

Abstract
Recent evidence indicates that p53 suppression increased the efficiency of induced pluripotent stem cell (iPSC) generation. This occurred even with the enforced expression of as few as two canonical transcription factors, Oct4 and Sox2. In this study, primary human keratinocytes were successfully induced into a stage of plasticity by transient inactivation of p53, without enforced expression of any of the transcription factors previously used in iPSC generation. These cells were later redifferentiated into neural lineages. The gene suppression plastic cells were morphologically indistinguishable from human ES cells. Gene suppression plastic cells were alkaline phosphatase-positive, had normal karyotypes, and expressed p53. Together with the accumulating evidence of similarities and overlapping mechanisms between iPSC generation and cancer formation, this finding sheds light on the emerging picture of p53 sitting at the crossroads between two intricate cellular potentials: stem cell vs. cancer cell generation. This finding further supports the crucial role played by p53 in cellular reprogramming and suggests an alternative method to switch the lineage identity of human cells. This reported method offers the potential for directed lineage switching with the goal of generating autologous cell populations for novel clinical applications for neurodegenerative diseases.
Keywords: cancer development, transdifferentiation, neural development
The ethical concerns and immune issue of human ES cells (hESCs) have motivated scientists to search for alternatives to generate patient-specific pluripotent cells that may enable cell therapy for many degenerative diseases. Human induced pluripotent stem cells (iPSCs) can be derived from reprogramming human fibroblasts, either by initially using the four transcriptional factors (OCT4, SOX2, KLF4, and c-MYC) (1) or by replacing KLF-4 and c-MYC with NANOG and LIN28 (2). The mechanisms involved in the reprogramming of differentiated cells into iPSCs remain poorly understood. This process may require epigenetic reprogramming (3). The overexpression of these defined transcription factors in somatic cells could result in other abnormal events, such as tumorigenesis (4, 5). It is well known that reprogramming has extremely low efficiency and kinetics with a rate-limiting step toward the establishment of a pluripotent state. To improve the efficiency, efforts to understand the nature and identity of the reprogramming have focused on the transcriptional and translational levels in the past few years (4, 6–8). However, pluripotent cells have limited therapeutical values because of the potential for teratoma formation and tendency for tumorigenesis. Lineage switching is a more practical and safe approach for cell therapy. Here, we report a method of switching keratinocytes to neural lineages through the transient suppression of p53.
p53 is a cellular protein of 53,000 Da, encoded by TP53 in humans (Trp53 in mice). It has been described as a multifunctional protein, dubbed the “molecular policeman” (9, 10), “tumor suppressor” (11), and tumor progressor. p53 functions as a master regulator of the transcriptional network, by positively and negatively regulating a plethora of genes (12). With increased activity, p53 acts as the guardian of genomic fidelity, effecting cell-cycle arrest to allow adequate opportunity for genetic repair mechanisms; senescence; early differentiation; and, with the anticipation of failure for all other mechanisms of control, initiation of apoptotic cell death (13). Furthermore, functional inactivation of p53 through mutation, loss of heterozygosity (LOH, allelic deletion), and/or inhibition is an obligate step for the formation of most human tumors. The molecular mechanisms exploited by tumors to achieve inactivation of the p53 network are complex and incompletely understood but offer intriguing therapeutical targets (14). Expression of p53 is induced by specific molecular signals that herald a cellular cry of “SOS,” such as genetic aberrations (genotoxic damage, oncogene activation, and telomere erosion), matrix alternation (loss of stromal support), and metabolic dysfunction (nutrient and oxygen deprivation) (15). Recently, p53 has also attracted attention for its novel role in cellular reprogramming (4, 6–8, 16), noting that the blocking of downstream effectors in the p53 pathway can improve measured efficiency rates of reprogramming by more than 100-fold compared with those in the absence of its inhibition (6). In our study, the transient ablation of p53 via shRNA resulted in activation of Oct4-GFP expression in fibroblasts isolated from Oct4-GFP transgenic mice. Subsequently, we applied the same method to human keratinocytes and successfully switched the lineage of these cells to provide abundant patient-specific neural progenitors. Our study suggests an alternative approach for generating specific cell populations suitable for personalized cell therapy approaches for the treatment of neurodegenerative diseases.
Results
Suppression of p53 Increased Oct4 Expression.
Our previous gene expression studies suggested that gene expression involved with the p53 network affects alterations in cell pluripotency (17). Specifically, we proposed that induced suppression of p53 could lead to a pluripotency state in these cells. Fibroblasts from Oct4-GFP transgenic mice were cultured and transduced with shRNA to suppress p53 expression (specifics provided in Materials and Methods). These Oct4-GFP fibroblasts do not express the pluripotency gene Oct4 under normal culture conditions. After transduction with this specific shRNA to suppress p53 expression, Oct4 gene was activated in some cells, as indicated by their expression of the GFP marker linked to Oct4 promoter in the transduced cells (Fig. 1). Some cells that expressed Oct4 also exhibited a change in morphology. In contrast, a subset of cells that also lost their original fibroblast morphology did not express Oct4-GFP (Fig. 1). The expression of Oct4 was not necessarily associated with changes in cell morphology, supporting the notion that cell morphology is not a sufficient marker for selecting pluripotent cells (18).
Fig. 1.

Suppression of p53 expression following shRNA transduction in mouse Oct4-GFP fibroblasts. After p53 suppression, the morphology of fibroblasts gradually changed to cells with undifferentiated round shapes. A subset of these cells also expressed GFP secondary to activation of the GFP-linked OCT4 promoter. (A) Five weeks after p53 suppression, some round-shaped cells appeared and only these cells expressed GFP. (B) Six weeks after p53 suppression, more cells expressed Oct4-GFP. (C) Seven weeks after p53 suppression, many round-shaped cells were apparent but not all of them were GFP-positive. (All images were taken with a magnification of 60×.)
Induction of Human Keratinocytes into hESC-Like Colonies.
We further tested whether suppression of p53 can change pluripotency in differentiated human cells. Human primary keratinocytes were purchased from the American Type Culture Collection and maintained as instructed. After transduction with shRNA lentiviral vector to suppress p53 expression, these cells were transferred onto mouse embryonic fibroblast (MEF) feeders with stem cell medium. Newly formed colonies appeared in these cultures (Fig. 2A) and could be expanded (Fig. 2B) into hESC-like colonies with distinct edges in 5 wk (Fig. 2C). These colonies were morphologically indistinguishable from normal hESC colonies. Furthermore, they tested positive with an alkaline phosphatase (AP) assay (Fig. 2D). These colonies maintained their morphology and AP-positive properties, even after five passages. The reprogramming efficiency was ∼0.025%. DNA finger printing was conducted with eight specific loci of these human keratinocytes to verify the lineage origin of these cells. The results showed that these colonies are derived from human keratinocytes rather than from MEFs. We further conducted the karyotype analyses on these colonies to screen for the appearance and numbers of the chromosomal anomalies. Chromosomal spread of gene suppression plastic cells (GSPCs) was assessed during the growth phase after 24 h of colcemid treatment. The chromosome count was based on G-banding obtained with Giemsa stain following digestion of chromosomes with trypsin. These results revealed normal 46XY karyotypes (Fig. 3C), the same as the cells from which they were derived.
Fig. 2.

Induction of human keratinocytes into GSPCs via p53 suppression. (A) Summary of the procedure for GSPC generation. After transducing shRNA to suppress p53 expression, keratinocytes were cultured on MEF feeder layers and maintained in hESC culture medium. The resultant hESC-like colonies appeared ∼5 wk posttransduction. (B) Newly formed colony. (C) Colony growing on MEFs. (D) Mature hESC-like colony with distinct edges from MEFs. (E) AP staining indicated that the colony was AP-positive. (Images of the colony were taken with a magnification of 10×.)
Fig. 3.

Cytogenetic analyses of GSPC. GSPCs were cultured to obtain cells in the interface (as indicated by arrows) in A (magnification, 10×) and B (magnification, 20×). (C) Karyotype analysis confirmed that GSPCs had the correct number of chromosomes, and no apparent structural abnormalities were observed.
Return of p53 Expression in GSPCs.
Previous reprogramming studies indicated that retroviral expression vectors were silenced after reprogramming (1, 2, 19). We performed quantitative RT-PCR to examine whether the p53 expression suppression was reversed in GSPCs. As shown in Fig. 4, p53 was expressed at a level comparable to that of hECSs (H9 strain). This indicates that p53 suppression was reversed and the shRNA was silenced in GSPC colonies. These data further suggest that similar to previously reported reprogramming factors, the suppression of p53 is lost during the process of reprogramming.
Fig. 4.

Semiquantitative RT-PCR confirmed that GSPCs expressed p53. Total RNAs were extracted from both GSPC colonies and H9 colonies and reverse-transcribed into cDNA. Quantified cDNA (50 ng) was used for PCR with p53-specific primers. GSPC colonies expressed p53 at a level comparable to H9 colonies. MW, molecular weight 1-kb DNA ladder (Invitrogen).
Neural Differentiation Potential of GSPCs in Vitro and ex Vivo.
Because pluripotent cells are capable of differentiating into various cell lineages, we tested whether GSPCs could be differentiated into neural lineages. GSPC colonies were placed into neural stem cell differentiation medium for neural differentiation as previously described (17). After switching to neural differentiation medium, GSPCs changed morphology and expressed nestin. As shown in Fig. 5, these GSPCs were positive for labeling with fluorescent-tag antibody against nestin, a marker for neural stem cell lineage. We further characterized the GSPCs for their neural differential potential and migration capacity within our proprietary ex vivo organotypic brain slice (OTS) system, a method we had established previously (20). In the coculture of GSPCs with the ex vivo organotypic slice, GSPCs introduced into the slice migrated out of the injection site along resident nerve fiber tracks (Fig. 6). These engrafted migratory GSPCs expressed human nestin, consistent with our previous observations (20, 21). The human origin of these cells was again confirmed, supporting their origin from GSPCs (Fig. 6). These data demonstrated that GSPCs have the ability to differentiate along neural lineages when introduced into an authentic brain environment. Such ability is critically important for potential clinical applications of these cells.
Fig. 5.
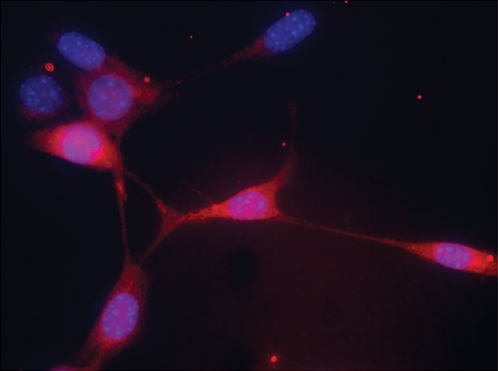
In vitro differentiation of GSPCs into nestin-positive neural stem cells in vitro. GSPC colonies were placed into neural stem cell differentiation medium. After 3 d in differentiation medium, immunochemical assays indicated that most of these cells were positive for nestin staining. Some bipolar cells were observed. (Magnification: 40×.)
Fig. 6.

Presumptive GSPCs expressed both a neural lineage biomarker and a human-specific antigen when transplanted into the ex vivo OTS culture system. (A) Schematic diagram of OTS system. (Inset) Cells were labeled with transient lentiviral-GFP vector and underwent live cell imaging. Nuclei (B; Hoechst dye, blue), human nuclei (C; human nuclei-specific antibody, green), and human nestin (D; human nestin-specific antibody, red) are shown. (E) Overlay of B, C, and D (note that there is no GFP labeling in B, C, and D). (Scale bars: 100 μm.)
Neural Differentiation Potential of GSPCs in Vivo.
To assess the in vivo developmental potentials of GSPCs further, we injected the GSPCs into SCID mice, a well-established assay testing for teratoma formation arising from pluripotent stem cell populations. In all three independent experiments, formation of teratoma-like tissue cysts was observed 5 wk postinjection. Morphologically, the neoplastic tissues were evident in these tissue cysts, suggesting the developmental plasticity of the GSPCs. Total RNA and proteins extracted from the tissue cysts were subjected to semiquantitative RT-PCR and Western blot analysis to determine p53 mRNA and protein expression, respectively. As shown in Fig. 7, both p53 RNA and protein were detected in GSPCs and tissue cysts generated from GSPCs. Immunohistochemical analysis of the tissue sections derived from these tissue cysts indicated that they contained early neural lineage cell types, staining positively for human-specific S-100 protein. S-100 protein is normally present in cells derived from the neural crest (22). The tendency of GSPCs to develop toward ectodermal fates suggests that suppression of p53 only increases the plasticity of the keratinocytes. The suppression of p53 in keratinocytes may increase the plasticity of these cells for their development toward a neural lineage (from which the keratinocytes are derived). The keratinocyte-derived GSPCs were not reprogrammed to the pluripotent stages more primitive than ectodermal origin. This GSPC feature of p53 expression and limited pluripotency may prove to be an asset for clinical application of GSPCs because it may represent a safety barrier restricting tumorigenicity of GSPCs.
Fig. 7.

In vivo characterization of GSPCs in the SCID mouse. (A) p53 mRNA was detected in the GSPC-derived immature teratoma-like cell masses. Total RNA from liver cells, teratoma-like GSPCs, and HeLa cells was extracted and converted into cDNA. p53 was detected in liver cells and teratoma-like GSPCs but not in HeLa cells. This result indicates that GSPCs expresses p53, whereas the cancer cell line HeLa does not express p53 at the mRNA level. (B) Western blot analyses indicate that p53 protein was detected in both keratinocytes and GSPCs. Cells were grown as described in Materials and Methods, and the lysate proteins were subjected to SDS/PAGE. The samples were subjected to immunoblot analyses as described in Materials and Methods. Equivalent amounts of total cell lysates were loaded to all the lanes. Note that anti-p53 mAb specifically binds to p53 protein, whereas anti-GAPDH antibody specifically recognizes GAPDH. These results further demonstrate that p53 is present at the protein level. (C) Western blot analyses show that p53 protein was detected in the GSPC-derived immature teratoma-like cell masses. Equivalent amounts of total cell lysates were loaded to all the lanes. Note that anti-p53 mAb specifically binds to p53 protein, whereas anti-α-actin antibody specifically recognizes α-actin. These results further demonstrate that p53 is present at the protein level. (D) Immature epithelial cells grow in solid nests in the right and left lower corners of this image. These cells are larger than the surrounding mature stromal cells (mostly fibroblasts and scattered groups of endothelial cells), with increased nuclear-to-cytoplasmic ratios and prominent nucleoli. Mitosis is present in the left lower corner within the area of the immature epithelial cells, suggesting rapid growth of the cells. Areas of necrosis (not shown) were appreciated. Foci of degenerated skeletal muscle cells with abundant pink cytoplasm are present in the center and upper portion of the image (H&E stain). (Magnification: 40×.) (E) Weak cytoplasmic positive staining of the immature epithelial cells with S-100, a neural marker, is observed, suggesting neural differentiation of these cells. (Magnification: 40×.)
Discussion
Our study suggests that transient suppression of p53 expression increases the pluripotency potential of somatic cells, such as keratinocytes. In our study, down-regulation of p53 triggers activation of Oct4 expression in mouse somatic cells (MEFs). Oct4 is a transcriptional factor required in early development and for the propagation of undifferentiated ES cells in culture (23, 24). The transcription factors Oct4, Sox2, and Nanog create a gene expression program necessary for pluripotency and self-renewal through regulating the Wnt pathway. This is accomplished via Tcf3, providing key developmental signals capable of tilting the balance of core regulatory circuitry of ES cells between pluripotency and differentiation (23). The finding that Oct4 expression is triggered by p53 suppression is in agreement with our previous studies supporting the hypothesis that the p53 gene network regulates cellular pluripotency (17). Genes often act in a network to regulate cell behavior. The perturbation of the p53 gene network may lead to the expression of various transcription factors, including Oct4. This finding could explain why p53 suppression leads to increased reprogramming efficiency as described in other studies.
In our study, we observed that GSPCs generated nestin-positive cells in vitro and in the ex vivo brain slice systems. These results demonstrated the neural lineage development potential of GSPCs for research and clinical application. In the in vivo studies, RT-PCR with human p53-specific primers and Western blot analysis with human p53 antibodies indicated that p53 was reexpressed at the mRNA and protein level in the tissue cysts from three independent experiments. Immunohistochemical analysis of these tissue cysts suggested that GSPCs differentiated toward ectodermal tissues positive for S-100. Because the GSPCs were generated from keratinocytes, which are themselves derived from ectodermal tissue, our directed suppression of p53 may not be sufficient to escape the ectodermal restriction of the developmental potential of these cells.
Multiple lines of independent studies agree with our study and show that p53 suppression increases the plasticity of cells in development. Yamanaka and colleagues (25) reported that using p53 KO mouse cells and retroviruses containing the four most commonly used transcription factors (Oct4, Sox2, Klf4, and c-Myc) generated up to 20% efficiency of reprogramming embryonic fibroblasts to form full-fledged iPSC populations. Deng's group (6) reported that knocking down p53 with RNAi in the presence of the transcription factor Utf1 increases the efficiency of generating iPSCs. Giorgetti et al. (26) successfully obtained iPSCs with only two transgenes (Oct4 and Sox2) when p53 levels were reduced. Hochedlinger's group (4) improved iPSC reprogramming rates by suppressing p53 or Ink4a/Arf, a locus that encodes two tumor suppressors that interact with p53. Other studies showed that the elimination of the p53 antitumor pathway improved the reprogramming of cells from older organisms as well as that of cells with heavy DNA damage or truncated telomeres (7, 27, 28).
Mutant p53 is a well-studied oncogene, and many cancer cells lack WT p53 function. Furthermore, “gain of function” characteristics associated with mutant p53 have been well documented (29). WT p53 protein contributes to G1 cell cycle arrest to allow DNA repair and, as a last resort, even the induction of apoptosis after DNA damage (30). The p53 KO mouse and rat lines have been generated to study the function of p53 in tumorigenesis (31, 32). Both the p53−/− mouse and rat develop normally, but the p53−/− mouse is more susceptible to cancer development (31, 33, 34). Our previous study (17) and our current study agree with these studies and may suggest a broader role for p53; the p53 gene network may prove pivotal in the regulation of self-renewal through regulation of the transcription factor Oct4 (and possibly others). Absence of functional p53 promotes enhanced cellular plasticity and flexibility in cell cycle progression. This may inadvertently enhance susceptibility and survivability to the accumulation of genetic alterations, thereby promoting cancer development. There is a therapeutical window of opportunity for the p53-regulated plasticity during development, which is consistent with all pluripotent stem cell potential for stem cell therapies (35).
Reprogramming with somatic cell nuclear transfer (36) and iPSC technology (1, 2) demonstrated that differentiated somatic cells can be reprogrammed into pluripotent cells. This finding provides novel approaches to treat degenerative diseases with patient-specific stem cells. Autologous pluripotent cell populations are needed as precursors for the generation of sufficient patient-specific cells required for realistic cell therapy strategies. However, these pluripotent cells also carry the risk for teratoma formation. Populations exhibiting sufficient plasticity to switch to a desired lineage, but not a fully pluripotent lineage, may prove more suitable for clinically applicable cell therapy. By suppressing p53 expression with shRNA in keratinocytes, we generated stem cell-like colonies (GSPCs) and subsequently differentiated the GSPCs toward neural lineages. These GSPCs were expanded in vitro for more than 10 passages while exhibiting normal karyotypes and can be differentiated into neural lineages. Healthy keratinocytes are readily obtained from most patients to serve as precursors for GSPCs.
In summary, our study suggests that perturbation of the p53 gene network may lead to expression of transcription factors and produce cells susceptible for lineage switch. We also demonstrated that p53 suppression offers a novel approach for regenerative medicine by producing expandable patient-specific GSPCs to be used after lineage switching.
Materials and Methods
Cell Culture.
All cultures were maintained in a humidified incubator at 37 °C, with 5% CO2/95% air. HEK293T cells were cultured with DMEM containing 10% FBS, 1% penicillin-streptomycin (Invitrogen). Fibroblast cells from Oct4-GFP transgenic mice were also cultured in DMEM. The human primary keratinocyte cell line was purchased from the American Type Culture Collection, and the cells were propagated and subcultured according to the provider's instructions.
Suppression of p53 by shRNA.
The Thermo Scientific Open Biosystems Lentiviral shRNAmir Library was used in this study to generate p53 shRNA according to the manufacturer's instructions. Briefly, five constructs containing hnRNAmir targeting different regions of human p53, TRCN0000003753, TRCN0000003754, TRCN0000003755, TRCN0000003756, and TRCN0000010814 were ordered from Open Biosystems. Replication-incompetent viral particles were produced using the Trans-Lentiviral packaging system (Open Biosystems), and the viral titers were determined. These viral particles were transduced into human primary keratinocytes to knock down p53 expression in human cells. For mouse Oct4-GFP MEFs, we used a shRNA vector from Open Biosystems targeted to the mouse p53 (NM_001127233). Viral production and transduction were carried out as instructed by the manufacturer.
Generation of GSPCs.
Human keratinocytes were transduced with viral particles containing shRNA targeted to knock down p53 expression. After transduction, cells were transferred to MEFs with stem cell medium. After 5 wk of culturing, hESC-like colonies were expanded. These colonies were then tested with AP assays for their pluripotency. AP-positive colonies were further selected against possible MEF cell contamination by DNA fingerprinting, and the clones of human keratinocyte lineage were called GSPCs.
Differentiation of GSPCs Toward Neural Lineage.
Proliferating cultures of GSPCs were differentiated by replacing 50% of the stem cell medium daily with 1:1 differentiation base medium supplemented with 1% FBS, 100 nM all-trans-retinoic acid (Sigma), 20 ng/mL BDNF (Chemicon), and 20 ng/mL neurotrophin-3 (Chemicon). Cells that differentiated into neural stem cells were identified by positive immunostaining of nestin, a biomarker for neural stem cells (37).
Immunostaining of Differentiated GSPCs.
After culturing GSPCs in the media for neural stem cell differentiation for 1 wk, cells were fixed for 20 min at room temperature with 4% paraldehyde. The cells were then washed three times with 1× PBS and permeabilized with 1× PBS containing 0.1% Triton for 20 min at room temperature. After rinsing cells three times with 1× PBS, cells were blocked with 1× PBS containing 10% goat serum for 1 h at room temperature. Cells were then washed three times with 1× PBS and stained with mouse–anti-human nestin mAb (LSBio) for 1 h at room temperature. After washing with 1× PBS three times, cells were incubated with the secondary antibody (phycoerythrin-conjugated anti-mouse IgG, 1:200; Santa Cruz) in 1× PBS containing 1% goat serum at 37 °C for 45 min. The unbound secondary antibodies were removed by washing three times in freshly made 1× PBS containing 0.5% Tween 20. The cells were then washed briefly in 1× PBS before mounting for fluorescent microscopy analysis.
Karyotyping of GSPCs.
GSPC confluent cultures were incubated in growth medium (GM) containing 10 ng/mL colcemid for 18 h. The cells were then lifted and centrifuged. Pellets were osmotically shocked with 0.075 M KCl and fixed with 3:1 methanol/glacial acetic acid. Standard cytogenetic analysis was performed by the Genetics Center (Orange, CA) (38).
Quantitative RT-PCR.
Total RNA was extracted with the SV Total RNA isolation system (Promega) as instructed from GSPC or H9 colonies, treated with DNA-free (Ambion), and reverse-transcribed with the Sensiscript RT Kit (Qiagen). The cDNAs were quantified with Nanodrop (Thermo Scientific), and PCR was performed with Crimson Taq DNA Polymerase (NE Biolabs). The p53-specific primers were as follows: forward primer, 5′-TTGGATCCATGTTTTGCCAACTGGCC-3′, and reverse primer, 5′-TTGAATTCAGGCTCCCCTTTCTTGCG-3′. The thermal cycle profile was 95 °C for 5 min, 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min for a total of 35 cycles (39).
OTSs.
OTSs were prepared from 4- to 9-d-old neonatal mice (CD-1; Charles River Laboratory) by modifying our previously published procedure (20). Briefly, mice were euthanized and sterilized with alcohol for cardiac perfusion. After decapitation, brains were placed in sterile-filtered slice culture medium on ice as described previously. Each brain was then embedded in 4% LMT agarose (Invitrogen), and slices ranging from 200–300 μm in thickness were generated with a vibratome (VT100S; Leica). Stem cells (500 ∼1 × 104 cells in 0.2 μL of PBS) labeled with a lentiviral construct carrying GFP were deposited onto the surface of the OTSs. Engrafted cells were cultured for 1 wk on OTSs. The GFP-labeled cells were observed and recorded daily using an Olympus IX-70 fluorescent microscope. For immunohistochemical analysis, at day 7, cells were fixed in freshly prepared 4% formaldehyde (from paraformaldehyde) in Dulbecco's PBS, pH 7.4. The formaldehyde was removed and washed with PBS, and saturated with 30% sucrose in PBS, followed by cryostat sectioning. The sections were rinsed in 0.1 mM Tris-buffered saline (TBS), rinsed, blocked in TBST (0.1 mM TBS, 5% donkey serum, 0.25% Triton X-100) for 1 h, and incubated overnight with primary antibodies (anti-human nestin antibody or anti-human nuclei antibody; Chemicon) in TBST at 4 °C. The sections were then rinsed and incubated with fluorescently labeled secondary antibodies [aminomethylcoumarin acetate (AMCA), AMCA, fluorescein isothiocyanate (FITC), FITC, Cy2, Cy3, and Rhodamine red-X (RRX); RRX; 1:250; Jackson Immunoresearch) overnight at 4 °C. Removal of the secondary antibodies was followed by additional washes. Some sections were stained with Hoechst 33342 (Sigma) before being rinsed and mounted. Images were taken on an Olympus IX-70 fluorescent microscope using objectives with a magnification of 4×, 10×, 20×, 40×, or 100×.
Teratoma Formation in SCID Mice.
GSPCs grown on MEFs were collected by collagenase treatment and injected into the hind limb muscle of 6-wk-old immunocompromised SCID-beige mice (Charles River Laboratory). We injected 100 μL of cell suspension (∼1 × 106 cells for each injection) into one of the hind limbs. Three mice were injected for three independent experiments. After ∼5 wk, teratoma-like tissue cysts were dissected and fixed in freshly prepared 4% formaldehyde (from paraformaldehyde) in Dulbecco's PBS, pH 7.4. The formaldehyde was removed and washed with PBS. Samples were embedded in paraffin and processed with H&E staining as well as S100 staining at the Histology Laboratory of the University of Southern California, School of Medicine, Los Angeles, CA (see below for details).
Western Blot Analysis.
We have previously described the Western blot analysis used in this study (40–43). The sample was subjected to immunoblot analysis with anti-p53 antibody (gift from Yun Yen, City of Hope Beckman Research Institute, Duarte, CA; BD Bioscience), anti-α-actin mAb (gift from Shan Li, City of Hope Beckman Research Institute, Duarte, CA; Sigma), and anti-GAPDH (gift from Yu-ting Lin, City of Hope Beckman Research Institute, Duarte, CA; Ambion). Equivalent amounts of total cell lysates were loaded into all the lanes.
Immunohistochemical Analysis.
Formalin-fixed 5-μm sections were taken from paraffin-embedded tissue cyst specimens and mounted on poly-l-lysine–coated slides. The slides were deparaffinized in xylene and washed with 100% ethanol, followed by rehydration in 95% ethanol; 3% hydrogen peroxide in absolute methanol was used to quench endogenous peroxidase. Antigen retrieval was done using citrate buffer (pH 6) and microwaving for 30 min, followed by cooling at room temperature for 20 min (44). The slides were then blocked with normal horse serum for 20 min, incubated for 1 h with primary antibody (monoclonal mouse–anti-human S-100; CellMarque Corporation), and then washed with PBS (Vector Laboratories, Inc.) for 10 min. They were then incubated with biotinylated horse anti-mouse secondary antibody, followed by avidin-biotin conjugation (Vector Laboratories, Inc.). Chromogen of 0.03% diaminobenzidine was then applied, with hematoxylin counterstaining. Negative controls consisting of diluents with no antibody and positive controls of human melanoma with heterogeneous immunoreactivity were used in all experiments.
Acknowledgments
We thank Brent A. Dethlefs; Philip H. Schwartz, PhD; John H. Weiss, MD, PhD; Hong Zhen Yin, MD; Alexander Stover; Vic Keschrumrus; Long Vu; Yuan Han Teh; and Shan Li, PhD, for their technical support. This study was supported by the Children's Hospital of Orange County (CHOC) Children's Foundation, CHOC Neuroscience Institute, Austin Ford Tribute Fund, W. M. Keck Foundation, Grant R21CA134391 from the National Institutes of Health, and Grant AW 0852720 from the National Science Foundation.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
References
- 1.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 3.Hochedlinger K, Plath K. Epigenetic reprogramming and induced pluripotency. Development. 2009;136:509–523. doi: 10.1242/dev.020867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Utikal J, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:1145–1148. doi: 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi K, Ichisaka T, Yamanaka S. Identification of genes involved in tumor-like properties of embryonic stem cells. Methods Mol Biol. 2006;329:449–458. doi: 10.1385/1-59745-037-5:449. [DOI] [PubMed] [Google Scholar]
- 6.Zhao Y, et al. Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell. 2008;3:475–479. doi: 10.1016/j.stem.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Marión RM, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–1153. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawamura T, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–1144. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lane DP. Worrying about p53. Curr Biol. 1992;2:581–583. doi: 10.1016/0960-9822(92)90154-3. [DOI] [PubMed] [Google Scholar]
- 10.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 11.Zuckerman V, Wolyniec K, Sionov RV, Haupt S, Haupt Y. Tumour suppression by p53: The importance of apoptosis and cellular senescence. J Pathol. 2009;219:3–15. doi: 10.1002/path.2584. [DOI] [PubMed] [Google Scholar]
- 12.Subramanian S, et al. Genome-wide transcriptome analyses reveal p53 inactivation mediated loss of miR-34a expression in malignant peripheral nerve sheath tumours. J Pathol. 2010;220:58–70. doi: 10.1002/path.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lozano G. Mouse models of p53 functions. Cold Spring Harb Perspect Biol. 2010;2:a001115. doi: 10.1101/cshperspect.a001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puca R, Nardinocchi L, Givol D, D'Orazi G. Regulation of p53 activity by HIPK2: Molecular mechanisms and therapeutical implications in human cancer cells. Oncogene. 2010;29:4378–4387. doi: 10.1038/onc.2010.183. [DOI] [PubMed] [Google Scholar]
- 15.Gottlieb E, Vousden KH. p53 regulation of metabolic pathways. Cold Spring Harb Perspect Biol. 2010;2:a001040. doi: 10.1101/cshperspect.a001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong H, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong JF, et al. Gene regulation networks related to neural differentiation of hESC. Gene Expr. 2007;14:23–34. doi: 10.3727/000000007783991781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong JF, Weiner LP, Jin Y, Lu W, Taylor CR. A real-time pluripotency reporter for human stem cells. Stem Cells Dev. 2010;19:47–52. doi: 10.1089/scd.2008.0363. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 20.Li SC, Loudon WG. A novel and generalizable organotypic slice platform to evaluate stem cell potential for targeting pediatric brain tumors. Cancer Cell Int. 2008;8:9. doi: 10.1186/1475-2867-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li SC, et al. Stem cell engineering for treatment of heart diseases: Potentials and challenges. Cell Biol Int. 2009;33:255–267. doi: 10.1016/j.cellbi.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Moore BW, Perez VJ, Gehring M. Assay and regional distribution of a soluble protein characteristic of the nervous system. J Neurochem. 1968;15:265–272. doi: 10.1111/j.1471-4159.1968.tb11610.x. [DOI] [PubMed] [Google Scholar]
- 23.Cole MF, Johnstone SE, Newman JJ, Kagey MH, Young RA. Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev. 2008;22:746–755. doi: 10.1101/gad.1642408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyer LA, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanatsu-Shinohara M, et al. Generation of pluripotent stem cells from neonatal mouse testis. Cell. 2004;119:1001–1012. doi: 10.1016/j.cell.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 26.Giorgetti A, et al. Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell Stem Cell. 2009;5:353–357. doi: 10.1016/j.stem.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–1139. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marion RM, et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. 2009;4:141–154. doi: 10.1016/j.stem.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 29.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 30.Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci USA. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donehower LA, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 32.Tong C, Li P, Wu NL, Yan Y, Ying QL. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature. 2010;467:211–213. doi: 10.1038/nature09368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kemp CJ, Donehower LA, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell. 1993;74:813–822. doi: 10.1016/0092-8674(93)90461-x. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto M, et al. p53 knockout mice (-/-) are more susceptible than (+/-) or (+/+) mice to N-methyl-N-nitrosourea stomach carcinogenesis. Carcinogenesis. 2000;21:1891–1897. doi: 10.1093/carcin/21.10.1891. [DOI] [PubMed] [Google Scholar]
- 35.Li SC, Han YP, Dethlefs BA, Loudon WG. Therapeutic window, a critical developmental stage for stem cell therapies. Curr Stem Cell Res Ther. 2010;5:297–303. [PMC free article] [PubMed] [Google Scholar]
- 36.Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KH. Viable offspring derived from fetal and adult mammalian cells. Nature. 1997;385:810–813. doi: 10.1038/385810a0. [DOI] [PubMed] [Google Scholar]
- 37.Gilyarov AV. Nestin in central nervous system cells. Neurosci Behav Physiol. 2008;38:165–169. doi: 10.1007/s11055-008-0025-z. [DOI] [PubMed] [Google Scholar]
- 38.Schwartz PH, et al. Isolation and characterization of neural progenitor cells from post-mortem human cortex. J Neurosci Res. 2003;74:838–851. doi: 10.1002/jnr.10854. [DOI] [PubMed] [Google Scholar]
- 39.Li S, Kirov I, Klassen HK, Schwartz PH. Characterization of stem cells using reverse transcriptase polymerase chain reaction. In: Loring JF, Wesselschmidt RL, Schwartz PH, editors. Human Stem Cell Manual: A Laboratory Guide. New York: Elsevier; 2007. pp. 127–148. [Google Scholar]
- 40.Ju X, et al. Akt1 governs breast cancer progression in vivo. Proc Natl Acad Sci USA. 2007;104:7438–7443. doi: 10.1073/pnas.0605874104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li S, Couet J, Lisanti MP. Src tyrosine kinases, Galpha subunits, and H-Ras share a common membrane-anchored scaffolding protein, caveolin. Caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. J Biol Chem. 1996;271:29182–29190. doi: 10.1074/jbc.271.46.29182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li S, et al. Evidence for a regulated interaction between heterotrimeric G proteins and caveolin. J Biol Chem. 1995;270:15693–15701. doi: 10.1074/jbc.270.26.15693. [DOI] [PubMed] [Google Scholar]
- 43.Flanagan LA, Ziaeian B, Palmer T, Schwartz PH. Immunocytochemical analysis of stem cells. In: Loring JF, Wesselschmidt RL, Schwartz PH, editors. Human Stem Cell Manual: A Laboratory Guide. New York: Elsevier; 2007. pp. 108–126. [Google Scholar]
- 44.Shi SR, Chaiwun B, Young L, Cote RJ, Taylor CR. Antigen retrieval technique utilizing citrate buffer or urea solution for immunohistochemical demonstration of androgen receptor in formalin-fixed paraffin sections. J Histochem Cytochem. 1993;41:1599–1604. doi: 10.1177/41.11.7691930. [DOI] [PubMed] [Google Scholar]