

Abstract
The synthesis and structure–activity relationships (SAR) of a series of benzothiophene piperazine and piperidine urea FAAH inhibitors is described. These compounds inhibit FAAH by covalently modifying the enzyme’s active site serine nucleophile. Activity-based protein profiling (ABPP) revealed that these urea inhibitors were completely selective for FAAH relative to other mammalian serine hydrolases. Several compounds showed in vivo activity in a rat complete Freund’s adjuvant (CFA) model of inflammatory pain.
Keywords: FAAH inhibitor, Fatty acid amide hydrolase, Urea, Activity-based protein profiling, Covalent inhibitor
Fatty acid amide hydrolase (FAAH) is an integral membrane enzyme that degrades the fatty acid amide family of signaling lipids, including the endocannabinoidanandamide.1 Genetic1g or pharmacological1f inactivation of FAAH leads to elevated endogenous levels of fatty acid amides and a range of behavioral effects including analgesic and anti-inflammatory phenotypes in rodents. Importantly, these behavioral phenotypes occur in the absence of alterations in motility, weight gain, or body temperature that are typically observed with direct cannabinoid receptor 1 (CB1) agonists, indicating that FAAH may represent an attractive therapeutic target for the treatment of inflammatory pain and related conditions.2 Several classes of FAAH inhibitors have been reported including electrophilic ketones (e.g., OL-1353), carbamates (e.g., URB5974 and SA-475) and, more recently, piperidine/piperazine ureas (e.g., PF-7506 and Takeda-25/JNJ-16610107) (Fig. 1). We recently reported that PF-750 inhibits FAAH by covalently modifying the enzyme’s active site serine nucleophile and is completely selective for FAAH relative to other mammalian serinehydrolases.6a In this Letter, we describe the synthesis and structure–activity relationships (SAR) of a series of benzothiophene piperazine and piperidine urea FAAH inhibitors.
Figure 1.

Structures of FAAH inhibitors.
A series of piperazine phenyl ureas, exemplified by 1, was discovered by high throughput screening of the Pfizer chemical file against human FAAH (Fig. 2). We prepared piperazine phenyl urea intermediate 2 and performed a series of reductive aminations with various aromatic aldehydes (Scheme 1, Eqs. 1 and 2). This allowed us to easily vary the aldehyde component and resulted in the identification of several bicyclic cores including PF-6226a and a series of benzothiophenes represented by 4h, which is the subject of this manuscript (Scheme 1, Eq. 2).
Figure 2.

Structures of HTS lead (1) and PF-622.
Scheme 1.

Synthesis of piperazine ureas 4 and 6: (a) PhNCO, CH2Cl2, 92%; (b) TFA, CH2Cl2, 80–99% (c) NaBH(OAc)3, DCE, 50–95%; (d) N-Boc-piperazine, NaBH(OAc)3, CH2Cl2; then TFA, CH2Cl2, 64%; (e) RNCO (R = alkyl, Ph, 3-pyr; ie. 4d–g, 4h, 6a), CH2Cl2, 50–90%; (f) RNHCO2Ph (R = heterocycle, ie. 4j–1, 6), DMSO, 60 °C, 50–80%.
We then explored the benzothiophene lead 4h in more detail. Therefore, the benzothiophene piperazine intermediate 5 was prepared according to Equation 3 and reacted with various commercially available alkyl and aryl isocyanates or phenyl carbamates of heterocyclic amines8 to give the corresponding piperazine ureas 4 and 6 (Scheme 1).
Scheme 2 and Scheme 3 outline the synthesis of representative piperidine urea FAAH inhibitors utilized in the current studies. Lithiation of benzothiophene 7 followed by reaction with Weinreb amide 8 provided ketone 9 in good yield.9 Ketone 9 was reduced with sodium borohydride to give alcohol 10 which underwent elimination when treated with p-toluene sulfonic acid (TsOH). The resulting double bond was hydrogenated to give piperidine 11 which was treated with isocyanate 12 as described previously to give the desired urea 13.8
Scheme 2.

Synthesis of piperidine urea 13: (a) n-BuLi, THF, −78 °C; then add 8, 70%, (b) NaBH4, EtOH, quant; (c) TsOH, toluene, quant; (d) H2(g), 20% Pd/C, MeOH, 87%; (e) 12, CH2Cl2, 60%.
Scheme 3.

Synthesis of linker analogs 15–18: (a) TFA, CH2Cl2, 99%; (b) 12, CH2Cl2, 85%; (c) Ph3PCH3Br, n-BuLi, THF, −20 °C to rt, 97%; d) H2(g), Pd/C, MeOH, 76%; (e) chiral chromatography (CHIRALCEL® OD); (f) NaBH4, EtOH, quant.
The linker analogs 15–18 were synthesized according to Scheme 3. Deprotection of Boc-piperidine 14 with TFA followed by reaction with isocyanate 12 afforded urea 15 with a ketone linker. Wittig olefination of ketone 15 provided compound 16 with a methylene substituted linker. Hydrogenation of the double bond gave 17 with a methyl substituted linker as the racemate and the enantiomers were separated by chiral chromatography. Furthermore, ketone 15 was reduced to give racemic 18 with a hydroxy substituted linker.
As previously reported, these piperidine/piperazine ureas inhibit FAAH by covalent carbamylation of the catalytic Ser241 nucleophile.6a,6b Therefore, the potency of these benzothiophene piperidine/piperazine urea inhibitors were determined by the second order rate constants kinact/Ki values using an enzyme-coupled FAAH assay as described previously.6b,10 Unlike IC50 values, kinact/Ki values do not change with various preincubation times and have been described as the best measure of potency for irreversible inhibitors.11
Table 1 shows the SAR of the right hand portion of the urea. The alkyl ureas (4a–g) had very low potency for FAAH, which is similar to what has been observed for N-alkylurea analogs of URB597.4a In contrast, we found that aryl ureas such as phenyl urea 4h were potent FAAH inhibitors. Next, we investigated a series of heteroaromatic ureas (4i–l, 6a) in part to avoid the generation of aniline upon covalent binding to FAAH. The 3-aminopyridyl group (6a) was preferred over the 2-aminopyridyl (4i) and 3-aminopyridazinyl (4j) groups in the benzothiophene series. The SAR of the heterocyclic group was highly sensitive. For example, isoxazole 4k retained potency, while the potency was lost with methylthiazole 4l, which is consistent with observations made by Keith et al. in a related thiadiazolopiperazinyl urea series.7b Boger and co-workers also observed that subtle changes in the central heterocycle of α-ketoheterocycle FAAH inhibitors greatly influence inhibitor activity.3c Also it is interesting to note that isoxazole 4k exhibited equal potency for hFAAH and rFAAH, while phenyl urea 4h and pyridazine urea 4j showed higher potency for hFAAH than rFAAH.
Table 1.
Human and rat FAAH potency (kinact/Ki values)15 for compounds 4a–l, 6a showing SAR of the right hand side of the urea
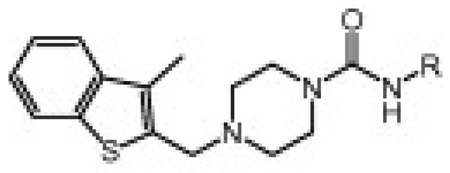 | |||
|---|---|---|---|
| R | hkinact/Ki (M−1 s−1) | rkinact/Ki (M−1 s−1) | |
| 4a | Butyl | 44 | 13 |
| 4b | CH2CH2OH | 42 | <10 |
| 4c | CH2C(O)NHMe | <10 | <10 |
| 4d | iPr | 70 | <10 |
| 4e | Cyclohexyl | <10 | <10 |
| 4f | tBu | <10 | <10 |
| 4g | Bn | <10 | <10 |
| 4h | Ph | 1918 | 762 |
| 4i | 2-pyr | 1002 | 635 |
| 6a | 3-pyr | 2401 | 1083 |
| 4j | 924 | 191 | |
| 4k | 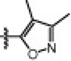 |
1559 | 1433 |
| 4l | <10 | <10 | |
Next, we investigated the effect of substituents at the 6-position of the 3-amino pyridine (Table 2). The compounds with 6-methoxy (6g), 6-amino (6i) and 6-NHAc (6n) substituents are roughly equipotent to 6a, while the 6-Cl (6d) and 6-NHMe (6j) groups result in compounds that are about twofold less potent and the potency is further diminished by the 6-NHEt (6l) substituent (fourfold less potent than 6a). Furthermore, 6-CF3 (6c) and 6-NEt2 (6m) substituents are detrimental to activity. Taken together with the results in Table 1, it is clear that the nature of the aromatic amine portion of the urea is critical for activity, but the potency does not appear to correlate with the leaving group ability. There appears to be a steric limitation at the 6-position as hFAAH potency generally decreases with the larger substituents (i.e., h kinact/Ki for NH2 > NHMe > NMe2 > NEt2). Interestingly, several 6-amino compounds (6j–m) actually show a preference for rFAAH over hFAAH.
Table 2.
Human and rat FAAH potency (kinact/Ki values)15 for compounds 6a–6n showing SAR of substituted pyridyl ureas
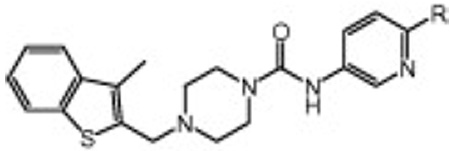 | |||
|---|---|---|---|
| R | hkinact/Ki (M−1 s−1) | rkinact/Ki (M−1 s−1) | |
| 6a | H | 2401 | 1083 |
| 6b | Me | 1481 | 390 |
| 6c | CF3 | 69 | <10 |
| 6d | CI | 1238 | 242 |
| 6e | Br | 645 | 157 |
| 6f | Ph | 631 | 1215 |
| 6g | OMe | 2931 | 1060 |
| 6h | O-(3-Pyr) | 605 | 101 |
| 6i | NH2 | 1871 | 1067 |
| 6j | NHMe | 1108 | 1513 |
| 6k | NMe2 | 892 | 1811 |
| 61 | NHEt | 597 | 1891 |
| 6m | NEt2 | 90 | 227 |
| 6n | NHAc | 2011 | 1719 |
Subsequent SAR studies focused on optimization of the benzothiophene substitution while maintaining the optimal 3-aminopyridyl urea (Table 3). It was found that incorporating an alkyl group at the 3-position of the benzothiophene (R1) led to improved potency. For example, compound 6a was 3–4-fold more potent than the unsubstituted benzothiophene 19. In most cases for hFAAH, the piperidine analogs were slightly more potent than the corresponding piperazine analogs (i.e., 20 > 6a), which is most likely a result of more favorable Van der Waals interactions of the piperidine with the acyl chain binding channel of FAAH.1b,6b Substitution was tolerated at the 4- and 5-position of the benzothiophene (24–36), but no significant improvement in potency was realized. Conversely, moving the substituent (R4) to the 6-position of the benzothiophene impaired potency (37–39), which is predicted to introduce steric clashes with the FAAH protein (Fig. 5).
Table 3.
Human and rat FAAH potency (kinact/Ki values)15 for compounds 13, 19–39 showing SAR of substituted benzothiophenes
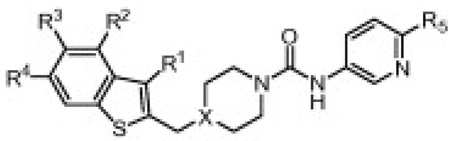 | ||||||||
|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | X | hkinact/Ki (M−1 s−1) | rkinact/Ki (M−1 s−1) | |
| 19 | H | H | H | H | H | N | 695 | 174 |
| 6a | Me | H | H | H | H | N | 2401 | 1083 |
| 20 | Me | H | H | H | H | C | 5129 | 2613 |
| 21 | Et | H | H | H | H | N | 1402 | 774 |
| 22 | Et | H | H | H | H | C | 1668 | 1013 |
| 6b | Me | H | H | H | Me | N | 1481 | 390 |
| 23 | Cl | H | H | H | Me | N | 900 | 133 |
| 24 | Me | F | H | H | H | N | 3321 | 1203 |
| 25 | Me | F | H | H | H | C | 4147 | 1547 |
| 26 | Me | F | H | H | Me | N | 1980 | 758 |
| 27 | Me | H | F | H | H | N | 3134 | 779 |
| 13 | Me | H | F | H | H | C | 4474 | 729 |
| 28 | Me | H | Me | H | H | N | 2635 | 557 |
| 29 | Me | H | Me | H | H | C | 3024 | 482 |
| 30 | Me | H | Cl | H | H | N | 2822 | 1133 |
| 31 | Me | H | CF3 | H | H | N | 1389 | 219 |
| 32 | Me | H | OMe | H | H | N | 1381 | 500 |
| 33 | Et | H | F | H | H | N | 1808 | 617 |
| 34 | Et | H | F | H | H | C | 2292 | 1141 |
| 35 | Me | H | F | H | Me | N | 1871 | 260 |
| 36 | Me | H | Me | H | Me | N | 1169 | 183 |
| 37 | Me | H | H | OMe | H | N | 73 | 113 |
| 38 | Me | H | H | OEt | H | N | 14 | <10 |
| 39 | Cl | H | H | Me | Me | N | 73 | 14 |
Figure 5.

Model of compound 38 covalently attached to Ser241 of h/rFAAH using coordinates from the PF-750-h/rFAAH crystal structure.6b Overlay with PF-750 shown in blue.
As shown in Table 4, the potency is maintained when a ketone or methylene is incorporated within the linker (15 and 16, Scheme 3) whereas the reduced compounds 17 and 18 with methyl and hydroxyl substituents lose much of their activity. The sp2 hybridization of the ketone and methylene linker compounds 15 and 16 imposes a conformational constraint that appears to help position the benzothiophene in a more favorable binding orientation. The SAR in this linker region is sensitive as further evidenced by the >10-fold difference in potency for the enantiomers of 17.
Table 4.
Human and rat FAAH potency (kinact/Ki values)15 for compounds 15–18 showing SAR of the piperidine linker
| hkinact/Ki (M−1 s−1) | rkinact/Ki (M−1 s−1) | |
|---|---|---|
| 15 | 3601 | 1226 |
| 16 | 5441 | 991 |
| 17-rac | 114 | 48 |
| 17-E1 | 34 | <10 |
| 17-E2 | 617 | 110 |
| 18-rac | 200 | 79 |
We have proposed that the urea may be activated upon binding in the active site of FAAH by undergoing a conformational change that diminishes conjugation of the nitrogen lone pair with the carbonyl, which would activate the urea toward nucleophile attack as shown in Figure 3.6a It appears that a cyclic secondary amine such as piperazine or piperidine is critical for activation of the urea as ureas prepared from primary amines are ineffective.4a
Figure 3.

Proposed mechanism of covalent inhibition of FAAH by piperidine ureas. The catalytic triad Ser-Ser-Lys is shown.
Compound 13 (Fig. 4) and compound 38 (Fig. 5) were modeled,12 using Sybyl 8.0, into the ‘humanized’ rat FAAH (h/rFAAH) crystal structure (2VYA).6b Compound 13 fit within the binding site in almost identical fashion to that of PF-750 with only very slight movement of the surrounding residue atoms, in essence, occupying virtually identical space. The green surface depicting the binding site volume and the space-filled representations were generated from the compound 13-h/rFAAH model and are virtually identical to those generated from the PF-750-h/rFAAH crystal structure. In contrast, when the model of compound 38-h/rFAAH was minimized, there was a significant shift in the position of the side chain of Met436. As shown in Figure 5, the ethoxy portion of compound 38 extends beyond the green surface generated from the compound 13 model illustrating the increased size of the ligand and the subsequent displacement (i.e., push) of the Met436 side chain. This steric interaction with Met436 could explain the decreased potency of compound 38.
Figure 4.

Model of compound 13 covalently attached to Ser241 of h/rFAAH using coordinates from the PF-750-h/rFAAH crystal structure.6b Overlay with PF-750 shown in blue.
To assess the selectivity of the benzothiophene piperidine/piperazine urea series, compounds 6b, 6g (PF-465), and 13 (PF-946) were profiled at 100 µM by competitiveactivity-based protein profiling (ABPP)13 in a number of different proteomes derived from mouse and human sources (Fig. 6). Competitive ABPP provides a global view of the proteome-wide selectivity of enzyme inhibitors as described previously.13b,13c In the case of FAAH inhibitors, we used a reporter-tagged fluorophosphonate (FP) ABPP probe,13c which serves as a general activity-based profiling tool for the serine hydrolase super family.13d,13e Serine hydrolases that show significant reductions in FP probe labeling intensity in the presence of inhibitor are scored as targets of the compound. The selectivity of compounds 6b, 6g, and 13 was compared to that of URB597, which was profiled at 10 and 100 µM. Gel images of soluble proteomes of mouse liver, mouse kidney, human liver, and human heart and the membrane proteome of human brain are shown in Figure 6. Consistent with previous reports,6a,14 multiple off-targets were observed in peripheral tissues for URB597 even at 10 µM, particularly amongst FP-labeled proteins migrating between 55 and 65 kDa. It is noteworthy that URB597, which has been shown to be highly selective for serine hydrolases in mouse brain proteomes, inhibits at least one additional hydrolase migrating at 55–60 kDa in the human brain membrane proteome. In contrast, no off-targets were observed for compounds 6b, 6g, and 13 even when tested at 100 µM (Fig. 6). Under the assay conditions, compounds 6b, 6g, 13, and URB597 completely inhibited FAAH from both mouse and human tissues.
Figure 6.

Selectivity profiling of FAAH inhibitors. Gel images of proteomes labeled with FP-rhodamine in the absence or presence of FAAH inhibitors (Lane 1, buffer; lane 2, 6b at 100 µM; lane 3, 6g at 100 µM; lane 4, 13 at 100 µM; lane 5, URB597 at 100 µM; lane 6, URB597 at 10 µM). Inhibited bands are shown with arrows. (A) soluble mouse liver proteome, (B) soluble mouse kidney proteome, (C) soluble human liver proteome, (D) soluble human heart proteome, and (E) human brain membrane proteome. Fluorescent gel signals shown in grayscale.
Next we selected compound 6g (PF-465) to assess its in vivo efficacy in a rat model of inflammatory pain. Subcutaneous injection of complete Freund’s adjuvant (CFA) into the plantar surface of the hind paw produced a significant decrease in mechanical paw weight threshold (PWT) at 5 days post injection (Fig. 7). Compound 6g dosed at 3, 10 and 30 mg/kg (ip) caused a dose-dependent inhibition of mechanical allodynia with a minimum effective dose (MED) of 10 mg/kg. At doses of 10 and 30 mg/kg, compound 6g inhibited pain responses to an equivalent degree as the nonsteroidal anti-inflammatory drug naproxen (10 mg/kg, ip).
Figure 7.

Anti-hyperalgesic effects of compound 6g (3–30 mg/kg, ip) in the CFA model of inflammatory pain. Compound 6g produces a dose-dependent reduction of mechanical allodynia (hyperalgesic) in rats (black bars). The effect of the non-steroidal anti-inflammatory drug naproxen (10 mg/kg, ip hatched bar) is shown for comparison. Anti-hyperalgesic responses were determined at 4 h following drug treatment and were significantly different between compound 6g- and vehicle-treated groups (p < 0.05). n = 8 rats/group.
In conclusion, we have described a series of benzothiophene piperazine/piperidine urea FAAH inhibitors that covalently modify the enzyme’s active site serinenucleophile. Activity-based protein profiling revealed that these urea inhibitors are highly selective for FAAH relative to other mammalian serine hydrolases. Furthermore, in vivo activity was demonstrated in a rat CFA model of inflammatory pain. Additional SAR development of this class of piperazine/piperidine urea FAAH inhibitors will be reported in due course.
References and notes
- 1.(a) Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Nature. 1996;384:83. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]; (b) Bracey MA, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Science. 2002;298:1793. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]; (c) Cravatt BF, Lichtman AH. Curr. Opin. Chem. Biol. 2003;7:469. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]; (d) McKinney MK, Cravatt BF. Annu. Rev. Biochem. 2005;74:411. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]; (e) Ahn K, McKinney MK, Cravatt BF. Chem. Rev. 2008;108:1687. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9371. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Nat. Med. 2003;9:76. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 2.(a) Lambert DM, Fowler CJ. J. Med. Chem. 2005;48:5059. doi: 10.1021/jm058183t. [DOI] [PubMed] [Google Scholar]; (b) Pacher P, Batkai S, Kunos G. Pharmacol. Rev. 2006;58:389. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Di Marzo V. Nat. Rev. Drug Disc. 2008;7:438. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]; (d) Seierstad M, Breitenbucher JG. J. Med. Chem. 2008;51:7327. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]
- 3.(a) Boger DL, Sato H, Lerner AE, Hedrick MP, Fecik RA, Miyauchi H, Wilkie GD, Austin BJ, Patricelli MP, Cravatt BF. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5044. doi: 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Leung D, Acevedo O, Guimaraes CRW, Jorgensen WL, Cravatt BF. J. Med. Chem. 2005;48:1849. doi: 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Garfunkle J, Ezzili C, Rayl TJ, Hochstatter DG, Hwang I, Boger DL. J. Med. Chem. 2008;51:4392. doi: 10.1021/jm800136b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D. J. Med. Chem. 2003;46:2352. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]; (b) Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. J. Med. Chem. 2004;47:4998. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]; (c) Tarzia G, Duranti A, Gatti G, Piersanti G, Tontini A, Rivara S, Lodola A, Plazzi PV, Mor M, Kathuria S, Piomelli D. Chem. Med. Chem. 2006;1:2352. doi: 10.1002/cmdc.200500017. [DOI] [PubMed] [Google Scholar]
- 5.Abouab-Dellah A, Burnier P, Hoornaert C, Jeunesse J, Puech F. WO 2004/099176. Patent. 2004 US 2006/0089344, 2006.
- 6.(a) Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, Cravatt BF. Biochemistry. 2007;46:13019. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]; (b) Mileni M, Johnson DS, Wang Z, Everdeen D, Liimatta M, Pabst B, Bhattacharya K, Nugent RA, Kamtekar S, Cravatt BF, Ahn K, Stevens RC. Proc. Natl. Acad. Sci. U.S.A. 2008;105:12820. doi: 10.1073/pnas.0806121105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Apodaca R, Breitenbucher JG, Pattabiraman K, Seierstad M, Xiao W. WO 2006/074025. Patent. 2006
- 7.(a) Matsumoto T, Kori M, Miyazaki J, Kiyota Y. WO 2006/054652. Patent. 2006 EP 1813606, 2007.; (b) Keith JM, Apodaca R, Xiao W, Seierstad M, Pattabiraman K, Wu J, Webb M, Karbarz MJ, Brown S, Wilson S, Scott B, Tham C-S, Luo L, Palmer J, Wennerholm M, Chaplan S, Breitenbucher JG. Bioorg. Med. Chem. Lett. 2008;18:4838. doi: 10.1016/j.bmcl.2008.07.081. [DOI] [PubMed] [Google Scholar]; (c) Karbarz MJ, Luo L, Chang L, Tham C-S, Palmer JA, Wilson SJ, Wennerholm ML, Brown SM, Scott BP, Apodaca RL, Keith JM, Wu J, Breitenbucher JG, Chaplan SR, Webb M. Anesthesia Analgesia. 2009;108:316. doi: 10.1213/ane.0b013e31818c7cbd. [DOI] [PubMed] [Google Scholar]
- 8.(a) Thavonekham B. Synthesis. 1997:1189. [Google Scholar]; (b) Swanson DM, Dubin AE, Shah C, Nasser N, Chang L, Dax SL, Jetter M, Breitenbucher JG, Liu C, Mazur C, Lord B, Gonzales L, Hoey K, Rizzolio M, Bogenstaetter M, Codd EE, Lee DH, Zhang S-P, Chaplan SR, Carruthers NI. J. Med. Chem. 2005;48:1857. doi: 10.1021/jm0495071. [DOI] [PubMed] [Google Scholar]
- 9.Matsunaga N, Kaku T, Itoh F, Tanaka T, Hara T, Miki H, Iwasaki M, Aono T, Yamaoka M, Kusaka M, Tasaka A. Bioorg. Med. Chem. 2004;12:2251. doi: 10.1016/j.bmc.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Ahn K. WO 2006/085196. Patent. 2006
- 11.Copeland RA. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis. 2nd ed. New York: Wiley-VCH; 2000. pp. 318–349. [Google Scholar]
- 12.All the hydrogens were added to the protein and each compound was covalently attached to the sidechain oxygen of Ser241. The protein/ligand structures were then minimized using the Sybyl forcefield with the backbone atoms held constant; charges were not used. These minimized structures were then compared to the PF-750-h/rFAAH crystal structure.
- 13.(a) Cravatt BF, Wright AT, Kozarich JW. Annu. Rev. Biochem. 2008;77:383. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]; (b) Kidd D, Liu Y, Cravatt BF. Biochemistry. 2001;40:4005. doi: 10.1021/bi002579j. [DOI] [PubMed] [Google Scholar]; (c) Leung D, Hardouin C, Boger DL, Cravatt BF. Nat. Biotechnol. 2003;21:687. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]; (d) Patricelli MP, Giang DK, Stamp LM, Burbaum JJ. Proteomics. 2001;1:1067. doi: 10.1002/1615-9861(200109)1:9<1067::AID-PROT1067>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (e) Liu Y, Patricelli MP, Cravatt BF. Proc. Natl. Acad. Sci. U.S.A. 1999;96:14694. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Alexander JP, Cravatt BF. Chem. Biol. 2005;12:1179. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang D, Saraf A, Kolasa T, Bhatia P, Zheng GZ, Patel M, Lannoye GS, Richardson P, Stewart A, Rogers JC, Brioni JD, Surowy CS. Neuropharmacol. 2007;52:1095. doi: 10.1016/j.neuropharm.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 15.The hFAAH (amino acids 32–579) and rFAAH (amino acids 30–579) constructs were generated as the N-terminal transmembrane-deleted truncated forms with N-terminal His6 tags, and were expressed in E. coli and purified as previously described.6b Both hFAAH and rFAAH enzymes used in the present study had purity greater than 95% based on SDS–PAGE visualized by Coomassie blue staining. The GDH-coupled FAAH assay was performed for determination of potencies (kinact/Ki values) of inhibitors in 384-well microplates with a final volume of 50 µL. The details on the assay and derivations of the overall potency, kinact/Ki value, have been described previously.6b In the 384-well format assay, the kinact/Ki values were generally calculated from the slope, kinact/[Ki (1 + [S]/Km)], which is obtained from the kobs versus [I] linear lines. The kinact/Ki values are averages of at least two independent experiments.