

Abstract
Vasospasm of the cerebrovasculature is a common manifestation of blast-induced traumatic brain injury (bTBI) reported among combat casualties in the conflicts in Afghanistan and Iraq. Cerebral vasospasm occurs more frequently, and with earlier onset, in bTBI patients than in patients with other TBI injury modes, such as blunt force trauma. Though vasospasm is usually associated with the presence of subarachnoid hemorrhage (SAH), SAH is not required for vasospasm in bTBI, which suggests that the unique mechanics of blast injury could potentiate vasospasm onset, accounting for the increased incidence. Here, using theoretical and in vitro models, we show that a single rapid mechanical insult can induce vascular hypercontractility and remodeling, indicative of vasospasm initiation. We employed high-velocity stretching of engineered arterial lamellae to simulate the mechanical forces of a blast pulse on the vasculature. An hour after a simulated blast, injured tissues displayed altered intracellular calcium dynamics leading to hypersensitivity to contractile stimulus with endothelin-1. One day after simulated blast, tissues exhibited blast force dependent prolonged hypercontraction and vascular smooth muscle phenotype switching, indicative of remodeling. These results suggest that an acute, blast-like injury is sufficient to induce a hypercontraction-induced genetic switch that potentiates vascular remodeling, and cerebral vasospasm, in bTBI patients.
Keywords: neurotrauma, mechanotransduction, tissue engineering, vascular mechanics
Blast traumatic brain injury (bTBI) is the hallmark injury among military personnel wounded in Afghanistan and Iraq (1, 2). While the injuries most commonly associated with TBI are diffuse axonal injury and compromise of the blood-brain barrier, cerebral vasospasm is a potentially lethal dysfunction whose incidence is elevated in bTBI, as compared to other forms of TBI (3). Cerebral vasospasm, characterized by chronic vascular hypercontraction followed by cell proliferation (4), extracellular matrix remodeling (5, 6), and arterial occlusion, is commonly diagnosed 3–7 d posttrauma (6), but its onset is often accelerated in bTBI patients (3). Traditionally cerebral vasospasm is attributed to subarachnoid hemorrhage (SAH), however, clinical reviews suggest that SAH is sufficient, but not necessary to potentiate cerebral vasospasm following bTBI (7). During blast injury, the cerebral vasculature likely bears significant acute pressure loads (8, 9). The role of chronically elevated luminal pressures in potentiating arterial thickening and stiffening to reequilibrate wall stress is well established (10); however, the vascular response to rapid acute pressure increases is unknown. The unique mechanics of blast pulse induced injury may contribute to the increased incidence of vasospasm in bTBI patients.
In vitro studies of vascular smooth muscle have revealed that direct stimulation of integrins induces enhanced calcium activity (11, 12) and myosin light chain phosphorylation (13). Cyclic mechanical stimulation within physiological ranges induces populations of vascular smooth muscle cells (VSMCs) to shift toward a contractile phenotype (14), while mechanical stresses of a higher magnitude, mimicking chronic hypertension, induce smooth muscle proliferation and extracellular matrix (ECM) remodeling (15). Following the acute mechanical injury impulse associated with an explosion, the cerebrovasculature may be exposed to a range of stimuli, including SAH (7), increased endothelial endothelin production (16), delayed systemic hypotension (17), and inflammatory responses (18). We hypothesized that, even in the absence of these stimuli, a single, low-magnitude mechanical impulse of sufficient energy, such as a blast wave, could initiate progression of cerebral vasospasm.
Here, we present evidence for mechanotransduced vasospasms in bTBI. We present a unique in vitro experimental approach that combines uniaxial high-speed stretching of micropatterned vascular tissue mimics, and muscular thin films (19, 20) to measure mechanically induced vascular hypercontractility. We find that simulated blast injury induces short-term endothelin-1 hypersensitivity followed by prolonged hypercontractility and phenotypic switching, indicative of remodeling. This data suggests that initiation of cerebral vasospasm can be mechanotransduced by blast wave associated pressure pulses, accounting for the increased incidence in bTBI patients.
Results
Simulated Blast in Engineered Arterial Lamellae.
A pressure pulse in an artery would primarily manifest as acute circumferential stretching caused by luminal expansion. To mimic this deformation, we developed engineered arterial lamellae (Fig. 1A) composed of a highly aligned monolayer of VSMCs (Fig. 1B) on an elastic substrate, which were exposed to a simulated blast consisting of a single axial high-velocity stretch (Fig. 1C, Movie S1). Due to the viscoelasticity (21) and strain stiffening (22) of arterial tissues, as well as the incompressibility of the surrounding brain tissue and cerebrospinal fluid (23), we assumed that the blast would result in a rapid, but low-magnitude strain. We therefore applied simulated blast at 5% and 10% strain at a strain rate of 1,000% strain/s (Fig. 1D, Movie S2). Evidence of acute and chronic vasospasm were measured 1–24 h postblast. Simulated blast did not cause obvious trauma such as altered tissue architecture (Fig. 1E), cell density, induced membrane poration, or increased apoptosis (Fig. S1)
Fig. 1.

In vitro model for bTBI in the vasculature. (A) Experimental model represents a single arterial lamella, isolated from surrounding tissue. (B) Phase contrast image of engineered lamella composed of micropatterned VSMCs. (Scale bar,100 μm). (C) Lamellar tissue is engineered on an elastic membrane, which is acutely stretched to mimic a blast pulse. (D) Lagrange strain during 5% and 10% simulated blast. Solid lines: axial strain. Dashed lines: transverse strain. (E) Simulated blast does not result in acute or delayed structural reorganization. Red: actin, Blue: nuclei. (F) Example single-cell cytosolic Ca2+ trace showing two transients, pre-ET-1 spontaneous transient and prolonged ET-1 induced transient. (G) Mean ET-1 induced temporal transients for blasted tissues. Black: control, Gray: 5% strain, Red: 10% strain. Inset: transient peaks (H) Single-cell ET-1 induced transient peak values. Mean + /- standard deviation. (I) Mean pre-ET-1 spontaneous transients.
Simulated Blast Induces Elevated Cytosolic Calcium Transients.
Recently, a study of controlled cortical impact injury in the rat demonstrated a reduced lumen diameter in the pial arterioles 1 h after injury, coincident with a reduction in ipsilateral cerebral blood flow (24). Subsequently, an additional study suggested L-type Ca2+ blockade would restore cerebral vasoreactivity following lateral fluid percussion injury in rats (25) . We asked if in our experimental model of blast-induced vascular injury, disrupted Ca2+ dynamics may play a role in initiating changes in vascular tone by contributing to the hypercontractility characteristic of acute vasospasm (26). We measured the temporal changes in cytosolic Ca2+ levels, or transients, in the engineered tissues 1 h after simulated blast (Fig. 1F, Movie S3). Endothelial injury following TBI can affect endothelin-1 (ET-1) production (16), and elevated cerebral spinal fluid levels of ET-1 have been implicated in cerebral vasospasm (27, 28), so we measured ET-1 induced calcium transients as well as spontaneous transients. Tissues exposed to simulated blast had transients with higher and more prolonged peaks following ET-1 stimulation than did control tissues (Fig. 1 G and H). This trend was also apparent for spontaneous transients (Fig. 1I). This elevated response suggests that blast mechanics predispose VSMCs to hypercontraction through increased Ca2+ uptake and may act synergistically with elevated ET-1 levels to accelerate vasospasm.
Simulated Blast Induces Dysfunctional Contractile Dynamics.
To characterize the functional effect of simulated blast, we measured contractile stress generation using a vascular muscular thin film (vMTF) assay (19, 20). The vMTFs are biohybrid constructs composed of a layer of polydimethylsiloxane (PDMS) and a layer of micropatterned VSMCs. When the muscle contracts, it bends the passive PDMS film whose curvature can be measured and used to calculate the tissue stress (Fig. 2B). The vMTFs were released from the stretchable substrate (Fig. 2A) and the temporal change in curvature (Fig. 2C, Movie S4) and tissue stress (Fig. 2D) were measured during stimulation with ET-1 and rho kinase (ROCK) inhibitor HA-1077, allowing characterization of the basal tone and contractility of the tissue (Fig. S2).
Fig. 2.

Simulated blast induces dysfunctional contractility, as measured with vMTFs. (A) Immediately prior to contraction experiment, vMTF is released from the membrane. (B) Stress is calculated from the radius of curvature ( ). (C-D) vMTFs were serially stimulated with ET-1 and HA-1077. (C) Single vMTF during experimental protocol. (
). (C-D) vMTFs were serially stimulated with ET-1 and HA-1077. (C) Single vMTF during experimental protocol. ( ). (D) Temporal change in vMTF stress due to serial stimulation, 1 h after simulated blast. (E–F) Normalized ET-1 induced contraction and basal tone 1 h after simulated blast. (G–H) Normalized induced contraction and basal tone 1 h after simulated blast for tissues pretreated with ROCK inhibitor. (I–J) Normalized induced contraction and basal tone 24 h after simulated blast. (K–L) Normalized induced contraction and basal tone 24 h after simulated blast for tissues treated ROCK inhibitor immediately following the blast. All graphs: mean + /-SEM.
). (D) Temporal change in vMTF stress due to serial stimulation, 1 h after simulated blast. (E–F) Normalized ET-1 induced contraction and basal tone 1 h after simulated blast. (G–H) Normalized induced contraction and basal tone 1 h after simulated blast for tissues pretreated with ROCK inhibitor. (I–J) Normalized induced contraction and basal tone 24 h after simulated blast. (K–L) Normalized induced contraction and basal tone 24 h after simulated blast for tissues treated ROCK inhibitor immediately following the blast. All graphs: mean + /-SEM.
An hour after simulated blast, tissues exhibited nearly twofold increases in ET-1 induced contraction (Fig. 2E), consistent with the observed elevated Ca2+, but no change in basal contractile tone (Fig. 2F). To test whether stress during the blast is responsible for this hypercontraction, we inhibited ROCK prior to simulated blast, to relieve cytoskeletal contractile tension during the blast. This stress relief during the blast mitigated the ET-1 hypersensitivity (Fig. 2G) with no change in basal tone (Fig. 2H), suggesting that cytoskeletal mechanical tension during blast contributes to postblast hypercontractility.
Because vasospasm is a chronic dysfunction, we also tested tissue function 24 h after simulated blast. At this later time point, mildly and severely injured tissues showed markedly different contractile dynamics (Fig. 2 I and J). Tissues strained 5% remained hypersensitive to ET-1 stimulation (Fig. 2I) and also developed elevated basal contractile tone (Fig. 2J), indicative of a chronic hypercontraction. However, the tissues strained 10% showed decreased ET-1 induced contraction (Fig. 2I) and no significant change in basal tone (Fig. 2J) compared to control.
Rho/ROCK signaling plays an important role in vasomotor tone. Inhibition of ROCK has been shown to relieve SAH induced vasospasm in vivo (29, 30). To test how ROCK signaling mediates the observed contractile behaviors in injured tissue, we inhibited ROCK immediately following simulated blast. We found that blasted tissues maintained ET-1 hypersensitivity, though at a decreased level, 1 h after blast (Fig. S3), but that 24 h postblast ROCK-inhibited tissues exhibited no differences in contractility between the blast exposed tissues and control (Fig. 2 K and L). This result suggests that Rho-mediated postblast hypercontraction potentiates chronic contractile variation between mildly and severely injured tissues.
Dynamic Phenotype Switching Predicts Long-Term Contractility and Stress Remodeling.
Given the reduction in contractile stress at 24 h in the severely injured tissues with respect to both control and mildly injured tissues, we reasoned that severely injured tissues were remodeling their stress state by changing the phenotype of a subpopulation of the VSMCs in the blast-induced hypercontractile tissues. We developed an elasticity-based computational model, based on previous models of stress-induced growth and remodeling (31, 32), to better understand how blast-induced hypercontraction potentiates the contractility observed 24 h postblast. The primary assumption of the model is that the tissue has an optimal stress state and actively remodels to return to this target value (33). Unlike previous models, we included a dynamic temporal interaction between tissue tension and VSMC phenotype (34), where for small perturbations in tissue tension, contractile adaptation dominates, while larger perturbations shift VSMCs toward a synthetic phenotype, facilitating large-scale remodeling (see Methods for details). This assumption is consistent with previous experimental reports that physiological levels of stress induce VSMCs to become more contractile (14), while pathological stresses lead to vessel thickening and ECM remodeling (15), indicative of synthetic VSMCs. We further assume that contractile cells generate contractile stress when stimulated, but do not contribute to tissue remodeling, while synthetic cells generate no contractile stress but more rapidly remodel the tissue.
As indicated by our cytosolic calcium measurements, we assumed that more severe blast results in increased hypercontraction (Fig. 3A), and thus, increased tissue stress (Fig. 3B), necessitating an adaptive response to reequilibrate the stress. To compare with the contractility results, we determined the simulated basal tone and induced contraction. The model predicts that mild blast leads to increased hypercontractility (Fig. 3C) and basal tone (Fig. 3D). Following severe injury, hypercontraction leads to large increases in tissue tension (Fig. 3B), which results in greatly decreased induced contraction (Fig. 3C) and basal tone (Fig. 3D) mediated by a phenotype shift toward a synthetic, noncontractile population (Fig. 3E). With time, the tissue returns to preblast equilibrium (Fig. 3 B–E); however, during early vasospasm development (e.g., 24 h postblast), the model qualitatively reproduces the experimentally observed contractility (Fig. 3 F and G, see Fig. 2 I and J for comparison) and predicts that mild injury induces a slightly more contractile population, while the more severe injury induces a more synthetic population (Fig. 3H). Phenotypic switching in VSMCs is necessary to maintain SAH induced vasospasm (35). Our model predicts that in bTBI vasospasm, stress-induced phenotype switching may drive postblast remodeling.
Fig. 3.

Theoretical model for stress-induced remodeling and phenotype switching. (A) Blast injury induces increased hypercontractility, characterized by greater stress-free shortening (λa). (See Methods for details). Left box: contour plot of temporal evolution of contractile shortening. The y-axis represents tissues with increasing blast injury. The x-axis represents the time after the blast. Right box: Temporal plots of stress-free shortening for the tissues indicated by the white lines in the left box. (B–E) Contour plots of the temporal evolution of tissue tension, induced contraction, basal tone, and phenotype population for varying magnitudes of simulated blast. (B) Temporal stress evolution of remodeling tissue, (C) model predicted contractility, and (D) model predicted basal contractile tone for varying blast-induced hypercontraction. (E) Predicted temporal change in fraction of contractile cells for varying blast-induced hypercontraction and tissue remodeling. (F–H) Time point snapshots of (F) induced contraction, (G) basal tone, and (H) fraction contractile cells at an early time in vasospasm development, indicated by the vertical line in (B), for mild injury, indicated by the lower horizontal line in (B), or a more severe injury, indicated by the upper horizontal line in (B). These graphs correspond with the 24 h contractility experiments in Fig. 3 F and G.
Simulated Blast Induces Dynamic Phenotype Switching in VSMCs.
To test in vitro whether blast forces can lead to phenotypic switch, we measured protein (Fig. 4 A and B) and mRNA (Fig. 4C) expression of two primary markers of contractile VSMCs, smooth muscle myosin heavy chain (SM-MHC) and smoothelin, 24 h after exposure to simulated blast. Tissues exposed to 5% strain exhibited nonsignificant increased SM-MHC transcripts but little change in smoothelin expression (Fig. 4 A–C), while severe (10% strain) simulated blast decreased expression of smoothelin (Fig. 4 A and B) and decreased mRNA expression of both markers (Fig. 4C). These data suggest that acute mechanical injury can potentiate a switch away from the contractile phenotype in VSMCs, as theoretically predicted. Further, if postblast tissue contraction is inhibited, contractile marker expression is unchanged in blasted tissues (Fig. 4D). These results demonstrate that mechanotransduced hypercontraction, due to acute mechanical injury, facilitates hastened VSMC phenotype switching, potentially accounting for accelerated onset of vasospasm in bTBI patients.
Fig. 4.

Simulated blast induces VSMC phenotype switching. (A) Representative Western blot of contractile phenotype markers, smooth muscle myosin heavy chain and smoothelin, 24 h after simulated blast. (B) Quantified protein expression of contractile markers (mean + /-SEM). (C) Quantified mRNA expression of SM-myosin heavy chain (MYH11) and smoothelin (SMTN) (mean + /-SEM). (D) Representative Western blot of contractile markers for tissues treated with ROCK inhibitor immediately following simulated blast.
Discussion
Vascular injury is an oft overlooked component of traumatic brain injury. But, local ischemia resulting from acute vasospasms may play a significant role in the delayed onset of clinical manifestations of bTBI. The normal progression of cerebral vasospasm is an initial hypercontraction (26) followed by vessel remodeling (4–6) and narrowing (9). Here, we find that within 24 h of blast-like injury, mechanotransduction of extreme mechanical forces can induce both hypercontractility and phenotypic switching indicative of the onset of remodeling.
Our theoretical model suggests that these phenomena are related by the stress-induced remodeling pathways that endow the vasculature with its robust mechanoadaptability (32, 34). Specifically, these results suggest that blast-induced hypercontraction leads to elevated tissue stress, which can, in cases of severe injury, induce maladaptive compensatory remodeling, facilitated by phenotype switching. The expression of contractile phenotype markers and the variable contractility suggests that in the 10% strain-injured tissue, there is a mixed population of hypercontractile and synthetic VSMCs.
The relief of short-term hypercontractility and long-term phenotype switching with inhibition of ROCK suggests that cytoskeletal tension and forces on integrins are playing a key role in vasospasm initiation. Integrins are the primary transducers of mechanical force to the cell’s cytoskeleton (36, 37), and integrin stimulation is involved in a wide range of cellular signaling, including altering calcium dynamics (11, 12) and phenotypic behavior (38–40) in VSMCs. The mechanical loading of the blast and the resulting hypercontractility both likely result in abnormal integrin stimulation, potentially mediating the observed vasospasm behavior.
While this study has focused on the effect of acute stretch on in vitro engineered arterial lamellae, in vivo the cerebrovasculature is exposed to a complex milieu of signals that are not accounted for in our model. The mechanotransduction pathways identified here likely act in concert with, for example, dysfunctional endothelial behavior (16) and SAH (7) to accelerate vasospasm onset in bTBI patients. Moreover, the cerebrovasculature is exposed to dynamically varying mechanical forces, such as delayed hypotension (17), that could also have an additional mechanotransductive effect on vascular remodeling.
As a result of the conflicts in Afghanistan and Iraq, the number of bTBI patients, for whom there are few effective therapies (41), is rising rapidly. To better understand how to treat these patients, it is important to understand the source and pathway of their dysfunction. Here, we identify a unique source of cerebral vasospasm; blast-induced mechanotransduced vasospasm independent of SAH. These results suggest that bTBI vasospasm may require different therapeutic strategies than those currently employed for SAH induced vasospasm.
Methods
Experimental methods are briefly described, a detailed description is provided in SI Text.
Tissue Construction.
Tissues were patterned using microcontact printed (42) 10 μm wide lines separated by 10 μm wide gaps. VSMCs were seeded at 25,000 cells/cm2, creating a confluent tissue. High-velocity acute uniaxial stretch (43, 44) was applied using a linear motor to displace one end of a 5 × 8 cm elastomer membrane while holding the other end fixed. The strain field (Fig. 1D) of the substrate was verified using high-speed video and a three-point strain algorithm (45).
Calcium Measurement.
To measure cytosolic calcium, Fluo-4 AM calcium indicating dye was added to culture for 15 min and the culture was allowed to equilibrate for 15 min post wash out. Fluorescent intensity was measured 1 frame/s for 10 min to acquire spontaneous transients and 20 min following ET-1 treatment. Normalized temporal intensities of individual regions were registered by onset time to determine the mean curves in Fig. 1.
Tissue Stress Measurement.
vMTF were constructed as previously published (19). The vMTF tissue stress was calculated as previously published (20). To inhibit ROCK during blast, tissues were treated with 100 μM HA-1077 for 30 min prior to simulated blast and immediately washed out following blast. To inhibit postblast ROCK activity, tissues were treated with 10 μM HA-1077 immediately following simulated blast. HA-1077 was washed out 30 min prior to vMTF experiments. Substrate and tissue thicknesses necessary for stress measurement were measured using stylus profilometry and confocal microscopy, respectively (Fig. S4).
Theoretical Model.
The model methods are presented briefly here, and in more extensive detail in SI Text, Figs. S5, S6, S7, and S8.
We employ finite elastic growth theory (46) adapted for pseudocontraction (47) to model the evolution of stress in the tissue. Within this framework, Cauchy stress (σ) is considered a function of elastic stretch ratio Fe such that
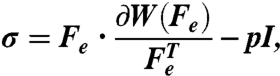 |
[1] |
where W is the strain-energy density function, and p is a Lagrange multiplier. Fe is a component of the total deformation (F) which is also composed of the remodeling deformation (Fg = diag(λg,1,1)), and active deformation (Fa = diag(λa,1,1)), which represent the change in the zero-stress configuration due to remodeling and contraction, respectively. (e.g., λa is the stretch ratio representing the stress-free shortening that the tissue would undergo if it was unconstrained.) Total deformation is defined as
| [2] |
The tissue is composed of a mixed population of contractile and synthetic cells and is treated as a constrained mixture (48) of both cell types. Total tissue tension and remodeling rate are dependent on the fraction of the population of each cell type, with synthetic cells being noncontractile (λa = 1), but remodeling more quickly. The total stress is given by
| [3] |
where ϕc and ϕs are the fraction of total cells expressing contractile or synthetic phenotypes, respectively and ϕc + ϕs = 1.
We assume that the tissue grows to reequilibrate both tissue stress and basal contraction (32). The relationship between the magnitude of the perturbation from a homeostatic tissue-level target stress and the rate of remodeling 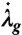 is given by
is given by
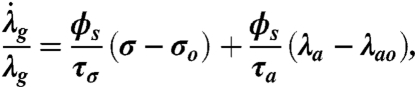 |
[4] |
where σo is the tissue’s target stress and τσ and τa are time constants (32, 33, 49). We further assume that perturbation from the target stress results in a change in the phenotype population, with small perturbations inducing a more contractile population and large perturbations causing the population to become more synthetic. We characterize this phenotype switching by the rate of change of contractile population fraction (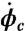 ) given by
) given by
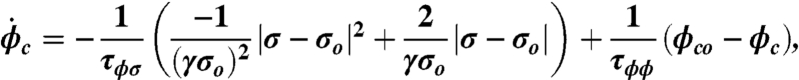 |
[5] |
where the first term represents the influence of stress perturbation on phenotype and the second is a corrective term that acts to bring the phenotype distribution back to equilibrium. γ is a constant, τϕσ and τϕϕ are time constants and ϕco is the homeostatic fraction of cells expression a contractile phenotype, here assumed to be 0.8.
Following the blast, we assume an acute hypercontraction followed by a temporal return to equilibrium, described by
| [6] |
where α is a constant that is dependent on the degree of hypercontraction, assumed to be proportional to the blast severity (Fig. 3A), β is a time constant and t is time after blast. In response to the perturbed contractile stress, the tissue temporally remodels and undergoes phenotype switching to reequilibrate the tissue tension.
To simulate the vMTF experiments, induced contraction and basal tone are calculated by setting λa = 0.6 (fully contracted) and λa = 1 (fully relaxed) respectively, and comparing the resulting stress to the unstimulated tissue.
Biochemistry.
Western blotting and RT-PCR were performed using standard methods detailed in the online supplement.
Supplementary Material
Acknowledgments.
The authors gratefully acknowledge the use of facilities at the Harvard Center for Nanoscale Systems. This work was funded by the Defense Advanced Research Projects Agency Preventing Violent Explosive Neurologic Trauma (PREVENT) N66001-08-C-2036 (K.K.P.) and Harvard School of Engineering and Applied Sciences.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1105860108/-/DCSupplemental.
References
- 1.Warden DL, French L. Traumatic brain injury in the war zone. N Engl J Med. 2005;353:633–634. [PubMed] [Google Scholar]
- 2.Bhattacharjee Y. Neuroscience. shell shock revisited: solving the puzzle of blast trauma. Science. 2008;319:406–408. doi: 10.1126/science.319.5862.406. [DOI] [PubMed] [Google Scholar]
- 3.Ling G, Bandak F, Armonda R, Grant G, Ecklund J. Explosive blast neurotrauma. J Neurotrauma. 2009;26:815–825. doi: 10.1089/neu.2007.0484. [DOI] [PubMed] [Google Scholar]
- 4.Borel CO, et al. Possible role for vascular cell proliferation in cerebral vasospasm after subarachnoid hemorrhage. Stroke. 2003;34:427–432. [PubMed] [Google Scholar]
- 5.Zhang ZD, Macdonald RL. Contribution of the remodeling response to cerebral vasospasm. Neurol Res. 2006;28:713–720. doi: 10.1179/016164106X151990. [DOI] [PubMed] [Google Scholar]
- 6.Humphrey JD, Baek S, Niklason LE. Biochemomechanics of cerebral vasospasm and its resolution: I. A new hypothesis and theoretical framework. Ann Biomed Eng. 2007;35:1485–1497. doi: 10.1007/s10439-007-9321-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armonda RA, et al. Wartime traumatic cerebral vasospasm: Recent review of combat casualties. Neurosurgery. 2006;59:1215–1225. doi: 10.1227/01.NEU.0000249190.46033.94. [DOI] [PubMed] [Google Scholar]
- 8.Cernak I, Wang Z, Jiang J, Bian X, Savic J. Ultrastructural and functional characteristics of blast injury-induced neurotrauma. J Trauma. 2001;50:695–706. doi: 10.1097/00005373-200104000-00017. [DOI] [PubMed] [Google Scholar]
- 9.Bauman RA, et al. An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J Neurotrauma. 2009;26:841–860. doi: 10.1089/neu.2008.0898. [DOI] [PubMed] [Google Scholar]
- 10.Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension. 2001;38:581–587. doi: 10.1161/hy09t1.096249. [DOI] [PubMed] [Google Scholar]
- 11.Wu X, et al. Modulation of calcium current in arteriolar smooth muscle by alphav beta3 and alpha5 beta1 integrin ligands. J Cell Biol. 1998;143:241–252. doi: 10.1083/jcb.143.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balasubramanian L, Ahmed A, Lo CM, Sham JS, Yip KP. Integrin-mediated mechanotransduction in renal vascular smooth muscle cells: activation of calcium sparks. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2007;293:R1586–1594. doi: 10.1152/ajpregu.00025.2007. [DOI] [PubMed] [Google Scholar]
- 13.Polte TR, Eichler GS, Wang N, Ingber DE. Extracellular matrix controls myosin light chain phosphorylation and cell contractility through modulation of cell shape and cytoskeletal prestress. Am J Physiol Cell Ph. 2004;286:C518–528. doi: 10.1152/ajpcell.00280.2003. [DOI] [PubMed] [Google Scholar]
- 14.Stegemann JP, Hong H, Nerem RM. Mechanical, biochemical, and extracellular matrix effects on vascular smooth muscle cell phenotype. J Appl Physiol. 2005;98:2321–2327. doi: 10.1152/japplphysiol.01114.2004. [DOI] [PubMed] [Google Scholar]
- 15.Chesler NC, Ku DN, Galis ZS. Transmural pressure induces matrix-degrading activity in porcine arteries ex vivo. Am J Physiol. 1999;277:H2002–2009. doi: 10.1152/ajpheart.1999.277.5.H2002. [DOI] [PubMed] [Google Scholar]
- 16.Petrov T, Steiner J, Braun B, Rafols JA. Sources of endothelin-1 in hippocampus and cortex following traumatic brain injury. Neuroscience. 2002;115:275–283. doi: 10.1016/s0306-4522(02)00345-7. [DOI] [PubMed] [Google Scholar]
- 17.DeWitt DS, Prough DS. Blast-induced brain injury and posttraumatic hypotension and hypoxemia. J Neurotrauma. 2009;26:877–887. doi: 10.1089/neu.2007.0439. [DOI] [PubMed] [Google Scholar]
- 18.Lenzlinger PM, Morganti-Kossmann MC, Laurer HL, McIntosh TK. The duality of the inflammatory response to traumatic brain injury. Mol Neurobiol. 2001;24:169–181. doi: 10.1385/MN:24:1-3:169. [DOI] [PubMed] [Google Scholar]
- 19.Feinberg AW, et al. Muscular thin films for building actuators and powering devices. Science. 2007;317:1366–1370. doi: 10.1126/science.1146885. [DOI] [PubMed] [Google Scholar]
- 20.Alford PW, Feinberg AW, Sheehy SP, Parker KK. Biohybrid thin films for measuring contractility in engineered cardiovascular muscle. Biomaterials. 2010;31:3613–3621. doi: 10.1016/j.biomaterials.2010.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Liu Y, Kassab GS. Viscoelasticity reduces the dynamic stresses and strains in the vessel wall: implications for vessel fatigue. Am J Physiol Heart C. 2007;293:H2355–2360. doi: 10.1152/ajpheart.00423.2007. [DOI] [PubMed] [Google Scholar]
- 22.Cox RH. Passive mechanics and connective tissue composition of canine arteries. Am J Physiol. 1978;234:H533–541. doi: 10.1152/ajpheart.1978.234.5.H533. [DOI] [PubMed] [Google Scholar]
- 23.Miller K, Chinzei K. Constitutive modelling of brain tissue: experiment and theory. J Biomech. 1997;30:1115–1121. doi: 10.1016/s0021-9290(97)00092-4. [DOI] [PubMed] [Google Scholar]
- 24.Golding EM, Robertson CS, Fitch JC, Goodman JC, Bryan RM., Jr Segmental vascular resistance after mild controlled cortical impact injury in the rat. J Cereb Blood Flow Metab. 2003;23:210–218. doi: 10.1097/01.WCB.0000044739.64940.B5. [DOI] [PubMed] [Google Scholar]
- 25.Maeda T, Lee SM, Hovda DA. Restoration of cerebral vasoreactivity by an L-type calcium channel blocker following fluid percussion brain injury. J Neurotrauma. 2005;22:763–771. doi: 10.1089/neu.2005.22.763. [DOI] [PubMed] [Google Scholar]
- 26.Baldwin ME, et al. Early vasospasm on admission angiography in patients with aneurysmal subarachnoid hemorrhage is a predictor for in-hospital complications and poor outcome. Stroke. 2004;35:2506–2511. doi: 10.1161/01.STR.0000144654.79393.cf. [DOI] [PubMed] [Google Scholar]
- 27.Masaoka H, et al. Raised plasma endothelin in aneurysmal subarachnoid haemorrhage. Lancet. 1989;2:1402. doi: 10.1016/s0140-6736(89)92019-9. [DOI] [PubMed] [Google Scholar]
- 28.Cosentino F, Katusic ZS. Does endothelin-1 play a role in the pathogenesis of cerebral vasospasm? Stroke. 1994;25:904–908. doi: 10.1161/01.str.25.4.904. [DOI] [PubMed] [Google Scholar]
- 29.Tachibana E, et al. Intra-arterial infusion of fasudil hydrochloride for treating vasospasm following subarachnoid haemorrhage. Acta Neurochir. 1999;141:13–19. doi: 10.1007/s007010050260. [DOI] [PubMed] [Google Scholar]
- 30.Sato M, Tani E, Fujikawa H, Kaibuchi K. Involvement of Rho-kinase-mediated phosphorylation of myosin light chain in enhancement of cerebral vasospasm. Circ Res. 2000;87:195–200. doi: 10.1161/01.res.87.3.195. [DOI] [PubMed] [Google Scholar]
- 31.Taber LA. A model for aortic growth based on fluid shear and fiber stresses. J Biomech Eng-Trans ASME. 1998;120:348–354. doi: 10.1115/1.2798001. [DOI] [PubMed] [Google Scholar]
- 32.Alford PW, Humphrey JD, Taber LA. Growth and remodeling in a thick-walled artery model: effects of spatial variations in wall constituents. Biomech Model Mechan. 2008;7:245–262. doi: 10.1007/s10237-007-0101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taber LA. Biomechanical growth laws for muscle tissue. J Theor Biol. 1998;193:201–213. doi: 10.1006/jtbi.1997.0618. [DOI] [PubMed] [Google Scholar]
- 34.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 35.Yamaguchi-Okada M, Nishizawa S, Mizutani A, Namba H. Multifaceted effects of selective inhibitor of phosphodiesterase III, cilostazol, for cerebral vasospasm after subarachnoid hemorrhage in a dog model. Cerebrovasc Dis. 2009;28:135–142. doi: 10.1159/000223439. [DOI] [PubMed] [Google Scholar]
- 36.Katsumi A, Orr AW, Tzima E, Schwartz MA. Integrins in mechanotransduction. J Biol Chem. 2004;279:12001–12004. doi: 10.1074/jbc.R300038200. [DOI] [PubMed] [Google Scholar]
- 37.Ingber DE. Cellular mechanotransduction: putting all the pieces together again. FASEB J. 2006;20:811–827. doi: 10.1096/fj.05-5424rev. [DOI] [PubMed] [Google Scholar]
- 38.Qin H, et al. Effects of extracellular matrix on phenotype modulation and MAPK transduction of rat aortic smooth muscle cells in vitro. Exp Mol Pathol. 2000;69:79–90. doi: 10.1006/exmp.2000.2321. [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto M, Yamamoto K, Noumura T. Type I collagen promotes modulation of cultured rabbit arterial smooth muscle cells from a contractile to a synthetic phenotype. Exp Cell Res. 1993;204:121–129. doi: 10.1006/excr.1993.1016. [DOI] [PubMed] [Google Scholar]
- 40.Peyton SR, Putnam AJ. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J Cell Physiol. 2005;204:198–209. doi: 10.1002/jcp.20274. [DOI] [PubMed] [Google Scholar]
- 41.Janowitz T, Menon DK. Exploring new routes for neuroprotective drug development in traumatic brain injury. Science Translational Medicine. 2010;2:27rv21. doi: 10.1126/scitranslmed.3000330. [DOI] [PubMed] [Google Scholar]
- 42.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 43.Lusardi TA, Rangan J, Sun D, Smith DH, Meaney DF. A device to study the initiation and propagation of calcium transients in cultured neurons after mechanical stretch. Ann Biomed Eng. 2004;32:1546–1558. doi: 10.1114/b:abme.0000049038.75368.75. [DOI] [PubMed] [Google Scholar]
- 44.Pfister BJ, Weihs TP, Betenbaugh M, Bao G. An in vitro uniaxial stretch model for axonal injury. Ann Biomed Eng. 2003;31:589–598. doi: 10.1114/1.1566445. [DOI] [PubMed] [Google Scholar]
- 45.Alford PW, Taber LA. Regional epicardial strain in the embryonic chick heart during the early looping stages. J Biomech. 2003;36:1135–1141. doi: 10.1016/s0021-9290(03)00089-7. [DOI] [PubMed] [Google Scholar]
- 46.Rodriguez EK, Hoger A, McCulloch AD. Stress-dependent finite growth in soft elastic tissues. J Biomech. 1994;27:455–467. doi: 10.1016/0021-9290(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 47.Ramasubramanian A, et al. On modeling morphogenesis of the looping heart following mechanical perturbations. J Biomech Eng-Trans ASME. 2008;130:061018-1–061018-11. doi: 10.1115/1.2978990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Humphrey JD, Rajagopal KR. A constrained mixture model for arterial adaptations to a sustained step change in blood flow. Biomech Model Mechan. 2003;2:109–126. doi: 10.1007/s10237-003-0033-4. [DOI] [PubMed] [Google Scholar]
- 49.Alford PW, Taber LA. Computational study of growth and remodelling in the aortic arch. Comput Methods Biome. 2008;11:525–538. doi: 10.1080/10255840801930710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.