
Commercially available Lindlar’s catalyst (5 wt.% Pd on CaCO3, poisoned with lead; 150 mg, 100 wt.%) was added to a solution of alkyne 4c (150 mg, 0.42 mmol) in dry ethyl acetate (15 mL). The flask was evacuated and flushed with hydrogen gas (two freeze/thaw cycles) and the mixture was stirred under hydrogen gas for 12 h. The resulting mixture was then filtered over a Celite cake and washed with ethyl acetate (2 × 30 mL). The solvent was removed under reduced pressure and the crude residue was purified by silica gel column chromatography eluting with ethyl acetate/dichloromethane/hexanes (1:1:5) to yield the title product as a yellow oil that solidified to a yellow powder (47 mg, 0.13 mmol, 31% yield). 1H NMR (450 MHz, CDCl3, 293 K): δ, ppm 8.73 (2H, s, NH), 6.33 (2H, s, CHCH), 4.26 (4H, q, CH2), 2.29 (6H, s, CH3), 1.99 (6H, s, CH3), 1.30 (6H, t, CH3). HRMS (ESI-TOF): m/z 359.1965 [M + H]+, calcd. for C20H27N2O4 359.1971.
1,2-bis[2-(5-benzyloxycarbonyl-3-ethyl-4-methylpyrrolyl)]-dodecaborane (7)
A mixture of dodecaborane (2.44 g, 3.0 mmol) and Et2S (9.0 mL, 37 mmol) in 20 mL of dry toluene was heated at 40 °C for 2 h under Ar, then raised to 60 °C for 3 h. Then 4a (1.06 g, 2 mmol) in 20 mL toluene was added into the mixture via a syringe, and the mixture was heated to 80 °C for 2 d under argon. After TLC indicated no 4a remained, the mixture was cooled to room temperature and excess decarborane was destroyed by adding methanol. The solvent was removed under reduced pressure and ethanol was added to allow codistillation of Et2S. The residue was purified on a silica gel column using 25% ethyl acetate in hexane as eluant, giving the title compound (0.363 g) in 29% yield as a light yellow power. 1H NMR (300 MHz, CDCl3): δ, ppm 8.60 (2H, s), 7.37–7.43 (10H, m), 5.27 (4H, s), 2.61–2.69 (4H, q), 2.18 (6H, s), 1.50–3.50 (br, 10H), 1.03–1.08 (6H, t). 13C NMR (CDCl3, 75 MHz): δ, ppm 161.8, 137.2, 133.1, 130.2, 129.9, 129.8, 129.5, 122.1, 121.6, 84.6, 67.9, 18.9, 16.5, 11.8. MALDI-TOF: [M]+ 624.76.
X-ray crystallographic data
Crystals of 4a, 4c, 5c, 5c–F− complex and 7 for X-ray crystallographic analysis were grown by slow evaporation of their dichloromethane solutions. Diffraction data were collected at low temperature on a Nonius KappaCCD diffractometer equipped with MoKα radiation (λ = 0.71073 Å) and an Oxford Cryosystems Cryostream chiller. Refinement was by full-matrix least squares using SHELXL, with H atoms in idealized positions, except for NH hydrogen atoms, which were located by difference maps and their positions refined. One of the ethyl groups in 4c was disordered into two orientations, as is one of the DCM molecules in the 5c fluoride complex. Crystallographic data: For 4a: C32H32N2O4, monoclinic space group P2/c, a = 9.870(2), b = 10.635(2), c = 13.545(2) Å, β = 105.469(10)°, V = 1370.3(4) Å3, T = 90.0(5) K, Z = 2, ρcalcd = 1.233 g cm−3, μ(MoKα) = 0.081 mm−1. A total of 20,271 data was collected at to θ = 28.8°. R = 0.041 for 2783 data with Fo2 > 2σ(Fo2) of 3549 unique data and 178 refined parameters, CCDC 821407. For 4c: C20H24N2O4, orthorhombic space group Pbca, a = 13.889(2), b = 14.845(4), c = 18.920(5) Å, V = 3901.0(16)Å3, T = 120.0(5) K, Z = 8, ρcalcd = 1.214 g cm−3, μ(MoKα) = 0.085 mm−1. 19,130 total data, θmax = 22.5°. R = 0.042 for 1838 data with Fo2 > 2σ(Fo2) of 3549 unique data and 264 refined parameters, CCDC 779360. For 5c: C26H29N3O4, triclinic space group P-1, a = 10.244(2), b = 10.789(2), c = 12.516(3) Å, α = 107.091(12), β = 111.886(10), γ = 97.204(13)°, V = 1183.1(5)Å3, T = 90.0(5) K, Z = 2, ρcalcd = 1.256 g cm−3, μ(MoKα) = 0.086 mm−1. 18,860 total data, θmax = 26.4°. R = 0.048 for 3352 data with Fo2 > 2σ(Fo2) of 4813 unique data and 312 refined parameters, CCDC 779361. For 5c–F− complex: (C16H36N) (C26H29N3O4)2F. 1.5(CH2Cl2) triclinic space group P-1, a = 8.7265(10), b = 15.106(2), c = 27.490(3) Å, α = 74.955(6), β = 84.843(7), γ = 78.574(7)°, V = 3427.4(7) Å3, T = 90.0(5) K, Z = 2, ρcalcd = 1.244 g cm−3, μ(MoKα) = 0.195 mm−1. 51,056 total data, θmax = 27.1°. R = 0.108 for 8547 data with Fo2 > 2σ(Fo2) of 14,946 unique data and 819 refined parameters, CCDC 779362. For 7: C32H42B10N2O4, triclinic space group P-1, a = 11.651(2), b = 11.903(2), c = 12.1444(17) Å, α = 96.959(10), β = 91.123(9), γ = 92.222(7)°, V = 1670.0(5)Å3, T = 115.0(5) K, Z = 2, ρcalcd = 1.246 g cm−3, μ(MoKα) = 0.075 mm−1. 35,739 total data, θmax = 26.0°. R = 0.044 for 4850 data with Fo2 > 2σ(Fo2) of 6566 unique data and 444 refined parameters, CCDC 821406.
Crystallographic data have been deposited at the Cambridge Crystallographic Data Center (CCDC) under above-cited deposition numbers. Copies can be obtained on request, free of charge, via www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44 1223-336-033 or email: deposit@ccdc.cam.ac.uk).
CONCLUSION
We have developed an efficient one-pot double Sonagashira coupling reaction from which a series of dipyrrolylethynes (4a–d) has been synthesized using an improved route from trimethylsilylethyne and various 2-halopyrroles. The resulting dipyrrolylethynes were further transformed into various novel dipyrroles (5–7) using the Larock indole synthesis, stereoselective catalytic hydrogenation, or reaction with B10H14. These novel dipyrroles should be valuable building blocks for the construction of elaborate pyrrole-containing functional molecules and materials. In addition, indolyl-dipyrroles 5 were found to selectively bind fluoride ions with high affinity, forming a 2:1 complex involving one pyrrolic NH and the indole NH.
Acknowledgments
The work described was supported by grants from the US National Institutes of Health, (CA 132861; KMS) and the US National Science Foundation (CHE 0911629; MGHV). The purchase of the X-ray diffractometer was made possible by Grant No. LEQSF(1999-2000)-ENH-TR-13, administered by the Louisiana Board of Regents.
Footnotes
Dedicated to Professor John A. Shelnutt on the occasion of his 65th birthday
References
- 1.a) Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. Vol. 6. Academic Press; San Diego: 2000. [Google Scholar]; b) Dismukes GC. Science. 2001;292:447. doi: 10.1126/science.1060222. [DOI] [PubMed] [Google Scholar]; c) Chang CJ, Chng LL, Nocera DG. J Am Chem Soc. 2003;125:1866. doi: 10.1021/ja028548o. [DOI] [PubMed] [Google Scholar]
- 2.a) Sessler JL, An D, Cho W-S, Lynch V. Angew Chem Int Ed. 2003;42:2278. doi: 10.1002/anie.200350941. [DOI] [PubMed] [Google Scholar]; b) Sessler JL, An D, Cho WS, Lynch V. J Am Chem Soc. 2003;125:13646. doi: 10.1021/ja038264j. [DOI] [PubMed] [Google Scholar]; c) Geier GR, III, Grindrod SC. J Org Chem. 2004;69:6404. doi: 10.1021/jo049131z. [DOI] [PubMed] [Google Scholar]; d) Jolicoeur B, Lubell WD. Org Lett. 2006;8:6107. doi: 10.1021/ol0625583. [DOI] [PubMed] [Google Scholar]; e) Roznyatovskiy VV, Roznyatovskaya NV, Weyrauch H, Pinkwart K, Tubke J, Sessler JL. J Org Chem. 2010;75:8355. doi: 10.1021/jo1016426. [DOI] [PubMed] [Google Scholar]; f) Jiao L, Yu C, Liu M, Cong K, Meng T, Wang Y, Hao E. J Org Chem. 2010;75:6035. doi: 10.1021/jo101164a. [DOI] [PubMed] [Google Scholar]; g) Sessler JL, Weghorn SJ. Expanded, Contracted and Isomeric Porphyrins. Pergamon Press; New York: 1997. [Google Scholar]
- 3.Jux N, Koch P, Schmickler H, Lex J, Vogel E. Angew Chem Int Ed. 1990;29:1385. [Google Scholar]
- 4.Cho DH, Lee JH, Kim BH. J Org Chem. 1999;64:8048. [Google Scholar]
- 5.Tu B, Ghosh B, Lightner DA. J Org Chem. 2003;68:8950. doi: 10.1021/jo030252t. [DOI] [PubMed] [Google Scholar]
- 6.Mio MJ, Kopel LC, Braun JB, Gadzikwa TL, Hull KL, Brisbois RG, Markworth CJ, Grieco PA. Org Lett. 2002;4:3199. doi: 10.1021/ol026266n. [DOI] [PubMed] [Google Scholar]
- 7.a) Black CB, Andrioletti B, Try AC, Ruiperez C, Sessler JL. J Am Chem Soc. 1999;121:10438. [Google Scholar]; b) Anzenbacher P, Jr, Try AC, Miyaji H, Jursíková K, Lynch VM, Marquez M, Sessler JL. J Am Chem Soc. 2000;122:10268. [Google Scholar]; c) Mizuno T, Wei W-H, Eller LR, Sessler JL. J Am Chem Soc. 2002;124:1134. doi: 10.1021/ja017298t. [DOI] [PubMed] [Google Scholar]; d) Sessler JL, Maeda H, Mizuno T, Lynch VM, Furuta H. J Am Chem Soc. 2002;124:13474. doi: 10.1021/ja0273750. [DOI] [PubMed] [Google Scholar]; e) Yoo J, Kim M-S, Hong S-J, Sessler JL, Lee C-H. J Org Chem. 2009;74:1065. doi: 10.1021/jo802059c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh T, Maiya BG, Wong MW. J Phys Chem A. 2004;108:11249. [Google Scholar]
- 9.Amendola V, Esteban-Gómez D, Fabbrizzi L, Licchelli M. Acc Chem Res. 2006;39:343. doi: 10.1021/ar050195l. [DOI] [PubMed] [Google Scholar]
- 10.a) Clark JC, Fabre B, Fronczek FR, Vicente MGH. J Porphyrins Phthalocyanines. 2005;9:803. [Google Scholar]; b) Fabre B, Clark JC, Vicente MGH. Macromolecules. 2006;39:112. [Google Scholar]; c) Hao E, Fabre B, Fronczek FR, Vicente MGH. Chem Commun. 2007:4387. doi: 10.1039/b710375a. [DOI] [PubMed] [Google Scholar]; d) Hao E, Fabre B, Fronczek FR, Vicente MGH. Chem Mater. 2007;19:6195. doi: 10.1039/b710375a. [DOI] [PubMed] [Google Scholar]
- 11.a) Lee DA, Xie H, Senge MO, Smith KM. J Chem Soc, Chem Commun. 1994:791. [Google Scholar]; b) Xie H, Lee DA, Wallace DM, Senge MO, Smith KM. J Org Chem. 1996;61:8508. [Google Scholar]
- 12.a) Jiao LJ, Hao EH, Fronczek FR, Vicente MGH, Smith KM. J Porphyrins Phthalocyanines. 2011;15:433. doi: 10.1142/S1088424611003355. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jiao L, Hao E, Vicente MGH, Smith KM. J Org Chem. 2007;72:8119. doi: 10.1021/jo701310k. [DOI] [PubMed] [Google Scholar]
- 13.Chinchilla R, Najera C. Chem Rev. 2007;107:874. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 14.a) Tanui H, Fronczek FR, Vicente MGH. Acta Crystallogr. 2007;E64:o75. doi: 10.1107/S1600536807061739. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tanui H, Fronczek FR, Vicente MGH. Acta Crystallogr. 2007;E64:o130. doi: 10.1107/S1600536807062964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larock RC, Yum EK. J Am Chem Soc. 1991;113:6689. [Google Scholar]
- 16.a) Lindlar H, Dubuis R. Org Synth. 1973;5:880. [Google Scholar]; b) Takeda H, Watanabe H, Nakada M. Tetrahedron. 2006;62:8054. [Google Scholar]
- 17.Luo F, Pan C, Wang W, Ye Z, Cheng J. Tetrahedron. 2010;66:1399. [Google Scholar]
- 18.Jiang W, Knobler CB, Hawthorne MF. Inorg Chem. 1996;35:3056. doi: 10.1021/ic951574l. [DOI] [PubMed] [Google Scholar]
- 19.Fery-Forgues S, Lavabre D. J Chem Educ. 1999;76:1260. [Google Scholar]