

Abstract
Trypanosoma cruzi is an intracellular parasite and the causative agent of Chagas disease. Previous work has shown that the chemokine receptor CCR5 plays a role in systemic T. cruzi protection. We evaluated the importance of CCR5 and CCL5 for mucosal protection against natural oral and conjunctival T. cruzi challenges. T. cruzi-immune CCR5−/− and wild-type C57BL/6 mice were generated by repeated infectious challenges with T. cruzi. CCR5−/− and WT mice developed equivalent levels of cellular, humoral and protective mucosal responses. However, CCR5−/−-immune mice produced increased levels of CCL5 in protected gastric tissues, suggesting compensatory signaling through additional receptors. Neutralization of CCL5 in CCR5−/−-immune mice resulted in decreased mucosal inflammatory responses, reduced T. cruzi-specific antibody secreting cells, and significantly less mucosal T. cruzi protection, confirming an important role for CCL5 in optimal immune control of T. cruzi replication at the point of initial mucosal invasion. To further investigate the mechanism responsible for mucosal protection mediated by CCL5-CCR5 signaling, we evaluated the effects of CCL5 on B cells. CCL5 enhanced proliferation and IgM secretion in highly purified B cells triggered by suboptimal doses of LPS. In addition, neutralization of endogenous CCL5 inhibited B cell proliferation and IgM secretion during stimulation of highly purified B cells, indicating that B cell production of CCL5 has important autocrine effects. These findings demonstrate direct effects of CCL5 on B cells, with significant implications for the development of mucosal adjuvants, and further suggest that CCL5 may be important as a general B cell co-activator.
Introduction
Trypanosoma cruzi is an intracellular parasite and the causative agent of Chagas disease, affecting approximately 8–11 million people in Latin America (1) of which ~10–40% develop chronic cardiac and/or gastrointestinal complications. Transmission of T. cruzi can occur cutaneously through exposure to parasites present in the reduviid vector excreta contaminating the reduviid bite site. Transmission also occurs through mucosal infection after oral and conjunctival exposure to infected reduviid excreta. In addition, T. cruzi can be transmitted through blood and organ donation from infected individuals. Although Chagas disease predominantly affects individuals in Latin America, an estimated 300,000 immigrants from endemic countries are chronically infected with T. cruzi and can transmit the parasite through blood and organ donation in the United States (2). Due to these risks, the WHO has established a new global effort to eliminate Chagas disease through prevention and control practices. As part of this new global initiative, it is important that a prophylactic and/or therapeutic vaccine against T. cruzi be generated in order to fully protect all those at risk of infection.
Chemokines interact with G-protein coupled receptors on leukocytes and are divided into four families (C, CC, CXC, CX3C) based on the position of the cysteine residues (3). Chemokines play important roles in both homeostatic and inflammatory conditions. CCR5 is a chemokine receptor expressed on subpopulations of lymphocytes, monocytes/macrophages, and NK cells, as well as endothelial and other nonhematopoietic cells (4–9). CCR5 is positively regulated by IL-12 (10), IFN-γ, TNF-α and IL-10 (11). CCR5+ cells migrate to both mucosal and systemic sites in response to the chemokines CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES). These ligands have been shown to preferentially attract activated and memory CD4+ and CD8+ T cells (12–14) due to the increased level of CCR5 expressed on these cells. CCL5 (RANTES) is a chemokine produced mainly by T cells, platelets, macrophages, endothelial and epithelial cells (15). CCL5 recruits T cells, dendritic cells, monocytes, NK cells and other cell types (16) to sites of inflammation and infection due to the cell surface expression of CCR1, CCR3 and/or CCR5. The CCR5-CCL5 ligand axis (the central signaling pair representing the predominant effects of interactions between CCL3, CCL4 and CCL5 and CCR1, CCR3 and CCR5) has been shown to play a role in lymphocyte activation (17–20), differentiation (21), polarization (22–25) and survival (26). CCL5 can induce T cell adhesion to VCAM-1, ICAM-1, laminin, collagen and fibronectin proteins in the extracellular matrix (27). CCL5 also plays a role in the initiation and enhancement of antigen-specific humoral and cellular immune responses through the activation of helper T cells which enhance B cell responses and the function of antigen presenting cells (28–30).
CCR5 and CCL5 have been studied in patients with Chagas disease and in systemic models of T. cruzi infection in mice. High numbers of CCR5+ T cells and levels of CCL5 mRNA and protein have been identified in the hearts of T. cruzi infected mice (31–34). Macrophages infected with T. cruzi have been shown to produce CCL5 mRNA and protein (35). CCL5 has been shown to induce the uptake and destruction of T. cruzi in macrophages in a nitric oxide-dependent manner (36, 37). CCR5−/− T. cruzi infected mice develop increased levels of blood parasitemia and acute cardiac parasitism that appears to correlate with reduced survival (31, 33). Furthermore, polymorphisms affecting CCR5 expression in humans have been associated with Chagas disease progression (38, 39). These studies have evaluated the role of CCR5 and CCL5 during T. cruzi systemic challenges. In this current work, we report the first investigations of the importance of CCR5 and CCL5 for mucosal T. cruzi protection.
Materials and Methods
Mice, parasites and challenge protocols
C57BL/6J wild-type and CCR5−/− female mice were obtained from Jackson Laboratories (Bar Harbor, ME, (40)) and bred at Saint Louis University. Female BALB/c mice were obtained from NCI-Charles River. Mice were bred and housed under pathogen free conditions and all studies were conducted with the approval of the Saint Louis University Animal Care Committee in an AAALAC accredited facility. Genotyping to confirm the knockout of ccr5 was done via PCR using primers specific for the ccr5 exon region reported to be deleted. Primers directed at a portion of a ccr5 intron were used as a control. All CCR5−/− mice were confirmed to have the ccr5 deletion (data not shown). The Tulahuèn strain of T. cruzi was used throughout these studies. Insect-derived metacyclic trypomastigotes (IMT) were obtained from T. cruzi-infected Dipetalogaster maximus triatomme insects as described previously (41). Oral and conjunctival infections were done as described previously (41, 42). Mice were initially infected with 2,000 IMT or 1–2×106 CMT (cultured-derived metacyclic tryptomastigotes; described previously (42)) either orally or conjunctivally. Starting 2 weeks after the primary T. cruzi infection, blood was removed from all infected mice and evaluated microscopically for parasitemia until positive. T. cruzi-immune CCR5−/− and wild-type mice were generated by repeated oral or conjunctival T. cruzi challenges. At least 1 month after the last challenge, T. cruzi-immune CCR5−/− and wild-type mice were orally or conjunctivally challenged with 2,000–12,500 IMT. Naïve CCR5−/− and wild-type mice were infected in parallel as primary controls. Mice were sacrificed 3–13 days post-challenge to evaluate immune and protection responses.
Antigen-specific IgA and IgG ELISA
Serum and fecal pellets were collected from individual mice to assess T. cruzi-specific circulating IgG and secretory IgA (sIgA), respectively, via enzyme-linked immunosorbent assay (ELISA). Fecal pellets were collected and added to PBS + 10% FCS (Sigma, St. Louis, MO). Samples were vortexed for 15 seconds and centrifuged at 20,000 x g for 10 minutes. Nunc PolySorp Immuno plates (Rochester, NY) were coated with 10 μg/mL of T. cruzi lysate overnight at 4°C. Plates were then washed four times with PBS-Tween (PBS-T) and blocked with PBS + 10% FCS for 2–4 hrs at room temperature. Serum and fecal extracts were serially diluted in PBS and incubated overnight at 4°C. Plates were then washed with PBS-T and incubated with either goat anti-mouse IgG-HRP or goat anti-mouse IgA-Biotin (SouthernBiotech, Birmingham, AL) diluted in PBS and incubated overnight at 4°C. Plates were washed and streptavidin-HRP (Jackson ImmunoResearch, West Grove, PA) was added in PBS and incubated for 90 min at room temperature. Plates were washed and developed by the addition of 3,3′,5,5′-tetramethylbenzidine substrate (Sigma, St. Louis, MO). ELISA plates were analyzed at 450 nm with a reference of 540 nm.
ELISPOT assay to assess IFN-γ, IgG and IgA producing cells
Millititer HA 96-well microtiter plates with nitrocellulose bases (Millipore, Bedford, MA) were coated with 10μg/mL of either a monoclonal antibody specific for murine IFN-γ (clone R46A2; Pharmingen, San Diego, CA) or recombinant trans-sialidase (rTS) (43) for up to 72 hrs at 4°C. Plates were washed with PBS four times and blocked with RPMI + 10% FCS at room temperature for at least 2 hrs. For IFN-γ ELISPOT analyses, spleen cells (5×105 cells/well) harvested on days 11, 12 and 13 post-challenge were added. Wells were stimulated with media alone, 10 μg/mL T. cruzi lysate, 10 μg/mL rTS, or 2.5 μg/mL of H-2Kb-restricted CD8 epitopes TSSA (ANYNFTLV (44, 45)), 77.2 (VDYNFTIV (46)) or ASP2 (VNHRFTLV (47)) (Sigma, St. Louis, MO) at 37°C in 5% CO2 overnight. For IgG and IgA ELISPOT analyses, 1×106 spleen cells were added to rTS coated plates and incubated overnight at 37°C, 5% CO2. Plates were washed with water, followed by PBS and incubated with either rat anti-mouse IFN-γ biotin (clone XMG1.2; BD Biosciences, San Diego, CA); goat anti-mouse IgA-biotin or goat anti-mouse IgG-biotin (SouthernBiotech, Birmingham, AL) for 2 hrs at room temperature in PBS or PBS+10% FCS, respectively. Plates were washed four times with PBS-T and streptavidin-HRP (Jackson ImmunoResearch, West Grove, PA) added for 2 hrs at room temperature in PBS. Plates were then washed three times with PBS-T and PBS and spots developed via 3-amino-9-ethylcarbazole substrate (AEC) precipitation. Spots were counted using a Cellular Technology Immunospot plate reader and scanning software (CTL, Cleveland, OH), and the results are reported as the number of spot-forming cells or antibody-secreting cells per million spleen cells.
Quantification of mucosal T. cruzi replication
Quantitative PCR (qPCR) and limiting dilution assays (LDA) were used to quantitate the levels of mucosal T. cruzi replication. T. cruzi challenged mice were individually assayed for both qPCR and LDA 11 to 13 days after the final IMT challenge. T. cruzi-specific qPCR assays were completed as described previously for both orally challenged (41) and conjunctivally challenged mice (42). In some instances, gastric tissue was placed in either RNAlater or Allprotect tissue reagent (Qiagen, Valencia, CA) in order to isolate both DNA and RNA. Tissue from the entire stomach of individual mice was homogenized and DNA/RNA isolated as per the AllPrep DNA/RNA or DNA/RNA/Protein kit manual (Qiagen, Valencia, CA). For parasite outgrowth LDA, spleen and draining lymph node cells from orally challenged mice (gastric lymph node) and conjunctivally challenged mice (submandibular/parotid lymph nodes) were serially diluted and plated in 96-well microtiter plates in parasite axenic medium as described previously (48). Plates were incubated at room temperature for 2.5 months and inspected by inverted light microscopy for parasite outgrowth of T. cruzi epimastigotes. Results are reported as the number of parasites per million cells.
In vivo neutralization of CCL5 in CCR5−/− T. cruzi-immune mice
A neutralizing CCL5 antibody (clone R6G9) was generated as described previously (49). We confirmed the neutralizing activity of this CCL5 antibody using an in vitro CCL5 transwell migration assay (data not shown). Beginning four days prior to oral T. cruzi rechallenge, CCR5−/− T. cruzi-immune mice were injected intraperitonally with 250 μg of either anti-CCL5 antibody or an IgG1κ isotype control antibody (Sigma, St. Louis, MO) suspended in 100 μl of sterile PBS. Mice were reinjected with the same antibodies every other day (d-4, d-2, d0, d2, d4, d6, d8, d10) and rechallenged orally with 1000–2000 IMT on d0. Twelve days after rechallenge, mice were sacrificed and assayed for immune and protective responses.
Gene expression profiling in the gastric mucosa of T. cruzi-immune mice
CCR5−/−, wild-type C57BL/6, and BALB/c T. cruzi-immune mice were sacrificed either the day of challenge or on days 3, 7 or 14 after oral IMT challenge, gastric tissue placed in RNAlater (Qiagen, Valencia, CA) and RNA isolated using the TRIzol reagent (Invitrogen, Carlsbad, CA). Anti-CCL5 or IgG1κ isotype control treated CCR5−/− T. cruzi-immune mice were sacrified 12 days post oral IMT challenge, gastric tissue placed in RNAlater (Qiagen, Valencia, CA) and RNA isolated using the AllPrep DNA/RNA isolation kit (Qiagen, Valencia, CA). Naïve, age-matched CCR5−/− or wild-type gastric tissue were used as controls. Genomic DNA was removed using the TURBO DNA-free™ kit (Ambion, Austin, TX) and RNA was cleaned using the RNeasy Mini Kit (Qiagen, Valencia, CA). One microgram of RNA was used to generate cDNA via the enhanced avian RT first strand synthesis kit (Sigma, St. Louis, MO). CCL4, CCL5, CCL19, CCL22, CCL25, CXCL5, CXCL9, CXCL10, CXCL11 and CXCL12 Taqman gene expression primer/probe sets were purchased from Applied Biosystems (Foster City, CA). Foxp3 and Gapdh primers and probes were designed by the Trudeau Institute (Saranac Lake, NY) and generated by Integrated DNA Technologies (Coralville, IA). IgA primers and probe were designed using the RealTime PCR design tool and generated by Integrated DNA Technologies (Coralville, IA). The sequences of the foxp3, gapdh and IgA primers and probes are as follows: foxp3 forward primer (5′-CCCAGGAAAGACAGCAACCTT-3′), reverse primer (5′-TTCTCACAACCAGGCCACTTG-3′), probe (5′-ATCCTACCCACTGCTGGCAAATGGAGTC-3′); gapdh forward primer (5′-CTCGTCCCGTAGACAAAATGG-3′), reverse primer (5′-AATCTCCACTTTGCCACTGCA-3′), probe (5′-CGGATTTGGCCGTATTGGGCG-3′); IgA forward primer (5′-GCGAGCTTTCAACCCTAA-3′), reverse primer (5′-ACACTAGGTAGCTTTCTGGG-3′), probe (5′-TGCGATGGCTGCATGGAAATGA- 3′). The Taqman gene expression master mix was used and samples were run in an ABI Prism 7700 sequence detector (Applied Biosystems, Foster City, CA) using the following conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. Data represent fold changes in gene expression compared to age-matched naïve wild-type or CCR5−/− mice calculated using the 2−ΔΔCt method with gapdh as a housekeeping gene control. Negative controls without added reverse transcriptase were included to confirm the removal of gastric DNA.
In vitro B cell stimulation
B cells were purified from the spleens of naïve C57BL/6 mice using the EasySep negative selection mouse B cell enrichment kit (Stemcell Technologies, Vancouver, British Columbia). Typically, B cell purity was > 95% as measured by flow cytometry. In order to further purify the B cells, we performed complement-mediated T cell depletion. Purified B cells were adjusted to 1×107 cells/mL in RPMI 1640 + 5% FCS, 50 U/mL Penicillin, 50 μg/mL Streptomycin, 2 mM L-glutamine and 50 μM 2-mercaptoethanol, and anti-Thy1.2 antibody (5 μg/mL; BD biosciences, San Diego, CA) was added for 45 min at 4°C. Cells were washed three times and Low-Tox®-M Rabbit Complement (Cedarlane, Burlington, NC) was added at a final dilution of 1:15. Cells were incubated for 1 hr at 37°C, washed three times, counted and adjusted to 1×106 cells/mL in RPMI 1640 + 10% FCS, Penicillin/Streptomycin, L-glutamine and 2-mercaptoethanol. 1×105 highly purified B cells (> 98% CD19+) were added to 96 well flat bottom plates. Lipopolysaccaride (LPS; 0111:B4; Sigma Aldrich, St. Louis, MO), recombinant mouse CCL5 (R&D Systems, Minneapolis, MN), anti-CCL5 mAb (gift from Thomas Lane, UC-Irvine) and IgG1κ isotype control mAb (Biolegend, San Diego, CA) were added as indicated in each figure legend. Cells were incubated at 37°C, 5% CO2 for 2–7 days. In order to functionally assess the depletion of T cells in highly purified B cell cultures, total spleen cells and purified B cells (1×105 cells/well) were added to anti-CD3/anti-CD28-coated (1 μg/mL each; BD biosciences) 96 well U-bottom plates for 2–7 days. Supernatants were collected for CCL5 and total IgM measurement, followed by the addition of 1 μCi/well [3H] thmidine (Perkin Elmer Wallac, (Waltham, MA) for 16 hrs. Samples were harvested as described previously (43).
Flow cytometry
In some instances, cells were labeled with CFDA-SE (Invitrogen, Carlsbad, CA) to measure B cell proliferation. Briefly, highly purified B cells were washed with PBS, resuspended in warm PBS at 5×107 cells/mL and CFDA-SE was added at a final concentration of 1 μM for 8 min at 37°C. Ice cold media was added to stop the reaction and cells were washed several times with complete medium, counted and adjusted to 1×106 cells/mL. Cells were stimulated as described above. On days 3, 5 and 7 after stimulation, cells were spun down and transferred to a 96 well V-bottom plate, anti-CD16/32 (Fc block) added in DPBS + 1% FCS + 0.01% sodium azide for 15 min at 4°C, and surface stained at 4°C for 30 min using CD19 PE-Cy7, CD8 APC, CD3 Alexa 700 and CD4 Pacific Blue (all antibodies purchased from BD biosciences). Cells were washed three times, fixed with 1% formaldehyde and analyzed with a LSR-II flow cytometer (BD) and FlowJo v7 software (Tree Star, Inc., Ashland, OR).
Total IgM ELISA
Culture supernatants from B cell stimulated cultures were collected at different time points and stored at −20°C until use. 96-well maxisorp plates (Nunc) were coated with 1μg/mL of goat anti-mouse Ig(H+L) purified antibody, incubated overnight at 4°C, washed 4 times with PBST and blocked with PBS + 10% FCS for 2 hrs at room temperature. Purified mouse IgM was used to generate a standard curve. Different dilutions of culture supernatant were added to PBS + 10% FCS and incubated overnight at 4°C. Plates were washed 4 times with PBST, goat anti-mouse IgM HRP added (1/5000 in PBS + 10% FCS) for 2 hours at room temperature before development with 3,3′,5,5′-tetramethylbenzidine substrate (Sigma). All antibodies purchased from Southern Biotech. % suppression of total IgM was calculated as follows: ((LPSalone- LPSantibody treated)/LPSalone) × 100.
CCL5 ELISA
Highly purified B cells (>98% CD19+) were cultured with medium alone, LPS (10μg/mL) or anti-CD3/CD28 (1 μg/mL each). Culture supernatants were taken at days 2, 3, 5 and 7 and frozen at −20°C until use. A DuoSet mouse CCL5/Rantes ELISA (R&D systems, Minneapolis, MN) was used to quantify CCL5 levels. 2 μg/mL of rat anti-mouse CCL5 in PBS was added to Nunc maxisorp plates and incubated overnight at 4°C. Plates were blocked with PBS + 1% BSA for 1 hour at RT. Recombinant mouse CCL5 and culture supernatants diluted 1:5 in PBS + 1% BSA were added for 2 hrs at RT. Plates were developed by sequential addition of biotinylated goat anti-mouse CCL5 antibody in PBS + 1% BSA for 2 hrs at RT, streptavidin-HRP for 20 min at RT in the dark, and 3,3′,5,5′-tetramethylbenzidine substrate (Sigma).
Statistics
Statistical analyses were performed with STATISTICA version 6, 8 or 9 software (StatSoft, Inc., Tulsa, OK). Mann-Whitney U tests or student t tests were used to compare responses between groups.
Results
Increased gene expression levels of several inflammatory chemokines and chemokine receptors in the gastric mucosa of T. cruzi-immune mice after oral T. cruzi challenge
Previous work has shown that several inflammatory chemokines, such as CCL3, CCL4 and CCL5 are upregulated in the hearts of T. cruzi-infected mice. We sought to identify whether these chemokines, as well as other inflammatory chemokines and chemokine receptors, were upregulated in the gastric mucosa of T. cruzi-immune mice after oral T. cruzi rechallenge as the gastric mucosa is the primary site of infection and parasite replication after oral infection (48). T. cruzi-immune mice were sacrificed on days 0, 3, 7 and 14 after oral T. cruzi rechallenge. Quantitative PCR was used to assess the relative mRNA levels of several chemokines and chemokine receptors in the gastric mucosa (Table I). Many of the chemokines and chemokine receptors studied were upregulated prior to rechallenge (d0) compared with naïve control mice. After oral rechallenge, the relative mRNA levels of several of these genes were even further upregulated at days 7 and 14. Most notably, CCL5 (RANTES), CCL19, CXCL9 (MIG), CXCL10 (IP-10), CXCL11 (I-TAC) and CXCR3 were highly upregulated compared with naïve, age-matched controls. These results demonstrate that after oral T. cruzi rechallenge, several inflammatory chemokines and chemokine receptors are upregulated in the gastric mucosa, facilitating the trafficking of lymphocytes into the gastric mucosa. Furthermore, high levels of inflammation in the gastric mucosa itself may directly enhance innate control of parasite replication.
Table I.
Kinetic quantification of chemokine and chemokine receptor gene expression in the gastric mucosa of T. cruzi-immune BALB/c mice.
| Fold change | ||||
|---|---|---|---|---|
| Chemokines | d0 Immune | d3 Immune | d7 Immune | d14 Immune |
| CCL3 | 2.55 ± 0.21* | 1.18 ± 0.04 | 3.97 ± 0.15** | 2.08 ± 0.13* |
| CCL4 | 2.31 ± 0.08** | 1.45 ± 0.06 | 5.65 ± 0.27** | 3.43 ± 0.13** |
| CCL5 | 6.63 ± 0.19*** | 5.12 ± 0.17*** | 19.62 ± 1.66** | 12.00 ± 0.85** |
| CCL19 | 7.68 ± 0.84* | 2.99 ± 0.23 | 10.57 ± 1.8* | 9.36 ± 3.07* |
| CXCL9 | 2.88 ± 0.32 | 9.36 ± 2.79** | 13.51 ± 3.11* | 8.84 ± 1.17* |
| CXCL10 | 1.91 ± 0.18 | 4.54 ± 1.17 | 6.35 ± 1.27* | 4.46 ± 0.38* |
| CXCL11 | 2.10 ± 0.14* | 4.08 ± 0.37** | 9.39 ± 2.55* | 5.88 ± 0.48* |
| Chemokines Receptors | d0 Immune | d3 Immune | d7 Immune | d14 Immune |
| CCR5 | 1.48 ± 0.14 | 1.72 ± 0.05 | 3.30 ± 0.38* | 1.76 ± 0.15 |
| CXCR3 | 6.12 ± 0.59* | 4.26 ± 0.25** | 19.04 ± 3.88** | 10.22 ± 1.52** |
BALB/c mice were orally infected 3 times with T. cruzi. Two to four months later, mice were rechallenged (or not) with T. cruzi orally and gastric RNA isolated 3, 7, or 14 days later. After genomic DNA removal and cleanup, real-time PCR was used to measure chemokine and chemokine receptor expression. Data represent fold changes (± standard error) in gene expression as compared to age-matched naïve BALB/c mice (calculated using ΔΔCt method with GAPDH as housekeeping gene). Non-RT controls were added to confirm the removal of genomic DNA. N= 4–16 per group.
p < 0.05,
p < 0.01,
p < 0.001; [Mann-Whitney U Test] comparing ΔCt of T. cruzi-immune mice to the ΔCt of naïve age-matched control BALB/c mice.
CCR5−/− mice develop equivalent levels of T. cruzi-specific antibodies after oral and conjunctival T. cruzi challenges
Based on previous work demonstrating the importance of CCR5 in systemic T. cruzi protection (31, 33) combined with our results demonstrating an increase in the relative levels of both CCR5 and CCL5 mRNA in the gastric mucosa of T. cruzi-immune BALB/c mice (Table I), we decided to evaluate whether CCR5 plays an important role in mucosal T. cruzi protection. We first evaluated whether immune responses in CCR5−/−versus wild-type C57BL/6 mice were different after mucosal challenge. T. cruzi-specific antibody responses were measured four weeks after T. cruzi challenge (Fig. 1). We have previously shown that after both oral and conjunctival T. cruzi challenges, T. cruzi-specific IgG and IgA can be detected in serum and fecal extracts, respectively (42, 50). There were similar levels of T. cruzi-specific serum IgG after secondary oral (Fig. 1A) and conjunctival (Fig. 1B) challenge in the CCR5−/− and wild-type mice. We also detected similar levels of T. cruzi-specific fecal IgA in the CCR5−/− and wild-type mice after secondary oral (Fig. 1C) and conjunctival (Fig. 1D) challenge. Thus, CCR5 expression is not required for B cell activation and differentiation into T. cruzi-specific plasma cells.
Figure 1.

CCR5−/− T. cruzi-immune mice develop similar levels of T. cruzi-specific serum IgG and fecal extract IgA as compared with wild-type mice. CCR5−/− and C57BL/6J wild-type mice were infected orally (Fig. 1A, C) or conjunctivally (Fig. 1B, D) with T. cruzi and challenged 8 weeks later via the same route. Four weeks later, serum (Fig. 1A, B) and fecal pellets (Fig. 1C, D) were collected from individual T. cruzi-immune (n=7–8/group) and naïve (n=3/group) mice, and T. cruzi-specific IgG (Fig. 1A, B) and IgA (Fig. 1C, D), respectively, studied via ELISA. No statistically significant differences were detected between T. cruzi-immune CCR5−/− and wild-type mice [Mann Whitney U-Test]. Immune = T. cruzi-immune mice; WT B6 = C57BL/6J mice
Next, the numbers of T. cruzi-specific IgG and IgA antibody secreting cells (ASC) were measured in the spleens of T. cruzi-immune CCR5−/− and wild-type mice 11–13 days after mucosal T. cruzi rechallenge via ELISPOT. There were no significant differences in the levels of T. cruzi-specific IgG ASC comparing the CCR5−/− and wild-type T. cruzi-immune mice after oral (Fig. 2A) or conjunctival (Fig. 2B) rechallenge. There also were no significant differences in the levels of T. cruzi-specific IgA ASC (data not shown). These results confirm the ELISA data showing no significant defect in the generation of T. cruzi-specific IgG or IgA responses in CCR5−/− compared with wild-type T. cruzi-immune mice.
Figure 2.

CCR5−/− mice have similar T. cruzi-specific IgG and IFN-γ ELISPOT responses as compared to wild-type mice in the spleen. CCR5−/− and wild-type C57BL/6 mice were orally or conjunctivally challenged several times with T. cruzi to generate T. cruzi-immune mice. Eleven- to thirteen-days post T. cruzi rechallenge, spleen cells from orally (Fig. 2A, C) and conjunctivally (Fig, 2B, D) challenged mice were isolated and evaluated for T. cruzi trans-sialidase (TS) specific IgG (Fig. 2A, B) and T. cruzi-specific IFN-γ (Fig. 2C, D) ELISPOT responses. 1×106 spleen cells (SC) were added per well to assess T. cruzi-specific IgG ASC responses (Fig. 2A, B). In order to detect T. cruzi-specific IFN-γ responses, SC (5×105) were pulsed with T. cruzi (Tc) lysate (10 μg/mL), trans-sialidase (TS; 10 μg/mL), or three individual H-2Kb CD8 epitopes (pep ASP2, VNHRFTLV; pep TSSA, ANYNFTLV; and pep 77.2, VDYNFTIV) at 2.5 μg/mL overnight at 37°C. No significant differences were detected between T. cruzi-immune CCR5−/− and wild-type mice [Mann-Whitney U Test]. TS = trans-sialidase, ASC = antibody secreting cell, SFC = spot-forming cell, SC = spleen cells. Immune = T. cruzi-immune mice. WT = C57BL/6 mice. n = 7–8 mice/group.
CCR5−/− mice generate equivalent levels of T. cruzi-specific IFN-γ-producing T cells
Next, we evaluated antigen-specific IFN-γ responses by T cells in CCR5−/− mice compared with wild-type mice in response to mucosal T. cruzi rechallenge. Previous work has shown that CCL5 can act through both G-protein coupled receptor (chemokine receptor) dependent and independent mechanisms to activate T cell (17). As CCL5 is a major ligand for CCR5, it was unclear whether there would be a difference in IFN-γ responses in CCR5−/− mice. Spleen cells (SC) harvested from T. cruzi-immune mice 11– 13 days after T. cruzi rechallenge were incubated with various peptides or proteins overnight to stimulate antigen-specific production of IFN-γ as measured by ELISPOT (Fig. 2C, D). IFN-γ spot-forming cells (SFC) were measured in response to three T. cruzi H-2Kb restricted CD8 T cell epitopes (ASP2, VNHRFTLV (47); TSSA, ANYNFTLV (44, 45); 77.2, VDYNFTIV (46)). All three peptides have been shown previously to be potent, if not immunodominant, epitopes in H-2b expressing mice infected with T. cruzi. Purified trans-sialidase (TS) protein and T. cruzi lysate were also used to stimulate antigen-specific IFN-γ responses. 11–13 days after mucosal T. cruzi rechallenge, there were no significant differences comparing the frequencies of antigen-specific IFN-γ responses in CCR5−/− and wild-type T. cruzi-immune mice that had been orally (Fig. 2C) or conjunctivally (Fig. 2D) rechallenged. In the conjunctivally challenged mice (Fig. 2D), there appeared to be a slight increase in the CD8+ T cell response against peptide TSSA in the CCR5−/− immune spleen cells. However, this difference was not statistically significant (p = 0.127; Mann-Whitney U Test). The most prominent antigen-specific IFN-γ responses were detected after stimulating spleen cells with T. cruzi lysate. In orally challenged mice (Fig. 2C), a slight decrease in the amount of T. cruzi-specific IFN-γ production in the CCR5−/−-immune mice was seen, but this was not statistically significant (p = 0.51; Mann-Whitney U Test). We also evaluated T. cruzi-specific IFN-γ responses during primary oral and conjunctival T. cruzi infection. There was no significant difference in the number of T. cruzi-specific IFN-γ spot-forming cells between the CCR5−/− and wild-type primary infected mice (p = 0.275 and 0.827 evaluating T. cruzi-lysate specific responses after oral and conjunctival infection respectively; Mann-Whitney U test; data not shown). Overall, these results demonstrate that CCR5−/− mice were able to generate similar levels of T. cruzi-specific IFN-γ responses as compared with wild-type mice.
CCR5−/− mice develop equivalent levels of mucosal T. cruzi protection
To evaluate mucosal protection at the initial point of invasion and within the draining lymph nodes, parasite replication was assessed after primary mucosal infection and rechallenge of T. cruzi-immune mice in both CCR5−/− and wild-type mice. One of the most sensitive methods to study parasite replication is through quantitative PCR (qPCR) analyses. The gastric mucosae from orally challenged mice and the nasal cavities from conjunctivally challenged mice were removed from primary infected and T. cruzi-immune CCR5−/− and wild-type mice 11–13 days after mucosal T. cruzi rechallenge. In both oral (Fig. 3A) and conjunctival (Fig. 3B) T. cruzi challenge models, there were no significant differences in parasite replication in the CCR5−/− mice compared with wild-type mice as detected by qPCR in either primary control or T. cruzi-immune mice (Fig. 3A, B). Both CCR5−/− and wild-type-immune mice were significantly more protected from mucosal T. cruzi rechallenge compared with matched primary control challenged mice (*p < 0.005; Mann-Whitney U Test). Next, we evaluated parasite outgrowth through the limiting dilution of cells from the spleens and draining lymph nodes (Fig. 3C–F). In the spleens of oral (Fig. 3C) or conjunctival (Fig. 3D) primary control infected mice, there were no significant differences in the levels of detectable parasites present in the CCR5−/− versus wild-type mice. We could not detect any parasites in 1×106 spleen cells from CCR5−/− and wild-type T. cruzi-immune mice (Fig. 3C, D). In the gastric lymph nodes of orally challenged mice (Fig. 3E) or the submandibular/parotid lymph nodes in conjunctivally challenged mice (Fig. 3F), there were no significant differences in the levels of detectable parasites in the primary control infected CCR5−/− versus wild-type control mice. In some of the T. cruzi-immune mice, we could detect parasites in the draining lymph nodes of both the orally and conjunctivally challenged mice, but the results were not significantly different between the CCR5−/− and wild-type mice (Fig. 3E, F). Overall, these results demonstrate that CCR5 is not required for the efficient development and recall of protective T. cruzi immunity at sites of mucosal T. cruzi challenge.
Figure 3.

CCR5 is not required for mucosal protection against T. cruzi. CCR5−/− and wild-type C57BL/6 mice were orally or conjunctivally challenged several times with T. cruzi to generate T. cruzi-immune mice. Eleven- to thirteen-days after T. cruzi rechallenge, mice were sacrificed, gastric DNA (Fig. 3A) and nasal cavity DNA (Fig. 3B) were isolated and the number of T. cruzi molecular equivalents were quantitated using a T. cruzi-specific qPCR assay. Spleen cells and draining lymph node cells were isolated and assessed for parasite outgrowth using a standard parasite limiting dilution assay (Fig. 3C-F). *p < 0.05, **p < 0.01, ***p < 0.005; [Mann-Whitney U Test]. Control = primary infected mice, Immune= T. cruzi-immune mice, SC = spleen cells, ME = T. cruzi molecular equivalents. ND = not detectable. n = 5–8/group.
CCL5 neutralization in CCR5−/− T. cruzi-immune mice results in decreased gastric inflammation and mucosal protection
We next evaluated the possibility that compensatory CCL5 signaling through other receptors in CCR5−/− mice could allow for the development of protective T. cruzi immunity. We examined chemokine gene expression levels in the gastric mucosa of T. cruzi-immune CCR5−/− and wild-type mice three days after oral T. cruzi rechallenge. There was a significant increase in the levels of CCL5 mRNA in the gastric mucosa of the CCR5−/− T. cruzi-immune mice compared with wild-type mice (Fig. 4A; *p <0.05; t test). These results implicated a possible CCL5-mediated compensatory mechanism that could allow for the development of equivalent levels of mucosal T. cruzi protection. Consistent with these results, CCL5 has been shown to play a role in the enhancement of both humoral and cellular mucosal immunity (28), and has also been shown to activate human macrophages to produce nitric oxide resulting in trypanocidal activity (36). To directly address the importance of CCL5 signaling in mucosal T. cruzi immunity, T. cruzi-immune CCR5−/− mice were treated with either an anti-CCL5 neutralizing monoclonal antibody (49) or IgG1κ isotype control antibody during oral T. cruzi rechallenge. Figure 4B demonstrates that there were significantly increased levels of T. cruzi DNA detectable in the gastric mucosa of CCR5−/− T. cruzi-immune mice treated with the neutralizing anti-CCL5 antibody 12 days after oral T. cruzi rechallenge (*p < 0.05; [Mann Whitney U test]). These results confirm that CCL5 signaling plays an important role in mucosal T. cruzi protection. Possible mechanisms for decreased mucosal protection include the lack of antigen-specific T cells and B cells migrating into the gastric mucosa and/or effects on innate immune cells, as CCL5 has been shown to induce trypanosomal activity in macrophages.
Figure 4.
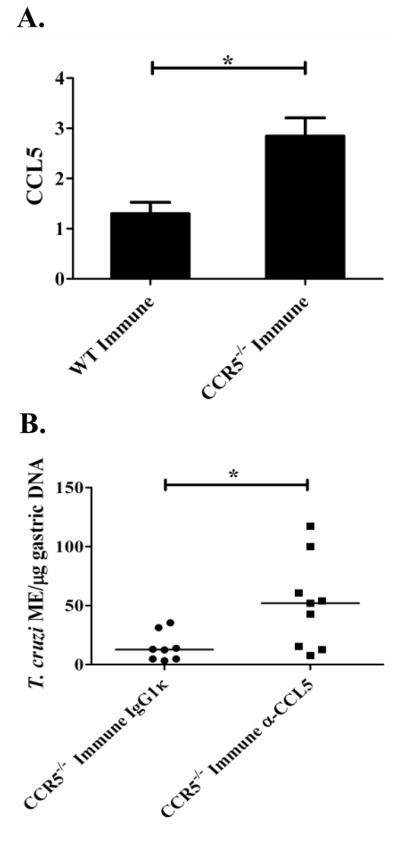
CCL5 neutralization in CCR5−/− T. cruzi-immune mice leads to decreased gastric mucosal T. cruzi protection. CCR5−/− and wild-type C57BL/6 mice were orally challenged several times with T. cruzi to generate T. cruzi-immune mice. Immune and naïve mice were then orally rechallenged with T. cruzi. In Figure 4A, T. cruzi-immune mice (n=3/group), along with naïve controls, were sacrificed on day 3 and gastric RNA isolated. Data represent fold changes (± standard error) in CCL5 gene expression compared with naïve CCR5−/− or wild-type mice (calculated using 2−ΔΔCt method with gapdh as a housekeeping gene). Negative controls without added reverse transcriptase were included to confirm the removal of gastric DNA (data not shown). *p < 0.05 [t test]. In Figure 4B, T. cruzi-immune CCR5−/− mice were treated with 250 μg of mouse neutralizing α-CCL5 or isotype control IgG1κ (Sigma) mAb I.P. every other day starting 4 days prior to oral T. cruzi challenge and continuing through day 10 after rechallenge. Total gastric DNA was isolated 12 days after rechallenge and assessed for the number of T. cruzi genomes (molecular equivalents; ME) using T. cruzi-specific real-time PCR. Lines represent median values. n=8–9/group. These results represent two independent experiments with the results pooled together. *p < 0.05 [Mann-Whitney U test].
Next, we wanted to determine if differences in T. cruzi-specific T or B cell responses could explain the reduced protection seen in CCR5−/−-immune mice treated with a neutralizing anti-CCL5 antibody. We did not detect any significant differences in T. cruzi-specific T cell responses in CCR5−/− immune mice treated with the anti-CCL5 antibody compared with mice treated with the IgG1κ isotype control antibody (data not shown). However, we did detect significant decreases in the levels of T. cruzi-specific IgG (Fig. 5A; *p < 0.05; Mann-Whitney U Test) and IgA (Fig. 5B; **p < 0.01; Mann-Whitney U Test) antibody secreting cells (ASC) detectable in the spleens from CCR5−/−-immune mice treated with the neutralizing anti-CCL5 antibody. We also saw a significant decrease in the levels of T. cruzi-specific IgA ASC (*p < 0.05; Mann-Whitney U Test) and a trend for a decrease in T. cruzi-specific IgG (p = 0.076; Mann-Whitney U test) ASC in wild-type immune mice treated with anti-CCL5 (data not shown). Despite these results, we did not detect significant differences in end-point titers of T. cruzi-specific IgG in the serum of the CCR5−/− mice treated with either the anti-CCL5 or isotype control (1:2.5×106 for each group). These apparently discrepant results are likely due to the fact that these mice had been challenged several times with T. cruzi prior to CCL5 neutralization and thus had already developed parasite-specific long-term plasma cells. The half-life of serum IgG has been reported to be between 1 to 4 weeks and thus would not be affected by a short-term treatment with this neutralizing antibody. However, we identified a 4.45 fold decrease in the level of T. cruzi-specific IgA in the fecal extracts of anti-CCL5 treated CCR5−/−-immune mice (1:8 dilution; * p <0.05 Mann-Whitney U test; Data not shown). The half-life of serum IgA has been reported to be markedly shorter (IgA t1/2 ~17–22 hours) than serum IgG (51) and is shed as well as proteolytically degraded over time in stool. These facts may explain why neutralization of CCL5 resulted in decreased levels of antigen-specific secretory IgA. Other than producing antibodies, B cells have been shown to play important roles as professional antigen presenting cells and cytokine producing cells able to skew immune responses (52, 53). Thus, a decrease in T. cruzi-specific antibody-secreting cells after CCL5 neutralization might specifically affect the ability to mount an effective immune response against T. cruzi through the inability of B cells to efficiently present antigen or produce cytokines. Thus, neutralization of CCL5 in CCR5−/− T. cruzi-immune mice results in decreased levels of T. cruzi-specific B cell responses, and decreased mucosal protection in these mice.
Figure 5.
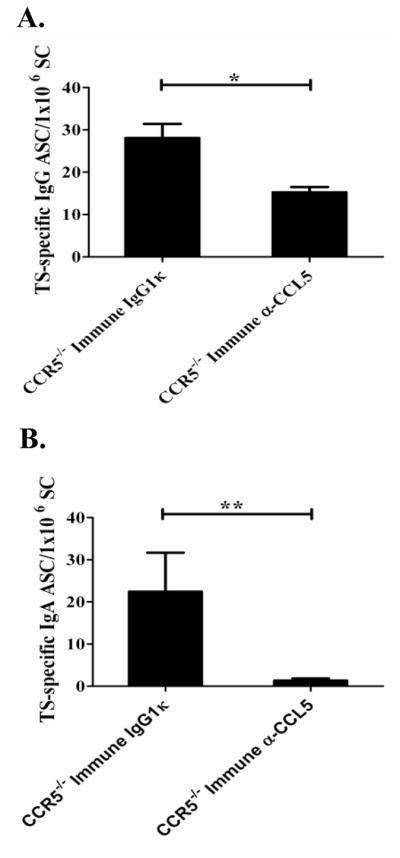
CCL5 neutralization in CCR5−/− T. cruzi-immune mice leads to decreased T. cruzi-specific IgG and IgA antibody secreting cells in the spleen. CCR5−/− mice were challenged multiple times orally with T. cruzi. Six weeks after the last challenge, mice were treated with 250 μg of neutralizing anti-CCL5 or isotype control IgG1κ mAb I.P. and rechallenged orally with T. cruzi as described in Figure 4. Twelve days after rechallenge, treated CCR5−/− mice (n=5/group), as well as naïve, age-matched CCR5−/− control mice (n=3), were sacrificed to assess T. cruzi-specific IgG (Fig. 6A) and IgA (Fig. 6B) antibody secreting cell (ASC) responses in the spleen. *p < 0.05, **p < 0.01 [Mann-Whitney U test]. These data represent 1 of 2 independent experiments with similar results. Immune = T. cruzi-immune mice; IgG1κ = IgG1κ isotype control antibody treated; α-CCL5 = anti-CCL5 neutralizing antibody treated. n=5/group.
We further evaluated the gene expression of several chemokines and other various genes in the gastric mucosa of these CCR5−/−-immune mice 12 days after oral T. cruzi rechallenge. We detected marked decreases in the levels of several inflammatory chemokines (CCL4, CCL5, CXCL9, CXCL10, CXCL11; Figure 6) in the gastric mucosa of CCR5−/−-immune mice treated with neutralizing anti-CCL5 antibody. There was also a decrease in the level of IgA mRNA in the gastric mucosa, consistent with the ELISPOT (Fig. 5B) and ELISA data showing decreased antigen-specific IgA. Overall, these results demonstrate that treatment of CCR5−/− T. cruzi-immune mice with a neutralizing anti-CCL5 antibody resulted in decreased T. cruzi-specific IgG and IgA ASC, decreased gastric inflammatory chemokine responses and decreased mucosal T. cruzi protection. These combined results indicate that CCL5 is likely to be important for both innate and adaptive immune responses required for control of T. cruzi mucosal infection.
Figure 6.
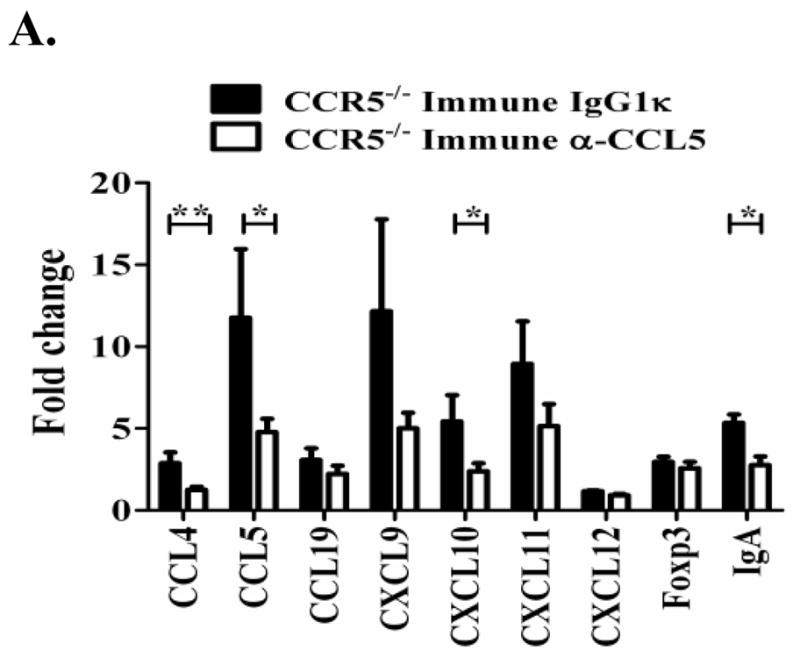
CCL5 neutralization in CCR5−/− T. cruzi-immune mice leads to decreased gastric inflammatory chemokines in the gastric mucosa. CCR5−/− mice were orally challenged multiple times with T. cruzi. Six weeks after the last challenge mice were treated with 250 μg neutralizing anti-CCL5 or isotype control IgG1κ mAb I.P. and rechallenged orally with T. cruzi as described in figure 4. Total gastric RNA was isolated 12 days post-challenge. After genomic DNA removal and cleanup, qRT-PCR was used to measure chemokine gene expression. Data represents fold changes (± standard error) in gene expression as compared to age-matched naïve CCR5−/− mice (n=3) (calculated using 2−ΔΔCt method with gapdh as a housekeeping gene). Negative controls without added reverse transcriptase were included to confirm the removal of gastric DNA. Immune = T. cruzi-immune, IgG1κ = IgG1κ isotype-control antibody treated. α-CCL5 = anti-CCL5 neutralizing antibody treated. *p < 0.05, **p < 0.01 [Mann-Whitney U Test]. n=5/group.
The direct effect of CCL5 on B cells
Neutralization of CCL5 in CCR5−/− and wild-type T. cruzi-immune mice resulted in decreased levels of T. cruzi-specific IgA and IgG antibody secreting cells (Figure 5 and data not shown). These results are consistent with previous results (28), indicating a role of CCL5 as a possible B cell adjuvant. To date, this adjuvant activity has been attributed to the effects of CCL5 on T cells as CCL5 has been shown to activate T cells (17). CCL5 upregulates CD28, CD30 and CD40L on DO11.10 CD4+ T cells after ovalbumin peptide stimulation (28), demonstrating that CCL5 can directly affect T cells. However, very little has been done to evaluate a potential direct effect of CCL5 on B cells. In our studies evaluating the effect of CCL5 neutralization in CCR5−/− mice, we did not observe any significant difference in effector T cells (data not shown). Thus, we sought to evaluate whether CCL5 could directly activate B cells. Addition of CCL5 to suboptimal concentrations of LPS increased B cell proliferation (Fig. 7A, 7B) and IgM secretion (Fig. 7C) in highly purified B cells. B cells themselves have been shown to produce CCL5 (54) and in Figure 7D, we show that 10 μg/mL of LPS can induce detectable levels of CCL5 in culture supernatants. Neutralization of CCL5 in B cells stimulated with a high concentration of LPS inhibited B cell proliferation (Fig. 7E) and total IgM secretion (Fig. 7F), suggesting that B cell production of CCL5 acts in an autocrine manner to increase B cell proliferation and total IgM secretion. CCL5 stimulation alone did not result in any detectable changes in B cell activation (Fig. 7) suggesting that CCL5 may be important as a general B cell co-activator. We have also seen similar results using purified B cells from CCR5−/− mice (data not shown). These data suggest that CCL5-mediated enhancement of B cell activation does not depend on direct CCL5-CCR5 signaling. CCL5 could signal through CCR1 or CCR3 in the absence of CCR5. In any case, these findings demonstrate a direct effect of CCL5 on B cells and have implications for the use of CCL5 as a mucosal adjuvant to directly enhance B cell responses.
Figure 7.

The direct effects of CCL5 on B cells. Highly purified B cells (> 98% CD19+) were isolated from naïve C57BL/6 mice and cultured with or without LPS (0.5 μg/mL) ± recombinant mouse CCL5 (400 pg/mL) and proliferative responses were measured via [3H]-thymidine incorporation (Fig. 7A) or CFSE dilution (Fig. 7B). Culture supernatants were taken 7 days after stimulation and assessed for total IgM secretion via ELISA (Fig. 7C). Culture supernatants were taken at days 2, 3, 5 and 7 after stimulation with or without LPS (10 μg/mL) or anti-CD3/CD28, and CCL5 protein production was measured via ELISA (Fig. 7D). In a separate experiment, highly purified B cells were stimulated with or without LPS (10 μg/mL) ± anti-CCL5 mAb or IgG1κ isotype control antibody and proliferative responses measured via [3H]-thymidine incorporation on days 2 and 3 post stimulation (Fig. 7E). Culture supernatants were taken 7 days after stimulation, total IgM secretion was measured via ELISA and the percent suppression was calculated ((LPSalone-LPSantibody treated)/LPSalone) x 100 = % suppression (Fig. 7F). Data represent two to three independent experiments with multiple triplicates of pooled samples with similar results obtained in each experiment.
Discussion
The role of CCR5 during T. cruzi infection has been studied in both humans and mice. Initially it was identified that T. cruzi sero-positive patients with a CCR5 59029 A→G mutation, resulting in decreased cell surface expression of CCR5, had less cardiac symptoms compared with cardiomyopathic patients (38, 39). Studies in mice have revealed increased levels of the CCR5 ligands, CCL3, CCL4 and CCL5, in the hearts of T. cruzi infected mice (31–34). Treatment of mice during the acute phase of T. cruzi infection with N-terminal-methionylated RANTES (Met-RANTES), a selective CCR1 and CCR5 antagonist, resulted in decreased levels of CD4+ and CD8+ T cells migrating into the heart, decreased fibronectin deposition and increased survival (32). Furthermore, CCR5−/− mice have been shown to be more susceptible to T. cruzi infection compared with wild-type mice (31, 33). Taken together, these studies have demonstrated that CCR5 plays a role in systemic protection and cardiac inflammation during T. cruzi infection.
In the present report, we evaluated whether CCR5 and CCL5 played a role in mucosal protection. CCR5 and CCL5 mRNA levels were upregulated at the initial site of T. cruzi infection and replication in T. cruzi immune mice several days after oral T. cruzi rechallenge. Our initial studies indicated that CCR5−/− mice had similar levels of T. cruzi replication in the nasal cavities after primary conjunctival infection as compared with wild-type mice (data not shown). We then wanted to evaluate whether CCR5−/− mice were able to develop protective memory immunity against T. cruzi. CCR5−/− and wild- type T. cruzi-immune mice had similar T. cruzi-specific IgG, IgA and IFN-γ responses and were similarly protected against T. cruzi mucosal infection. These results suggested that CCR5 does not play a nonredundant critical role in mucosal T. cruzi protection. We then wanted to evaluate possible compensatory mechanisms that allowed the CCR5−/−mice to develop equivalent levels of protective mucosal immunity against T. cruzi. There was an increase in CCL5 mRNA in the gastric mucosa of CCR5−/− T. cruzi-immune mice three days after oral T. cruzi rechallenge as compared with wild-type controls. We then hypothesized that increased levels of CCL5 may 1) increase the recruitment and/or activation of memory T cells and B cells into the gastric mucosa, or 2) increase killing of T. cruzi through the direct activation of macrophages. Neutralization of CCL5 in these CCR5−/− T. cruzi-immune mice did not affect antigen-specific IFN-γ production by T cells, but resulted in decreased mucosal inflammatory responses, T. cruzi-specific IgG and IgA secreting cells and mucosal T. cruzi protection. To further evaluate possible mechanisms for this decreased mucosal protection, we studied the direct effects of CCL5 on B cells. Proliferative and IgM responses triggered by suboptimal doses of LPS in highly purified B cells were enhanced by CCL5. In addition, neutralization of CCL5 in purified B cell cultures stimulated with LPS inhibited their proliferative responses. Thus, neutralization of CCL5 in CCR5−/− T. cruzi-immune mice could inhibit B cell activation (both naïve and memory B cells), decreased induction of antigen-specific plasma cells, leading to decreased mucosal T. cruzi protection.
Our data indicate that CCR5 does not play a nonredundant critical role in mucosal T. cruzi protection. One possible explanation for this finding is that CCR5 may not be involved in trafficking of lymphocytes to mucosal sites and thus CCR5 would not be important for mucosal immunity. However, many investigators have reported the presence of CCR5+ T cells and CCR5 ligands in mucosal tissues (55–59). In one study evaluating long-term non-progression of SIV infection in Chinese rhesus macaques, early restoration of CD4+CCR5+ T cells into the gut correlated with decreased viral load and a positive clinical outcome (60). In children infected with H. pylori, there were high levels of CCR5+ cells detected in the gastric lamina propria, suggesting that CCR5 played a role in gastric immune responses (56). Increased levels of CCR5 ligands have been reported in the gastric mucosa during H. pylori infection (57–59). Overall, these studies have demonstrated that both CCR5 and CCL5 play important roles during inflammatory reactions in mucosal tissues during infection.
There is a large amount of redundancy in the chemokine and chemokine receptor system. It has been demonstrated that multiple chemokines can bind to and signal through multiple chemokine receptors. This redundancy may allow for fine control over immune responses due not only to interactions between chemokines and chemokine receptors on leukocytes, but also the effects on adhesion molecule interactions, glycosaminoglycans and different oligomerization states of ligands and their receptors (3). CCL3, CCL4 and CCL5 have been shown to signal through CCR5. In addition, CCL3 can signal through CCR1 and CCR3. Furthermore, CCL5 can signal through CCR1, CCR3 and CCR5 (61). Even with this large amount of redundancy, CCR5 has been shown to be important in systemic protection against T. cruzi (31–33). Since it did not appear that CCR5 played a nonredundant critical role in mucosal protection against T. cruzi, one could hypothesize that CCL5 signaling through CCR1 and/or CCR3 may compensate for the absence of CCR5. However, deletion of both components of the CCR5-CCL5 ligand axis does appear to have important effects on mucosal T. cruzi protection. We did not study the role of CCL3 or CCL4 in this study as CCL5 mRNA levels were much higher compared to CCL3 and CCL4 in the gastric mucosa of T. cruzi-immune mice (Table I). However, CCL3 and CCL4 may also play a role in mucosal protection against T. cruzi. In normal mice, CCR5 may play a role in mucosal protection against T. cruzi. However, the large amount of redundancy may compensate for the lack of CCR5 in the CCR5−/− mice.
IFN-γ, IL-12 and β2-microglobulin have been shown to play critical roles in protection against T. cruzi as mice deficient in these molecules fail to develop mucosal and systemic immunity (62). We have previously shown the importance of Th1 T cells in protection against T. cruzi. Mice immunized with recombinant IL-12 plus an anti-IL-4 antibody to bias for Th1 responses in vivo had decreased blood parasitemia levels and 100% survival after a normally lethal T. cruzi systemic challenge. Adoptive transfer studies showed that CD4+ T cells are necessary, but not sufficient, for the development of immunity protective against T. cruzi challenge. However, these CD4+ T cells were not required for memory immune effector functions protective against T. cruzi rechallenge (63). Others have shown that CD8+ T cells from mice immunized with T. cruzi antigens can transfer systemic protection to naïve mice (64). Collectively, these results demonstrate that both type 1-skewed CD4+ and CD8+ T cells are necessary for the development of protective immunity, but that CD8+ T cells alone are sufficient for protective T. cruzi effector functions.
B lymphocyte responses specific for T. cruzi have also been shown to be important for protection. T. cruzi-specific serum IgG antibodies have been shown to play a role in complement activation and lysis (65–68), opsonization (69), and antibody-dependent cell cytotoxicity (ADCC) (70). T. cruzi mucosal infection induces protection against rechallenge, associated with T. cruzi-specific IgA (48). Fecal extracts containing T. cruzi-specific secretory IgA from T. cruzi-immune mice provide protective opsonization activity both in vitro and in vivo against T. cruzi cellular invasion (42). In addition to the effects of antibodies secreted by B cells, the antigen presentation function of B cells has been shown to be important for T. cruzi protective immunity (43). μMT−/− mice which lack mature B cells developed lower numbers of CD4+ and CD8+ central and effector memory T cells, as well as increased susceptibility after systemic T. cruzi infection (71, 72). These studies have demonstrated that B cells play an important role in systemic protection against T. cruzi through participating in the generation of central and effector memory T cells and antibodies. Overall, CD4+ T cells, CD8+ T cells and B cells play critical roles in the establishment of T. cruzi protective immunity.
Previously, CCL5 has been shown to enhance humoral and cellular immune responses after protein immunization as well as mucosal bacterial infection (28–30). High concentrations of CCL5 have been shown to induce T cell activation through both G-protein coupled receptor (GPCR)-dependent and – independent mechanisms (17, 73). Intranasal OVA plus CCL5 immunization resulted in increased levels of OVA-specific antibodies, CD4+ T cell proliferation and IL-2, IFN-γ, IL-5 and IL-6 cytokine production (28). The authors of this later report hypothesized that CCL5 could augment immune responses by enhancing antigen presentation or by activating host immune cells. Neutralization of CCL5 in mice mucosally challenged with Streptococcus pneumoniae resulted in decreased numbers of leukocytes in multiple tissues, decreased antigen- specific IgG and IgA responses, and decreased IFN-γ, TNF-α, IL-6, IL-4 production as well as CD4+ T cell proliferation in spleen cells (29). Interestingly, there was an increased level of IL-10 production in CD4+ T cells from the spleen, cervical lymph nodes and NALT harvested from anti-CCL5 treated mice after antigenic restimulation, suggesting that neutralization of CCL5 may result in conditions conducive for the generation of Th2 cells or IL-10+ regulatory T cells (Tr1) (29). Neutralization of CCL5 during mucosal Chlamydia muridarum infection resulted in increased vaginal shedding of the bacterium up to 42 days after challenge, indicating that CCL5 is important in chlamydial immunity (30). Others have studied the use of chemokines as adjuvants to modulate immune responses during DNA vaccination. CCL5 administration during DNA vaccination has been shown to polarize CD4+ T cells to become Th1 cells (74) and induce potent CD8+ CTL responses (75). Our data reinforces much of these previous infectious studies in that neutralization of CCL5 in T. cruzi-immune CCR5−/− mice resulted in decreased parasite-specific IgA and IgG antibody-secreting cells in the spleen, decreased secretory IgA responses and decreased mucosal T. cruzi protection. We did not detect any significant defect in the frequency of IFN-γ producing antigen-specific T cells after CCL5 neutralization (data not shown). However, we evaluated antigen-specific IFN-γ responses 12 days after oral T. cruzi rechallenge, as this is the optimal time point to evaluate T. cruzi mucosal replication. We cannot rule out a delayed onset of IFN-γ responses in the CCL5 neutralized mice, associated with reduced mucosal T. cruzi protection at earlier time points after rechallenge, and this should be investigated in future studies. Our data indicate that increased levels of CCL5 in the gastric mucosa plays a role in mucosal protection by the activation and/or recruitment of antigen-specific B cells, and perhaps also T cells and innate immune cells such as macrophages.
We have shown that CCL5 can directly affect B cells increasing B cell proliferation and total IgM secretion (Fig. 7). As discussed above, these effects could be useful for mucosal adjuvant effects. However, these results also suggest that CCL5 may be important as a general B cell co-activator with implications in several different fields of medicine. CCL5 has been shown to be upregulated in several autoimmune diseases, such as lupus nephritis and Rheumatoid Arthritis (76, 77). Increased levels of CCL5 in the serum and urine of lupus patients have been shown to correlate with disease severity (78, 79). Since B cells play an important role in these diseases and B cells have been shown to produce CCL5 (54), autocrine effects of CCL5 on B cells in these patients may result in increased B cell activation and the formation of autoimmune antibodies. Thus, CCL5 may be important as a general B cell co-activator that could be the target for novel treatment strategies for autoimmune and other inflammatory diseases.
In conclusion, we have shown that CCR5 does not play a nonredundant critical role in mucosal protection against T. cruzi. CCR5−/− T. cruzi-immune mice developed similar levels of T. cruzi-specific antibody, IFN-γ, and protection compared with wild-type mice. However, neutralization of CCL5 in CCR5−/− T. cruzi-immune mice resulted in decreased T. cruzi-specific antibody responses, decreased inflammatory chemokine gene expression in gastric mucosa, and decreased overall mucosal T. cruzi protection. We also demonstrate direct effects of CCL5 on B cells, suggesting a role for CCL5 as a B cell co-activator. Overall, these results indicate that CCL5 is necessary for optimal control of parasite replication in the gastric mucosa via either 1) the recruitment and/or activation of memory T cells and B cells likely by signaling through a redundant set of receptors (i.e. CCR1, CCR3 and/or CCR5), or 2) direct activation of innate immune cells allowing for parasite control in the initial site of infection.
Acknowledgments
We thank Richard DiPaolo for helpful discussions and Mark Ebel for technical assistance in performing the in vitro chemotaxis assays.
Abbreviations used in this paper
- T. cruzi
Trypanosoma cruzi
- IMT
T. cruzi insect-derived metacyclic tryptomastigote
- CMT
T. cruzi cultured-derived metacyclic tryptomastigote
- MT
metacyclic tryptomastigote
- TS
T. cruzi trans-sialidase protein antigen
- TSSA
tryptomastigote small surface antigen
- ASP2
amastigote surface protein 2
- LPS
lipopolysaccaride
- WT
wild type
- ASC
antibody secreting cells
- SFC
spot forming cells
- sIgA
secretory IgA
- LDA
limiting dilution assay
- qPCR
quantitative PCR
- TcME
T. cruzi molecular equivalents
- SC
spleen cells
- LN
lymph node
Footnotes
This work was supported by a grant from the National Institutes of Health (R01 AI040196 to D.F.H.).
References
- 1.Centers for Disease, C., and Prevention. Chagas Disease; Epidemiology & Risk Factors. 2007 http://www.cdc.gov/chagas/epi.html.
- 2.Bern C, Montgomery SP. An estimate of the burden of Chagas disease in the United States. Clinical Infectious Diseases. 2009;49:e52–e54. doi: 10.1086/605091. [DOI] [PubMed] [Google Scholar]
- 3.Allen SJ, Crown SE, Handel TM. Chemokine: receptor structure, interactions, and antagonism. Annual Review of Immunology. 2007;25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- 4.Rottman JB, Ganley KP, Williams K, Wu L, Mackay CR, Ringler DJ. Cellular localization of the chemokine receptor CCR5. Correlation to cellular targets of HIV-1 infection. American Journal of Pathology. 1997;151:1341–1351. [PMC free article] [PubMed] [Google Scholar]
- 5.Honczarenko M, Le Y, Glodek AM, Majka M, Campbell JJ, Ratajczak MZ, Silberstein LE. CCR5-binding chemokines modulate CXCL12 (SDF-1)-induced responses of progenitor B cells in human bone marrow through heterologous desensitization of the CXCR4 chemokine receptor. Blood. 2002;100:2321–2329. doi: 10.1182/blood-2002-01-0248. [DOI] [PubMed] [Google Scholar]
- 6.Yurchenko E, Tritt M, Hay V, Shevach EM, Belkaid Y, Piccirillo CA. CCR5-dependent homing of naturally occurring CD4+ regulatory T cells to sites of Leishmania major infection favors pathogen persistence. Journal of Experimental Medicine. 2006;203:2451–2460. doi: 10.1084/jem.20060956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaufmann A, Salentin R, Gemsa D, Sprenger H. Increase of CCR1 and CCR5 expression and enhanced functional response to MIP-1 alpha during differentiation of human monocytes to macrophages. Journal of Leukocyte Biology. 2001;69:248–252. [PubMed] [Google Scholar]
- 8.Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachy J, Bruhl H, Frink M, Anders HJ, Vielhauer V, Pfirstinger J, Stangassinger M, Schlondorff D. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. Journal of Immunology. 2001;166:4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- 9.Edinger AL, Mankowski JL, Doranz BJ, Margulies BJ, Lee B, Rucker J, Sharron M, Hoffman TL, Berson JF, Zink MC, Hirsch VM, Clements JE, Doms RW. CD4-independent, CCR5-dependent infection of brain capillary endothelial cells by a neurovirulent simian immunodeficiency virus strain. Proceedings of the National Academy of Sciences. 1997;94:14742–14747. doi: 10.1073/pnas.94.26.14742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwasaki M, Mukai T, Nakajima C, Yang YF, Gao P, Yamaguchi N, Tomura M, Fujiwara H, Hamaoka T. A mandatory role for STAT4 in IL-12 induction of mouse T cell CCR5. Journal of Immunology. 2001;167:6877–6883. doi: 10.4049/jimmunol.167.12.6877. [DOI] [PubMed] [Google Scholar]
- 11.Juffermans NP, Paxton WA, Dekkers PE, Verbon A, de Jonge E, Speelman P, van Deventer SJ, van der PT. Up-regulation of HIV coreceptors CXCR4 and CCR5 on CD4(+) T cells during human endotoxemia and after stimulation with (myco)bacterial antigens: the role of cytokines. Blood. 2000;96:2649–2654. [PubMed] [Google Scholar]
- 12.Schall TJ, Bacon K, Camp RD, Kaspari JW, Goeddel DV. Human macrophage inflammatory protein alpha (MIP-1 alpha) and MIP-1 beta chemokines attract distinct populations of lymphocytes. Journal of Experimental Medicine. 1993;177:1821–1826. doi: 10.1084/jem.177.6.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taub DD, Conlon K, Lloyd AR, Oppenheim JJ, Kelvin DJ. Preferential migration of activated CD4+ and CD8+ T cells in response to MIP-1 alpha and MIP-1 beta. Science. 1993;260:355–358. doi: 10.1126/science.7682337. [DOI] [PubMed] [Google Scholar]
- 14.Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. 2006;440:890–895. doi: 10.1038/nature04651. [DOI] [PubMed] [Google Scholar]
- 15.Appay V, Rowland-Jones SL. RANTES: a versatile and controversial chemokine. Trends in Immunology. 2001;22:83–87. doi: 10.1016/s1471-4906(00)01812-3. [DOI] [PubMed] [Google Scholar]
- 16.Levy JA. The unexpected pleiotropic activities of RANTES. Journal of Immunology. 2009;182:3945–3946. doi: 10.4049/jimmunol.0990015. [DOI] [PubMed] [Google Scholar]
- 17.Bacon KB, Premack BA, Gardner P, Schall TJ. Activation of dual T cell signaling pathways by the chemokine RANTES. Science. 1995;269:1727–1730. doi: 10.1126/science.7569902. [DOI] [PubMed] [Google Scholar]
- 18.Taub DD, Turcovski-Corrales SM, Key ML, Longo DL, Murphy WJ. Chemokines and T lymphocyte activation: I. Beta chemokines costimulate human T lymphocyte activation in vitro. Journal of Immunology. 1996;156:2095–2103. [PubMed] [Google Scholar]
- 19.Taub DD, Ortaldo JR, Turcovski-Corrales SM, Key ML, Longo DL, Murphy WJ. Beta chemokines costimulate lymphocyte cytolysis, proliferation, and lymphokine production. Journal of Leukocyte Biology. 1996;59:81–89. doi: 10.1002/jlb.59.1.81. [DOI] [PubMed] [Google Scholar]
- 20.Camargo JF, Quinones MP, Mummidi S, Srinivas S, Gaitan AA, Begum K, Jimenez F, VanCompernolle S, Unutmaz D, Ahuja SS, Ahuja SK. CCR5 expression levels influence NFAT translocation, IL-2 production, and subsequent signaling events during T lymphocyte activation. Journal of Immunology. 2009;182:171–182. doi: 10.4049/jimmunol.182.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luther SA, Cyster JG. Chemokines as regulators of T cell differentiation. Nature Immunology. 2001;2:102–107. doi: 10.1038/84205. [DOI] [PubMed] [Google Scholar]
- 22.Olszewski MA, Huffnagle GB, McDonald RA, Lindell DM, Moore BB, Cook DN, Toews GB. The role of macrophage inflammatory protein-1 alpha/CCL3 in regulation of T cell-mediated immunity to Cryptococcus neoformans infection. Journal of Immunology. 2000;165:6429–6436. doi: 10.4049/jimmunol.165.11.6429. [DOI] [PubMed] [Google Scholar]
- 23.Karpus WJ, Lukacs NW, Kennedy KJ, Smith WS, Hurst SD, Barrett TA. Differential CC chemokine-induced enhancement of T helper cell cytokine production. Journal of Immunology. 1997;158:4129–4136. [PubMed] [Google Scholar]
- 24.Lillard JW, Jr, Singh UP, Boyaka PN, Singh S, Taub DD, McGhee JR. MIP-1alpha and MIP-1beta differentially mediate mucosal and systemic adaptive immunity. Blood. 2003;101:807–814. doi: 10.1182/blood-2002-07-2305. [DOI] [PubMed] [Google Scholar]
- 25.Zou W, Borvak J, Marches F, Wei S, Galanaud P, Emilie D, Curiel TJ. Macrophage-derived dendritic cells have strong Th1-polarizing potential mediated by beta-chemokines rather than IL-12. Journal of Immunology. 2000;165:4388–4396. doi: 10.4049/jimmunol.165.8.4388. [DOI] [PubMed] [Google Scholar]
- 26.Pinto LA, Williams MS, Dolan MJ, Henkart PA, Shearer GM. Beta-chemokines inhibit activation-induced death of lymphocytes from HIV-infected individuals. European Journal of Immunology. 2000;30:2048–2055. doi: 10.1002/1521-4141(200007)30:7<2048::AID-IMMU2048>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 27.Lloyd AR, Oppenheim JJ, Kelvin DJ, Taub DD. Chemokines regulate T cell adherence to recombinant adhesion molecules and extracellular matrix proteins. Journal of Immunology. 1996;156:932–938. [PubMed] [Google Scholar]
- 28.Lillard JW, Jr, Boyaka PN, Taub DD, McGhee JR. RANTES potentiates antigen-specific mucosal immune responses. Journal of Immunology. 2001;166:162–169. doi: 10.4049/jimmunol.166.1.162. [DOI] [PubMed] [Google Scholar]
- 29.Palaniappan R, Singh S, Singh UP, Singh R, Ades EW, Briles DE, Hollingshead SK, Royal W, III, Sampson JS, Stiles JK, Taub DD, Lillard JW., Jr CCL5 modulates pneumococcal immunity and carriage. Journal of Immunology. 2006;176:2346–2356. doi: 10.4049/jimmunol.176.4.2346. [DOI] [PubMed] [Google Scholar]
- 30.Sakthivel SK, Singh UP, Singh S, Taub DD, Igietseme JU, Lillard JW., Jr CCL5 regulation of mucosal chlamydial immunity and infection. BMC Microbiology. 2008;8:136. doi: 10.1186/1471-2180-8-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Machado FS, Koyama NS, Carregaro V, Ferreira BR, Milanezi CM, Teixeira MM, Rossi MA, Silva JS. CCR5 plays a critical role in the development of myocarditis and host protection in mice infected with Trypanosoma cruzi. Journal of Infectious Diseases. 2005;191:627–636. doi: 10.1086/427515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marino AP, da SA, dos SP, Pinto LM, Gazzinelli RT, Teixeira MM, Lannes-Vieira J. Regulated on activation, normal T cell expressed and secreted (RANTES) antagonist (Met-RANTES) controls the early phase of Trypanosoma cruzi-elicited myocarditis. Circulation. 2004;110:1443–1449. doi: 10.1161/01.CIR.0000141561.15939.EC. [DOI] [PubMed] [Google Scholar]
- 33.Hardison JL, Wrightsman RA, Carpenter PM, Kuziel WA, Lane TE, Manning JE. The CC chemokine receptor 5 is important in control of parasite replication and acute cardiac inflammation following infection with Trypanosoma cruzi. Infection and Immunity. 2006;74:135–143. doi: 10.1128/IAI.74.1.135-143.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Medeiros GA, Silverio JC, Marino AP, Roffe E, Vieira V, Kroll-Palhares K, Carvalho CE, Silva AA, Teixeira MM, Lannes-Vieira J. Treatment of chronically Trypanosoma cruzi-infected mice with a CCR1/CCR5 antagonist (Met-RANTES) results in amelioration of cardiac tissue damage. Microbes and Infection. 2009;11:264–273. doi: 10.1016/j.micinf.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 35.Aliberti JC, Machado FS, Souto JT, Campanelli AP, Teixeira MM, Gazzinelli RT, Silva JS. beta-Chemokines enhance parasite uptake and promote nitric oxide-dependent microbiostatic activity in murine inflammatory macrophages infected with Trypanosoma cruzi. Infection and Immunity. 1999;67:4819–4826. doi: 10.1128/iai.67.9.4819-4826.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villalta F, Zhang Y, Bibb KE, Kappes JC, Lima MF. The cysteine-cysteine family of chemokines RANTES, MIP-1alpha, and MIP-1beta induce trypanocidal activity in human macrophages via nitric oxide. Infection and Immunity. 1998;66:4690–4695. doi: 10.1128/iai.66.10.4690-4695.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lima MF, Zhang Y, Villalta F. Beta-chemokines that inhibit HIV-1 infection of human macrophages stimulate uptake and promote destruction of Trypanosoma cruzi by human macrophages. Cell Mol Biol. 1997;43:1067–1076. [PubMed] [Google Scholar]
- 38.Calzada JE, Nieto A, Beraun Y, Martin J. Chemokine receptor CCR5 polymorphisms and Chagas’ disease cardiomyopathy. Tissue Antigens. 2001;58:154–158. doi: 10.1034/j.1399-0039.2001.580302.x. [DOI] [PubMed] [Google Scholar]
- 39.Fernandez-Mestre MT, Montagnani S, Layrisse Z. Is the CCR5–59029-G/G genotype a protective factor for cardiomyopathy in Chagas disease? Human Immunology. 2004;65:725–728. doi: 10.1016/j.humimm.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 40.Kuziel WA, Dawson TC, Quinones M, Garavito E, Chenaux G, Ahuja SS, Reddick RL, Maeda N. CCR5 deficiency is not protective in the early stages of atherogenesis in apoE knockout mice. Atherosclerosis. 2003;167:25–32. doi: 10.1016/s0021-9150(02)00382-9. [DOI] [PubMed] [Google Scholar]
- 41.Hoft DF, Eickhoff CS. Type 1 immunity provides optimal protection against both mucosal and systemic Trypanosoma cruzi challenges. Infection and Immunity. 2002;70:6715–6725. doi: 10.1128/IAI.70.12.6715-6725.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giddings OK, Eickhoff CS, Smith TJ, Bryant LA, Hoft DF. Anatomical route of invasion and protective mucosal immunity in Trypanosoma cruzi conjunctival infection. Infection and Immunity. 2006;74:5549–5560. doi: 10.1128/IAI.00319-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoft DF, Eickhoff CS, Giddings OK, Vasconcelos JR, Rodrigues MM. Trans-sialidase recombinant protein mixed with CpG motif-containing oligodeoxynucleotide induces protective mucosal and systemic Trypanosoma cruzi immunity involving CD8+ CTL and B cell-mediated cross-priming. Journal of Immunology. 2007;179:6889–6900. doi: 10.4049/jimmunol.179.10.6889. [DOI] [PubMed] [Google Scholar]
- 44.Katae M, Miyahira Y, Takeda K, Matsuda H, Yagita H, Okumura K, Takeuchi T, Kamiyama T, Ohwada A, Fukuchi Y, Aoki T. Coadministration of an interleukin-12 gene and a Trypanosoma cruzi gene improves vaccine efficacy. Infection and Immunity. 2002;70:4833–4840. doi: 10.1128/IAI.70.9.4833-4840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyahira Y, Takashima Y, Kobayashi S, Matsumoto Y, Takeuchi T, Ohyanagi-Hara M, Yoshida A, Ohwada A, Akiba H, Yagita H, Okumura K, Ogawa H. Immune responses against a single CD8+-T-cell epitope induced by virus vector vaccination can successfully control Trypanosoma cruzi infection. Infection and Immunity. 2005;73:7356–7365. doi: 10.1128/IAI.73.11.7356-7365.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wizel B, Nunes M, Tarleton RL. Identification of Trypanosoma cruzi trans-sialidase family members as targets of protective CD8+ TC1 responses. Journal of Immunology. 1997;159:6120–6130. [PubMed] [Google Scholar]
- 47.Low HP, Santos MA, Wizel B, Tarleton RL. Amastigote surface proteins of Trypanosoma cruzi are targets for CD8+ CTL. Journal of Immunology. 1998;160:1817–1823. [PubMed] [Google Scholar]
- 48.Hoft DF, Farrar PL, Kratz-Owens K, Shaffer D. Gastric invasion by Trypanosoma cruzi and induction of protective mucosal immune responses. Infection and Immunity. 1996;64:3800–3810. doi: 10.1128/iai.64.9.3800-3810.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glass WG, Hickey MJ, Hardison JL, Liu MT, Manning JE, Lane TE. Antibody targeting of the CC chemokine ligand 5 results in diminished leukocyte infiltration into the central nervous system and reduced neurologic disease in a viral model of multiple sclerosis. Journal of Immunology. 2004;172:4018–4025. doi: 10.4049/jimmunol.172.7.4018. [DOI] [PubMed] [Google Scholar]
- 50.Schnapp AR, Eickhoff CS, Scharfstein J, Hoft DF. Induction of B- and T-cell responses to cruzipain in the murine model of Trypanosoma cruzi infection. Microbes and Infection. 2002;4:805–813. doi: 10.1016/s1286-4579(02)01600-3. [DOI] [PubMed] [Google Scholar]
- 51.Vieira P, Rajewsky K. The half-lives of serum immunoglobulins in adult mice. European Journal of Immunology. 1988;18:313–316. doi: 10.1002/eji.1830180221. [DOI] [PubMed] [Google Scholar]
- 52.Hoglund P, Ohlen C, Carbone E, Franksson L, Ljunggren HG, Latour A, Koller B, Karre K. Recognition of beta 2-microglobulin-negative (beta 2m-) T-cell blasts by natural killer cells from normal but not from beta 2m- mice: nonresponsiveness controlled by beta 2m- bone marrow in chimeric mice. Proceedings of the National Academy of Sciences USA. 1991;88:10332–10336. doi: 10.1073/pnas.88.22.10332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wojciechowski W, Harris DP, Sprague F, Mousseau B, Makris M, Kusser K, Honjo T, Mohrs K, Mohrs M, Randall T, Lund FE. Cytokine-Producing Effector B Cells Regulate Type 2 Immunity to H. polygyrus. Immunity. 2009;30:421–433. doi: 10.1016/j.immuni.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mueller CG, Boix C, Kwan WH, Daussy C, Fournier E, Fridman WH, Molina TJ. Critical role of monocytes to support normal B cell and diffuse large B cell lymphoma survival and proliferation. Journal of Leukocyte Biology. 2007;82:567–575. doi: 10.1189/jlb.0706481. [DOI] [PubMed] [Google Scholar]
- 55.Lundgren A, Suri-Payer E, Enarsson K, Svennerholm AM, Lundin BS. Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infection and Immunity. 2003;71:1755–1762. doi: 10.1128/IAI.71.4.1755-1762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krauss-Etschmann S, Sammler E, Koletzko S, Konstantopoulos N, Aust D, Gebert B, Luckow B, Reinhardt D, Schendel DJ. Chemokine receptor 5 expression in gastric mucosa of Helicobacter pylori-infected and noninfected children. Clinical and Diagnostic Laboratory Immunology. 2003;10:22–29. doi: 10.1128/CDLI.10.1.22-29.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohtani N, Ohtani H, Nakayama T, Naganuma H, Sato E, Imai T, Nagura H, Yoshie O. Infiltration of CD8+ T cells containing RANTES/CCL5+ cytoplasmic granules in actively inflammatory lesions of human chronic gastritis. Laboratory Investigation. 2004;84:368–375. doi: 10.1038/labinvest.3700039. [DOI] [PubMed] [Google Scholar]
- 58.Kusugami K, Ando T, Ohsuga M, Imada A, Shinoda M, Konagaya T, Ina K, Kasuga N, Fukatsu A, Ichiyama S, Nada T, Ohta M. Mucosal chemokine activity in Helicobacter pylori infection. Journal of Clinical Gastroenterology. 1997;25(Suppl 1):S203–S210. doi: 10.1097/00004836-199700001-00032. [DOI] [PubMed] [Google Scholar]
- 59.Kikuchi T, Kato K, Ohara S, Sekine H, Arikawa T, Suzuki T, Noguchi K, Saito M, Saito Y, Nagura H, Toyota T, Shimosegawa T. The relationship between persistent secretion of RANTES and residual infiltration of eosinophils and memory T lymphocytes after Helicobacter pylori eradication. Journal of Pathology. 2000;192:243–250. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH688>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 60.Ling B, Veazey RS, Hart M, Lackner AA, Kuroda M, Pahar B, Marx PA. Early restoration of mucosal CD4 memory CCR5 T cells in the gut of SIV-infected rhesus predicts long term non-progression. AIDS. 2007;21:2377–2385. doi: 10.1097/QAD.0b013e3282f08b32. [DOI] [PubMed] [Google Scholar]
- 61.Stein JV, Nombela-Arrieta C. Chemokine control of lymphocyte trafficking: a general overview. Immunology. 2005;116:1–12. doi: 10.1111/j.1365-2567.2005.02183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoft DF, Eickhoff CS. Type 1 immunity provides both optimal mucosal and systemic protection against a mucosally invasive, intracellular pathogen. Infection and Immunity. 2005;73:4934–4940. doi: 10.1128/IAI.73.8.4934-4940.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoft DF, Schnapp AR, Eickhoff CS, Roodman ST. Involvement of CD4+ Th1 cells in systemic immunity protective against primary and secondary challenges with Trypanosoma cruzi. Infection and Immunity. 2000;68:197–204. doi: 10.1128/iai.68.1.197-204.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wizel B, Nunes M, Tarleton RL. Identification of Trypanosoma cruzi trans-sialidase family members as targets of protective CD8+ TC1 responses. Journal of Immunology. 1997;159:6120–6130. [PubMed] [Google Scholar]
- 65.Mota I, Umekita LF. The effect of C3 depletion on the clearance of Trypanosoma cruzi induced by IgG antibodies. Immunology Letters. 1989;21:223–225. doi: 10.1016/0165-2478(89)90108-9. [DOI] [PubMed] [Google Scholar]
- 66.Brodskyn CI, Silva AM, Takehara HA, Mota I. IgG subclasses responsible for immune clearance in mice infected with Trypanosoma cruzi. Immunology and Cell Biology. 1989;67 ( Pt 6):343–348. doi: 10.1038/icb.1989.50. [DOI] [PubMed] [Google Scholar]
- 67.Spinella S, Liegeard P, Hontebeyrie-Joskowicz M. Trypanosoma cruzi: predominance of IgG2a in nonspecific humoral response during experimental Chagas’ disease. Experimental Parasitology. 1992;74:46–56. doi: 10.1016/0014-4894(92)90138-z. [DOI] [PubMed] [Google Scholar]
- 68.Lages-Silva E, Ramirez LE, Krettli AU, Brener Z. Effect of protective and non-protective antibodies in the phagocytosis rate of Trypanosoma cruzi blood forms by mouse peritoneal macrophages. Parasite Immunology. 1987;9:21–30. doi: 10.1111/j.1365-3024.1987.tb00485.x. [DOI] [PubMed] [Google Scholar]
- 69.Scott MT, Moyes L. 75Se-methionine labelled Trypanosoma cruzi blood trypomastigotes: opsonization by chronic infection serum facilitates killing in spleen and liver. Clinical and Experimental Immunology. 1982;48:754–757. [PMC free article] [PubMed] [Google Scholar]
- 70.Lima-Martins MV, Sanchez GA, Krettli AU, Brener Z. Antibody-dependent cell cytotoxicity against Trypanosoma cruzi is only mediated by protective antibodies. Parasite Immunology. 1985;7:367–376. doi: 10.1111/j.1365-3024.1985.tb00083.x. [DOI] [PubMed] [Google Scholar]
- 71.Cardillo F, Postol E, Nihei J, Aroeira LS, Nomizo A, Mengel J. B cells modulate T cells so as to favour T helper type 1 and CD8+ T-cell responses in the acute phase of Trypanosoma cruzi infection. Immunology. 2007;122:584–595. doi: 10.1111/j.1365-2567.2007.02677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kumar S, Tarleton RL. The relative contribution of antibody production and CD8+ T cell function to immune control of Trypanosoma cruzi. Parasite Immunology. 1998;20:207–216. doi: 10.1046/j.1365-3024.1998.00154.x. [DOI] [PubMed] [Google Scholar]
- 73.Appay V, Dunbar PR, Cerundolo V, McMichael A, Czaplewski L, Rowland-Jones S. RANTES activates antigen-specific cytotoxic T lymphocytes in a mitogen-like manner through cell surface aggregation. International Immunology. 2000;12:1173–1182. doi: 10.1093/intimm/12.8.1173. [DOI] [PubMed] [Google Scholar]
- 74.Frauenschuh A, DeVico AL, Lim SP, Gallo RC, Garzino-Demo A. Differential polarization of immune responses by co-administration of antigens with chemokines. Vaccine. 2004;23:546–554. doi: 10.1016/j.vaccine.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 75.Kim JJ, Yang JS, Dentchev T, Dang K, Weiner DB. Chemokine gene adjuvants can modulate immune responses induced by DNA vaccines. Journal of Interferon and Cytokine Research. 2000;20:487–498. doi: 10.1089/10799900050023906. [DOI] [PubMed] [Google Scholar]
- 76.Tian S, Li J, Wang L, Liu T, Liu H, Cheng G, Liu D, Deng Y, Gou R, Wan Y, Jia J, Chen C. Urinary levels of RANTES and M-CSF are predictors of lupus nephritis flare. Inflammation Res. 2007;56:304–310. doi: 10.1007/s00011-007-6147-x. [DOI] [PubMed] [Google Scholar]
- 77.Yao TC, Kuo ML, See LC, Ou LS, Lee WI, Chan CK, Huang JL. RANTES and monocyte chemoattractant protein 1 as sensitive markers of disease activity in patients with juvenile rheumatoid arthritis: a six-year longitudinal study. Arthtitis and Rheumatism. 2006;54:2585–2593. doi: 10.1002/art.21962. [DOI] [PubMed] [Google Scholar]
- 78.Vila LM, Molina MJ, Mayor AM, Cruz JJ, Rios-Olivares E, Rios Z. Association of serum MIP-1alpha, MIP-1beta, and RANTES with clinical manifestations, disease activity, and damage accrual in systemic lupus erythematosus. Clinical Rheumatology. 2007;26:718–722. doi: 10.1007/s10067-006-0387-y. [DOI] [PubMed] [Google Scholar]
- 79.Rovin BH, Song H, Birmingham DJ, Hebert LA, Yu CY, Nagaraja HN. Urine chemokines as biomarkers of human systemic lupus erythematosus activity. Journal of the American Society of Nephrology. 2005;16:467–473. doi: 10.1681/ASN.2004080658. [DOI] [PubMed] [Google Scholar]