

Abstract
Cartilage oligomeric matrix protein (COMP) is a pentameric ∼524 kDa multidomain extracellular matrix protein and is the fifth member of the thrombospondin family. COMP is abundantly expressed in proliferating and hypertrophic chondrocytes of the growth plate, articular cartilage, synovium, tendon, and ligament. The spatial localization of COMP highlights its importance in the phenotypes of pseudoachondroplasia (PSACH) and multiple epiphyseal dysplasia (MED), COMP disorders that are characterized by disproportionate short stature, brachydactyly, scoliosis, early-onset osteoarthritis, and joint hypermobility. In this study, the role of COMP in ligament was investigated with a series of cell attachment assays using ligament cells binding to COMP. A dose-dependent cell attachment activity was found, which was inhibited by a peptide containing the SFYVVMWK amino acid sequence derived from the globular C-terminal domain of COMP. This activity was independent of the recently described RGD-dependent attachment activity. Function-blocking antibodies to CD47 and αVβ3 integrin reduced cell attachment to COMP, implicating the participation of these cell surface molecules in COMP cell binding. Immunofluorescence studies showed that cell attachment to COMP induced the formation of lamellae containing F-actin microspikes associated with fascin. We propose that COMP promotes cell attachment via two independent mechanisms involving cell surface CD47 and αVβ3 integrin and that a consequence of cell attachment to COMP is the specific induction of fascin-stabilized actin microspikes.
Keywords: COMP, Extracellular matrix, Cell adhesion, Signaling, Actin cytoskeleton
Introduction
Cartilage oligomeric matrix protein (COMP) is an extracellular matrix (ECM) protein that resides in cartilage, tendon, and other connective tissues. Despite the attention that it has received in the contexts of human skeletal dysplasias and different forms of arthritis, its physiological function remains poorly understood. In this report, we explore cell binding and possible signaling functions of COMP.
Cartilage oligomeric matrix protein is a multidomain ECM protein that forms a disulfide-bonded homopentamer of ∼524 kDa [1, 2]. It is comprised of an amino-terminal pentamerization domain, four type 2 (epidermal growth factor (EGF)-like) domains, eight type 3 calcium-binding (calmodulin-like) domains, and a globular carboxyl-terminal domain [3]. This modular structure places COMP within the thrombospondin (TSP) family of proteins and it is also known as thrombospondin 5 (TSP-5). COMP is abundantly expressed in articular cartilage as well as in proliferating and hypertrophic chondrocytes of the growth plate [4], suggesting its importance in endochondral ossification and articular cartilage. COMP is also expressed in tendon and ligament, in synovial cells and in dermal fibroblasts. Interaction studies have shown that COMP binds to a plethora of ECM proteins, including matrilin-1 and collagens I, II, III, and IX [5–7]. In addition to acting as a bridging molecule within the ECM, COMP can also bind to chondrocytes and rheumatoid arthritis synovial fibroblasts via interactions with integrins [8–10], an activity that suggests a possible signaling function for COMP.
Mutations in the COMP gene cause a well-described spectrum of osteochondrodysplasias from severe PSACH (MIM#177170) to mild MED (MIM#132400) [11, 12]. PSACH is characterized by moderate short stature, short limbs and small hands, scoliosis, loose joints, genu varum and genu valgum, and early-onset osteoarthrosis [13]. MED is characterized by mild short-limb short stature, prominent joints, small irregular epiphyses, and mild metaphyseal abnormalities, however, the vertebrae are usually unaffected. Electron microscopy of chondrocytes from PSACH patients reveals inclusions in the rough endoplasmic reticulum (RER) that have a characteristic lamellar appearance [14]. The RER inclusions contain COMP as well as type IX collagen and matrilin-3 [15]. The surrounding ECM is generally poorly assembled, although type II collagen and aggrecan synthesis and secretion do not appear to be disrupted.
The mutations found in both PSACH and MED are located in exons encoding two of the domains in the COMP protein [16, 17]. Most are found within the calcium-binding type 3 domains and <15% are found in the carboxyl-terminal domain. Mutations that have been described include point mutations that cause single amino acid substitutions and small in-frame deletions or duplications and are predicted to affect the overall conformation and structure of COMP [16, 18, 19]. The disease phenotype may be a composite of different disturbances related to the synthesis of abnormal COMP [20]. For example, alteration of the structure of the type 3 domain decreases the ability of COMP to bind calcium and leads to a less packed conformation of the protein [21–24] and, although COMP accumulates in the RER of PSACH patients, in cell culture some pentameric COMP is secreted [14, 25, 26]. Decreased levels of conformationally unstable COMP in the ECM may produce the observed disorganized ECM in PSACH. Also mutations have been described within the regions of COMP that bind to other ECM proteins such as collagens type I and IX [7] fueling speculation that altered interactions between mutant COMP and the ECM may also reduce the structural integrity of the ECM. The accumulation of structurally defective COMP in the RER inclusions has also been proposed as one causative factor in the disease and has been shown in cell culture models to lead to decreased cell proliferation and increased apoptosis [27, 28].
The role of COMP binding directly to cells and the effect of mutations on this binding is only recently beginning to be characterized. The binding of COMP to chondrocytes was first documented in 1984 by DiCesare et al. [8]. More recently, COMP has been shown to interact with chondrocytes and rheumatoid arthritis synovial fibroblasts, with the RGD triplet of the fourth type 3 domain proposed to interact with either α5β1 and αVβ3 integrins, respectively, and, in the latter case, with a concomitant induction of galectin-3 protein [9, 10]. The carboxyl-terminal domain of COMP has also been suggested to contain a cell interaction site [13, 29]. This site has been suggested to include the amino acid sequence FYVVMWK that is conserved throughout the TSP family, and peptides and recombinant proteins including this motif from TSP-1 have been shown to bind to cell surface receptors [30, 31].
Materials and methods
Cell culture and proteins
Recombinant mouse COMP was expressed using COMP cDNA constructs in a mammalian expression vector transfected into 293 cells as described previously [32]. COMP was purified from media using Talon Ni2+ resin (Clontech Laboratories, Mountain View, CA, USA) and eluted with phosphate buffer containing 150 mM imidazole. Recombinant protein was dialyzed into Tris-buffered saline and quantified using the BCA protein assay kit (Pierce, Rockford, IL, USA). Normal human ligament cells were obtained from the International Skeletal Dysplasia Registry cell collection and cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (FCS), 1% penicillin/streptomycin, and 1% sodium pyruvate. Chondrocytes were isolated from human fetal cartilage that was digested overnight in DMEM containing 0.3% collagenase at 37°C. The cells were collected by centrifugation and resuspended in DMEM supplemented with 10% FCS, 1% penicillin/streptomycin, and 1% sodium pyruvate. The suspension was allowed to settle, and cells in the supernatant were removed, counted, and seeded at 3×106 cells per 25 cm2 flask. Chondrocytes were grown for 24 h and then used for cell attachment assays.
Cell attachment assays
Cell attachment assays were performed according to the well-defined methodologies [33]. COMP was diluted into phosphate-buffered saline with cations (PBS+) (Invitrogen, Carlsbad, CA, USA), and 100 μl aliquots of known concentration was pipetted onto wells of a 96-well plate (Costar, Corning Inc., Acton, MA, USA). Human fibronectin (Sigma, St. Louis, MO, USA) was used as a positive control. The plate was incubated at 4°C overnight or for 4 h at room temperature. The unbound protein ligand was removed by aspiration, and unbound plastic was blocked for 1.5 h with 200 μl 10 mg/ml heat-denatured bovine serum albumin (BSA) in PBS+. Cultured ligament cells or primary chondrocytes at 70% confluence were trypsinized and quenched with 30 ml FCS-media. Cells were collected by centrifugation at 290×g and resuspended in media containing 25 mM HEPES, 2.5 g/l glucose. Cell density was counted and adjusted to 1 × 105/ml for ligament cells and 2 × 105/ml for chondrocytes. The cells were allowed to recover for 10 min at 37°C. In the peptide inhibition studies, cells were incubated at this point with the wild-type (VV) peptide, KSSFYVVMWKQK, or control (GG) peptide, KSSFYGGMWKQK, at 50 μg/ml or with GRGDTP or GRADSP peptides at 1 mg/ml. Lysine residues were incorporated at the ends of the peptide to increase solubility. In a second parallel set of attachment assays, the wild-type peptide was replaced with a more COMP-specific sequence, KSSFYVVMWKQME (VV-2). Known amounts of cells were added to unblocked wells in triplicate to generate a standard curve to estimate values of cell attachment. 100 μl aliquots of cells was pipetted into wells containing the test proteins. After incubation at 37°C, 5% CO2 for 40 min, test wells were washed once in PBS+ to remove unbound cells, and the attached cells were fixed with 100 μl aliquots of 5% (w/v) glutaraldehyde in PBS+. Wells were washed three times with 300 μl phosphate-buffered saline without cations (PBS), and cells were stained with 100 μl of 0.1% (w/v) crystal violet in 0.2 M MES, pH 6, for 1 h at room temperature. Wells were washed five times with 300 μl dH2O to remove excess crystal violet, and 100 μl 10% acetic acid was added for 10 min to solubilize the stain which was measured at 570 nm using a plate reader (Molecular Devices Corp., Sunnyvale, CA, USA). In the peptide inhibition studies, an F test was performed to determine sample variance and then significance was determined using a two-tailed t test, assuming equal variances (VV-2 vs. untreated cells; VV vs. GRGDTP + VV) or assuming unequal variance (GRGDTP vs. GRGDTP + VV). Cell attachment assays were also repeated using function-blocking antibodies to CD47 (clone B6H12, Santa Cruz Biotechnologies Inc., Santa Cruz, CA, USA) and αVβ3 integrin (MAB1976, Chemicon International, Temecula, CA, USA) or normal mouse IgG (Sigma, St. Louis, MO, USA). The cells were preincubated with these antibodies at 25 μg/ml for 10 min prior to plating on the COMP protein as described above. The significance of the inhibitory effect of the function-blocking antibodies was determined using a two-tailed t test, assuming unequal variances, according to a preliminary F test to determine sample variance.
Human COMP purification
Media from human ligament cells was collected and dialyzed into 20 mM Tris, 70 mM NaCl, and pH 7.6 in the presence of protease inhibitors. The media was filtered over a 0.45-μm vacuum filter and applied over a HiPrep 16/10 DEAE FF column (Amersham Biosciences, Piscataway, NJ, USA). Fractions were eluted with 20 mM Tris, 1 M NaCl, pH 7.6 and those containing COMP were dialyzed into 20 mM Tris, 150 mM NaCl, pH 7.6 and run over a HiTrap 5 ml Heparin HP column to remove TSP and fibronectin. The flow-through was dialyzed into 20 mM Tris, pH 7.6, and then applied to a HiTrap 1 ml Q HP column. Eluted fractions that contained COMP were lyophilized to concentrate the sample and resuspended into 20 mM Tris, 140 mM NaCl, pH 7.6, for size exclusion chromatography on a 16/100 column containing Superdex 200 resin. Fractions from each step were analyzed for COMP by western blotting. BioRad protean III apparatus (BioRad, Hercules, CA, USA) was used for SDS-PAGE using standard protocols. Primary antibodies for COMP were a kind gift from Vladimir Vilim (Institute of Rheumatology, Prague, Czech Republic). Secondary antibodies were supplied from Jackson ImmunoResearch Laboratories, West Grove, PA, USA.
Immunofluorescence
Cell attachment assays were repeated using 50 nM solutions of COMP, fibronectin, and TSP-1 aliquoted onto 8-well cell culture slides. In order to determine whether fascin was required for actin microspike formation, cell attachment assays were repeated with cells treated with DMSO or 50 nM phorbol 12-myristate 13-acetate (PMA) in DMSO prior to attachment to COMP. Following cell attachment, slides for actin visualization were fixed in 4% formaldehyde in PBS+ for 20 min. The slides were washed in PBS+ and blocked in 0.5% BSA in PBS+ for 30 min. The cells were then stained with Alexa Fluor 488-conjugated phalloidin (Invitrogen, Carlsbad, CA, USA) for 1 h at room temperature, washed in 0.5% BSA in PBS+, and then mounted in VectaShield mounting solution containing DAPI (Vector Laboratories Inc., Burlingame, CA, USA) for nuclei staining. Slides for fascin visualization were fixed in methanol for 20 min, blocked as described above, and then stained with an anti-fascin antibody (Affinity Bioreagents Inc., Golden, CO, USA) and an Alexa Fluor 488-conjugated secondary antibody.
Results
Ligament cells and chondrocytes attach to COMP
The attachment of ligament cells directly to plates coated with recombinant mouse COMP was first tested. Cell attachment was ligand-concentration dependent, with minimal attachment at 0.1 μg/ml COMP, and appearing to plateau at between 10–25 μg/ml. An average of about 50% of ligament cells attached to 25 μg/ml COMP (Fig. 1a). This is compared with about 70% of cells attaching to fibronectin and ∼10% cells attaching to heat-denatured BSA. In order to determine whether COMP has a cell attachment role in cartilage, a major site of COMP expression, human fetal articular chondrocytes, was isolated from distal femur and used in cell attachment assays. Chondrocytes from three different fetuses were tested and the data pooled. Figure 1b shows that freshly prepared chondrocytes bound to recombinant COMP at similar levels to ligament cells.
Fig. 1.
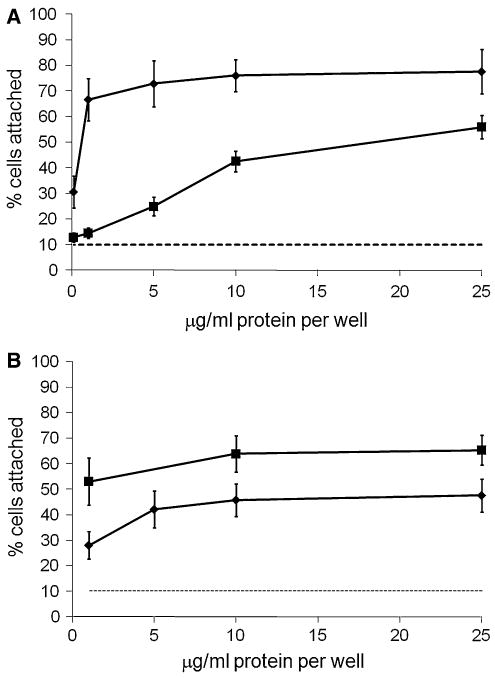
Recombinant COMP promotes ligament cell attachment. a Ligament cells were plated on wells coated with recombinant COMP (filled squares) or fibronectin (filled diamonds) at various concentrations as described in “Materials and methods” section. Cell adhesion is expressed as a percentage of total number cells aliquoted per well. b Pooled data for articular chondrocytes isolated from three different fetal sources and plated on wells coated with recombinant COMP (filled squares) or fibronectin (filled diamonds) at varying concentrations as described in “Materials and methods” section. Background binding to 10 mg/ml BSA (- - -). Results are means ±2 × SEM; a n ≥ 21; b n ≥ 9
Cell attachment to COMP is inhibited by peptides derived from the COMP carboxyl-terminal domain
In order to investigate whether the conserved FYVVMWQ peptide sequence was involved in cell attachment, plates were coated with protein as before but the ligament cells were pre-incubated with either wild-type peptide (VV) consisting of 12 amino acids surrounding the core conserved sequence or a negative control peptide (GG) that had the central valine pair replaced with a glycine pair. Pre-incubation of ligament cells with the VV peptide reduced COMP-mediated cell attachment by about 50% (Fig. 2a). Pre-incubation of ligament cells with the GG peptide had minimal effect on cell attachment to COMP, indicating that the consecutive valine residues within the FYVVMWK amino acid sequence derived from the carboxyl-terminal domain of COMP are involved, at least in part, in inhibiting cell attachment to COMP. The inhibitory effect of the VV peptide on cell attachment to COMP was also tested on freshly prepared chondrocytes. Similar to ligament cells, chondrocyte cell attachment to COMP was inhibited to about half the levels of normal chondrocyte attachment by the VV peptide (Fig. 2b), but the GG peptide had an insignificant effect on cell attachment.
Fig. 2.
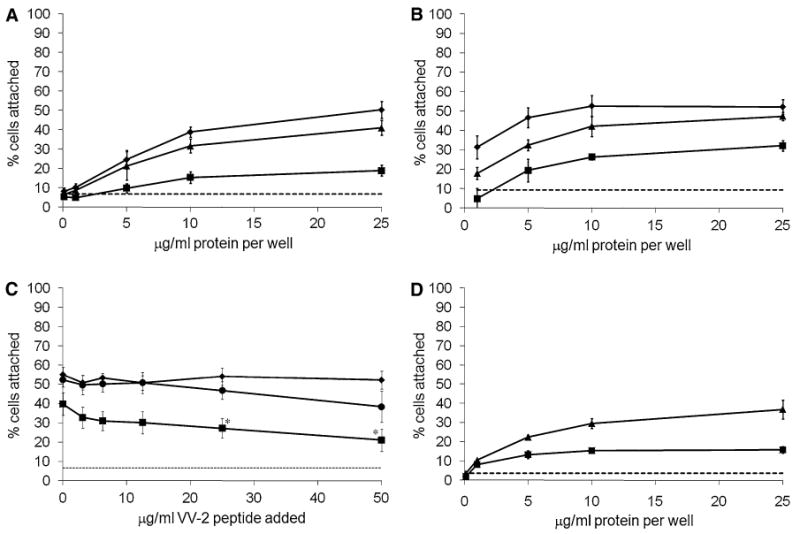
The VV-containing peptide specifically inhibits cell attachment to COMP. a Ligament cells were preincubated with carrier alone or inhibitory peptides at 50 μg/ml and were plated on wells coated with recombinant COMP at various concentrations as described in “ Materials and methods” section. Carrier alone (filled diamonds); GG peptide (filled triangles); VV peptide (filled squares); background binding to 10 mg/ml BSA (- - -). b Freshly prepared articular chondrocytes were preincubated with carrier alone (filled diamonds), GG peptide (filled triangles), or VV peptide (filled squares) (at 50 μg/ml) and plated on wells coated with recombinant COMP. c Ligament cells were preincubated with increasing concentrations of the VV-2 peptide and were plated on wells coated with equimolar concentrations of recombinant COMP (filled squares), fibronectin (filled diamonds) or TSP-1 (filled circles) as described in “Materials and methods” section. Background binding to 10 mg/ml BSA (- - -). d Ligament cell attachment to purified human COMP, with (filled squares) or without (filled triangles) preincubation with the VV peptide; background binding to 10 mg/ml BSA (- - -). Cell adhesion is expressed as a percentage of total number cells aliquoted per well. Results are means ±2 × SEM; a n ≥ 9; b n ≥ 3; c n ≥ 9, * P < 0.005, significant difference comparing VV-2 treated cells with untreated cells; d n ≥ 3
A second peptide, VV-2, containing the more COMP-specific sequence SSFYVVMWQME equally inhibited ligament cell attachment to COMP. The inhibitory effect was significant at 25–50 μg/ml (∼35 μM, P < 0.00005), but also appeared to have an effect at as little as 3.125 μg/ml (∼2 μM) (Fig. 2c). In comparison, the VV-2 peptide had no significant effect on cell attachment to fibronectin. The VV-2 peptide was also tested for inhibition of cell attachment to TSP-1 plated at an equivalent molar concentration to COMP and only had significant inhibitory effect compared to control cell attachment at the maximum concentration of 50 μg/ml (P < 0.05).
Ligament cells attach to human COMP
In order to determine whether human COMP had similar cell attachment properties as compared with recombinant mouse COMP, human COMP was purified from media of ligament primary cell cultures using a series of separations by anion-exchange and size exclusion chromatography. SDS-PAGE and silver stain were used to visualize the purity of the preparation, which was confirmed by mass spectrometry (data not shown). Ligament cells attached to purified human COMP at almost identical levels to cells attaching to recombinant mouse COMP (Fig. 2d), and the VV peptide also inhibited this cell attachment by about 50%. These data demonstrate that human COMP acts similarly to recombinant mouse COMP in promoting human ligament cell attachment.
Ligament cell attachment to COMP involves two independent mechanisms
A recent study has described the involvement of the RGD triplet within a calcium-binding motif of COMP in the attachment of chondrocytes to COMP under certain conditions [9]. In order to investigate whether the RGD triplet was involved in ligament cell attachment to COMP, cell attachment assays were repeated as before with cells pre-incubated with either GRGDTP or GRADSP peptides before attachment to the protein ligand on the plates. Pre-incubation of ligament cells with the GRGDTP peptide significantly decreased cell attachment to COMP (Fig. 3), whereas pre-incubation with the GRADSP peptide had a much lesser effect. In order to test the efficacy of the GRGDTP peptide under the assay conditions, ligament cell attachment to fibronectin was also tested with either the GRGDTP or GRADSP peptide. Ligament cells pre-incubated with the GRGDTP peptide showed decreased cell attachment to fibronectin compared to untreated cells or cells pre-incubated with GRADSP. In order to determine if cell attachment to COMP involved two independent mechanisms, cells were pre-incubated with both the GRGDTP and VV peptides combined. Cell attachment was significantly decreased to levels below those of cells pre-incubated with GRGDTP or VV alone (P = 0.004 or P = 0.002, respectively) (Fig. 3), indicating that the effect of each peptide was additive and implying that cell attachment to COMP independently involved cell attachment sites inhibited by the RGD- and the SFYVVMWK-containing peptides.
Fig. 3.
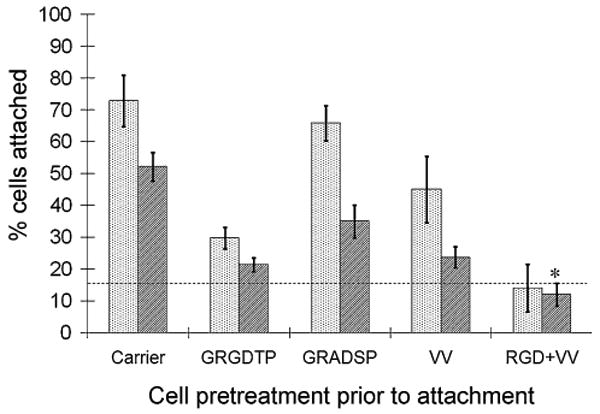
Ligament cell attachment to COMP is independently inhibited by either the VV-containing peptide or an RGD peptide. Ligament cells were preincubated with carrier alone or inhibitory peptides (GRGDTP or GRADSP at 1 mg/ml; VV at 50 μ/ml) and were plated on wells coated with recombinant COMP or fibronectin (10 μg/ml or 1 μg/ml, respectively) as described in “Materials and methods” section. Dotted bars fibronectin, shaded bars COMP, background binding to 10 mg/ml BSA (- - -). Results are means ±2 × SEM; n ≥ 12. * P < 0.005, significant difference compared with inhibition of cell attachment by GRGDTP or VV alone
CD47 (integrin associated protein) and αVβ3 integrin are involved in cell attachment to COMP
Function-blocking antibodies to CD47 and αVβ3 integrin (B6H12 and MAB1976, respectively) were used to determine if either of these proteins were involved in the ligament cell attachment to COMP. Pre-incubating ligament cells with the CD47 antibody at different concentrations demonstrated significant inhibition of cell attachment from 2.5 to 25 μg/ml (P < 0.005, at each concentration) indicating that the function-blocking CD47 antibody has a dose-dependent inhibitory effect on cell attachment (Fig. 4a). Cell attachment was reduced to background levels at the maximum concentration of 25 μg/ml (Fig. 4a, b). In contrast, the αVβ3 antibody had a more modest effect on cell attachment, reducing cell attachment to about 65% of the normal levels at the maximum concentration of 25 μg/ml (P < 0.05; Fig. 4b). Normal mouse IgG had no significant inhibitory effect on cell attachment to COMP.
Fig. 4.
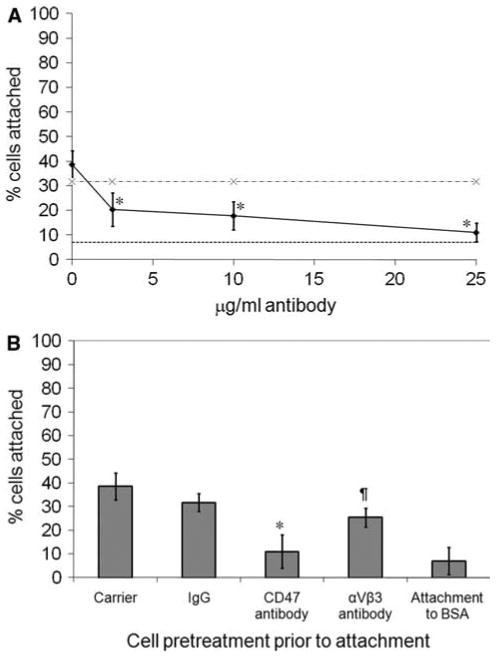
Ligament cell attachment to COMP is inhibited by function-blocking antibodies to CD47 and αVβ3 integrin. a Ligament cells were preincubated with different concentrations of CD47 antibody and plated on wells coated with 26.2 μg/ml (50 nM) COMP as described in “Materials and methods” section. Cells preincubated with normal mouse IgG (·−·); cells preincubated with CD47 antibody (filled diamonds); background binding to 10 mg/ml BSA (- - -). b Ligament cells were preincubated with carrier alone, function-blocking antibody or normal mouse IgG at 25 μ/ml and were plated on wells coated with recombinant COMP (26.2 μg/ml) as described previously. a, b Results are means ±2 × SEM; n ≥ 18. a, b * P < 0.005, significant difference comparing CD47 antibody inhibition with control untreated cells. b ¶ P < 0.05, significant difference comparing αVβ3 antibody inhibition with control untreated cells
Cell attachment to COMP induces the formation of fascin-associated actin microspikes
Immunofluorescence studies were used to observe changes in the actin cytoskeleton arising from cells attaching to COMP. In contrast to the spread cell morphology and organized actin microfilaments of cells attached to fibronectin, phalloidin labeling demonstrated that cells attached to COMP had multiple finger-like protrusions with actin microspikes radiating from the protrusion tips (Fig. 5a, c). In order to determine if fascin-1 was involved in the formation of the actin microspikes, cells were fixed and labeled in parallel with antibodies to fascin-1. As alternative fixation methods were required to visualize actin or fascin (involving formaldehyde or methanol, respectively), co-labeling of both within the same field was not possible. Figure 5b and d shows that in contrast to the diffuse distribution of fascin-1 seen throughout the cells attached to fibronectin, fascin was also localized in microspikes radiating from the tips of the cellular protrusions that formed in cells attached to COMP. Cells were preincubated with phorbol 12-myristate 13-acetate (PMA) to determine if fascin was critical for the formation of the COMP-induced actin microspikes. PMA is a potent protein kinase C activator that induces phosphorylation of fascin and inhibits its actin-bundling activity, reducing the number of actin microspikes able to form [34]. Immunofluorescence showed that cells treated with PMA were able to attach to COMP, but the fascin localization to the microspikes was no longer apparent and fascin was redistributed to a diffuse perinuclear region (compare Fig. 6a with b). Phalloidin labeling demonstrated that the change in fascin distribution was accompanied by a significant decrease in actin microspikes, with a concomitant redistribution of actin into a unique radial appearance at the plasma membrane (compare Fig. 6c with d).
Fig. 5.
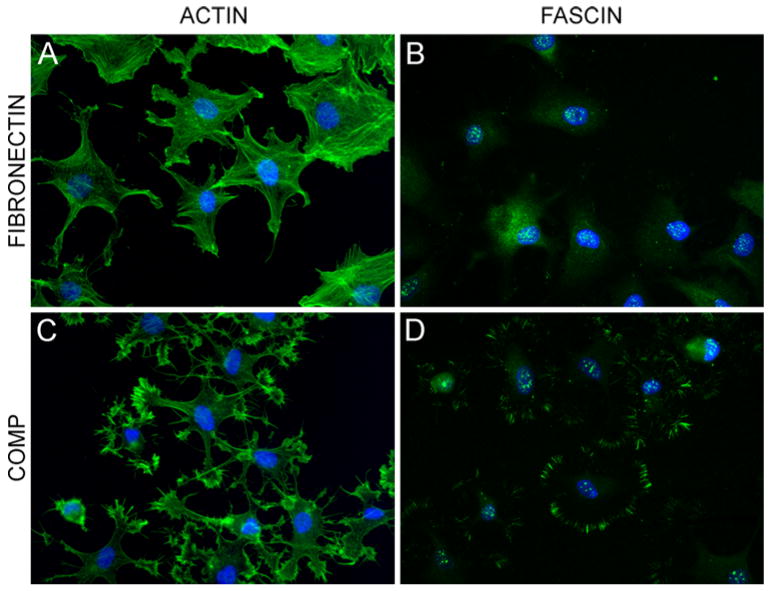
Ligament cell attachment to COMP induces actin and fascin microspikes. Ligament cells were attached to equimolar concentrations of fibronectin (panels a, b), or COMP (panels c, d) as described in “Materials and methods” section. Cells were stained with phalloidin-conjugated FITC for actin (panels a, c) and with an antibody against fascin (panels c, d). All cells were stained with DAPI for nuclei
Fig. 6.
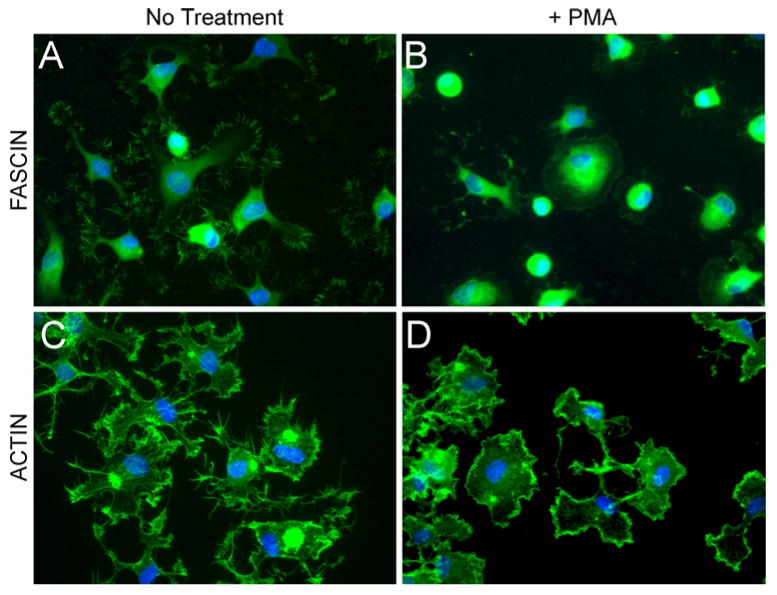
COMP-induced actin microspikes are dependent on fascin. Ligament cells were pretreated with PMA for 15 min to induce phosphorylation of fascin and then attached to equimolar concentrations of COMP as described in “ Materials and methods” section. Cells were stained with an antibody against fascin (panels a, b) or phalloidin-conjugated FITC for actin (panels c, d). All cells were stained with DAPI for nuclei
Discussion
The studies presented here have shown that COMP promotes attachment of cultured cells isolated from ligament, a major site of COMP expression, and that cell attachment involves CD47 on the cell surface. A consequence of cell attachment to COMP is the formation of fascin-stabilized actin microspikes that radiate from multiple lamellipodia elaborated by the cell upon COMP binding. Further experiments have also shown that COMP promotes attachment of freshly isolated chondrocytes, contributing to previously published data from attachment studies of rheumatoid arthritis synovial fibroblasts, and monolayer cultured chondrocytes suggest that COMP may provide a structural scaffold for cell attachment in the cartilage ECM [9, 10].
The carboxyl-terminal domain of COMP contains an amino acid sequence, FYVVMWK, which is conserved throughout the TSP family. There has been speculation that COMP may bind to cells via this motif [13, 29], and indeed, previous studies have shown that peptides and recombinant proteins derived from the TSP-1 carboxyl-terminal domain promote cell attachment and spreading [30, 31, 35, 36]. In this study, peptides containing the SFYVVMWK sequence specifically inhibited cell attachment to COMP, whereas a control peptide containing -GG- in place of -VV-, previously reported to have no cell attachment activity [30, 31], had a much reduced effect. Furthermore, the COMP-derived peptide had only a modest effect on cell attachment to TSP-1. The data demonstrate that the SFYVVMWK peptide plays a major role in inhibiting cell attachment to COMP and, more specifically, that the consecutive valine residues are critical for this activity. Dual inhibition with an RGD peptide in addition to the VV-containing peptide reduced cell attachment to COMP to background levels, suggesting that COMP promotes cell attachment via two different receptor sites that are independently inhibited by RGD- or SFYVVMWK-containing peptides. Attempts to further characterize the roles of the RGD and SFYVVMWK motifs in cell attachment by site-directed mutagenesis of these sites were unsuccessful since as is the case for many COMP mutations, amino acid changes at these sites lead to ER retention of the mutated protein. It is also worth noting that the potential physiological importance of COMP-mediated attachment of cells via the RGD motif is strengthened by the fact that this motif in COMP is conserved across all species with the exception of rat COMP, wherein the arginine is replaced by phenylalanine (QGD). Recent work on other proteins containing integrin binding RGD sites has demonstrated that the arginine within the RGD motif is not obligatory and that substitution of this residue can modulate integrin-binding specificity [37].
CD47 has been shown to consistently bind to peptides containing the RFYVVMWK amino acid sequence of TSP-1 [30, 31, 38]. In the study presented here, a function-blocking antibody to CD47 was used to competitively decrease cell attachment to COMP to background levels. A second function-blocking antibody to αVβ3 integrin reduced cell attachment to COMP by 35%. In conjunction with the peptide inhibition studies, the data are compatible with a model in which COMP promotes cell attachment primarily via cell surface CD47 in a mechanism that can be inhibited by the SFYVVMWK peptide and via αVβ3 integrin in a mechanism that can be inhibited by the RGD peptide. However, a direct interaction between COMP and CD47 has yet to be demonstrated, so it is possible that additional proteins may also be involved.
This model complements published data indicating that COMP interacts with αVβ3 [10] or α5β1 [9] integrins. Neidhart et al. also demonstrated an increase in intracellular galectin-3 subsequent to cell attachment to COMP, implicating COMP as a protein able to cause downstream signaling effects in cells. The immunofluorescence studies presented here demonstrate that another downstream effect of COMP binding is the induction of multiple lamellipodia with fascin-dependent actin microspikes. While the elucidation of the immediate consequences of this microspike assembly for ligament cell function was beyond the scope of this manuscript, it is worth noting that the actin cytoskeletal organization is strikingly distinct from that observed in cells attached to fibronectin and is reminiscent of fascin-dependent actin microspikes seen in skeletal myoblasts attached to TSP-1 [39, 40]. This finding demonstrates conservation of signaling activities among TSP family members. Together these data are consistent with the hypothesis that members of the TSP family may act as “matricellular modulators of cell function” [41, 42].
Recently, a crystal structure of a fragment of TSP-1 containing three type 3 repeats and the carboxyl-terminal domain has been determined [43]. Kvansakul et al. suggest that both the RGD site and the RFYVVMWK site are of limited availability for interactions with integrins or CD47, respectively; however, previous studies with the TSP-1 RFYVVMWK sequence and the current study certainly implicate the (R/S)FYVVMWK motif in promoting cell attachment via the CD47/IAP receptor [31, 36, 38]. The discrepancy between the TSP-1 crystal structure and the data presented here needs to be clarified and the precise mechanism by which COMP promotes cell attachment warrants further investigation. Dynamic modulation of the structure could allow sites apparently unavailable based on the crystal structure to become exposed and functional.
Cartilage oligomeric matrix protein has traditionally been described as a structural ECM protein whose importance is highlighted by its involvement in two inherited skeletal diseases, PSACH and MED [11]. The structural mutations in COMP that produce these phenotypes disrupt the structure of the cartilage ECM and alter interactions between COMP and other ECM molecules [5, 7]. In addition, the accumulation of structurally defective COMP within the RER may also contribute to phenotypic expression by disrupting the normal progression of chondrocytes from the resting through hypertrophic stages within the growth plate [27]. Combined with recent studies showing that COMP is involved in cell attachment [9, 10], the data presented here suggest the intriguing possibility that COMP plays a previously understated role as an anchor for cells in the ECM. It is possible that decreased interactions of mutant COMP with chondrocytes and ligament cells may also contribute to the epiphyseal and metaphyseal abnormalities and ligamentous laxity observed in PSACH, either by a reduced amount of COMP in the ECM available for cell attachment or by a reduced ability of structurally abnormal COMP to bind to cells [14, 16, 20].
In summary both ligament cells and chondrocytes have been shown to attach to COMP. COMP promotes cell attachment via two independent mechanisms involving CD47 and integrins at the cell surface. A downstream effect of cell attachment to COMP is the formation of lamellipodia and the reorganization of actin into fascinstabilized microspikes. The data highlight the importance of COMP as a structural scaffold for cell attachment in the ECM of ligament and cartilage, and suggest that COMP plays an important role in matrix-cell signaling affecting the organization of the cytoskeleton. Future studies can now be directed toward understanding the signaling cascades that are initiated upon adhesion of cells to COMP and determining the consequences of fascin microspike assembly to cell function.
Acknowledgments
This study was supported in part by grants from the National Institutes of Health (AR493139 and HD22657 (DHC) and the Shriners Hospitals for Children (WAH). Daniel Cohn is a Winnick Family Clinical Research Scholar supported by the General Clinical Research Center at Cedars-Sinai under NCRR grant M01-RR00425. Paul Holden was a recipient of a Shriners Research Fellowship Award. We thank Deborah Krakow for assistance with obtaining cartilage samples, Vincent Funari for help with statistical analysis, Vladimir Vilim for COMP monoclonal antibodies, and MaryAnn Weiss and David Eyre for analyzing COMP purity by mass spectrometry.
Contributor Information
Matthew J. Rock, Medical Genetics Institute, Steven Spielberg Pediatric Research Center, Los Angeles, CA 90048, USA
Paul Holden, Research Center, Shriners Hospitals for Children, Portland, OR 97239, USA; Department of Molecular and Medical Genetics, Oregon Health & Science University, Portland, OR 97239, USA.
William A. Horton, Email: wah@shcc.org, Research Center, Shriners Hospitals for Children, Portland, OR, 97239, USA; Department of Molecular and Medical Genetics, Oregon Health & Science University, Portland, OR 97239, USA.
Daniel H. Cohn, Medical Genetics Institute, Steven Spielberg Pediatric Research Center, Los Angeles, CA 90048, USA; Department of Human Genetics, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA; Department of Pediatrics, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA
References
- 1.Hedbom E, Antonsson P, Hjerpe A, Aeschlimann D, Paulsson M, et al. Cartilage matrix proteins. An acidic oligomeric protein (COMP) detected only in cartilage. J Biol Chem. 1992;267:6132–6136. [PubMed] [Google Scholar]
- 2.Oldberg A, Antonsson P, Lindblom K, Heinegard D. COMP (cartilage oligomeric matrix protein) is structurally related to the thrombospondins. J Biol Chem. 1992;267:22346–22350. [PubMed] [Google Scholar]
- 3.Newton G, Weremowicz S, Morton CC, Copeland NG, Gilbert DJ, et al. Characterization of human and mouse cartilage oligomeric matrix protein. Genomics. 1994;24:435–439. doi: 10.1006/geno.1994.1649. [DOI] [PubMed] [Google Scholar]
- 4.DiCesare PE, Morgelin M, Carlson CS, Pasumarti S, Paulsson M. Cartilage oligomeric matrix protein: isolation and characterization from human articular cartilage. J Orthop Res. 1995;13:422–428. doi: 10.1002/jor.1100130316. [DOI] [PubMed] [Google Scholar]
- 5.Holden P, Meadows RS, Chapman KL, Grant ME, Kadler KE, et al. Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J Biol Chem. 2001;276:6046–6055. doi: 10.1074/jbc.M009507200. [DOI] [PubMed] [Google Scholar]
- 6.Mann HH, Ozbek S, Engel J, Paulsson M, Wagener R. Interactions between the cartilage oligomeric matrix protein and matrilins. Implications for matrix assembly and the pathogenesis of chondrodysplasias. J Biol Chem. 2004;279:25294–25298. doi: 10.1074/jbc.M403778200. [DOI] [PubMed] [Google Scholar]
- 7.Thur J, Rosenberg K, Nitsche DP, Pihlajamaa T, Ala-Kokko L, et al. Mutations in cartilage oligomeric matrix protein causing pseudoachondroplasia and multiple epiphyseal dysplasia affect binding of calcium and collagen I, II, and IX. J Biol Chem. 2001;276:6083–6092. doi: 10.1074/jbc.M009512200. [DOI] [PubMed] [Google Scholar]
- 8.DiCesare PE, Mörgelin M, Mann K, Paulsson M. Cartilage oligomeric matrix protein and thrombospondin 1. Purification from articular cartilage, electron microscopic structure, and chondrocyte binding. Eur J Biochem. 1994;223:927–937. doi: 10.1111/j.1432-1033.1994.tb19070.x. [DOI] [PubMed] [Google Scholar]
- 9.Chen FH, Thomas AO, Hecht JT, Goldring MB, Lawler J. Cartilage oligomeric matrix protein/thrombospondin 5 supports chondrocyte attachment through interaction with integrins. J Biol Chem. 2005;280:32655–32661. doi: 10.1074/jbc.M504778200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neidhart M, Zaucke F, von Knoch R, Jungel A, Michel BA, et al. Galectin-3 is induced in rheumatoid arthritis synovial fibroblasts after adhesion to cartilage oligomeric matrix protein. Ann Rheum Dis. 2005;64:419–424. doi: 10.1136/ard.2004.023135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Briggs MD, Chapman KL. Pseudoachondroplasia and multiple epiphyseal dysplasia: mutation review, molecular interactions, and genotype to phenotype correlations. Hum Mutat. 2002;19:465–478. doi: 10.1002/humu.10066. [DOI] [PubMed] [Google Scholar]
- 12.Cohn DH, Briggs MD, King LM, Rimoin DL, Wilcox WR, et al. Mutations in the cartilage oligomeric matrix protein (COMP) gene in pseudoachondroplasia and multiple epiphyseal dysplasia. Ann N Y Acad Sci. 1996;785:188–194. doi: 10.1111/j.1749-6632.1996.tb56258.x. [DOI] [PubMed] [Google Scholar]
- 13.Cohn DH. COMP and pseudoachondroplasia. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn errors of development. Oxford University Press; New York: 2003. pp. 970–976. [Google Scholar]
- 14.Delot E, Brodie SG, King LM, Wilcox WR, Cohn DH. Physiological and pathological secretion of cartilage oligomeric matrix protein by cells in culture. J Biol Chem. 1998;273:26692–26697. doi: 10.1074/jbc.273.41.26692. [DOI] [PubMed] [Google Scholar]
- 15.Vranka J, Mokashi A, Keene DR, Tufa S, Corson G, et al. Selective intracellular retention of extracellular matrix proteins and chaperones associated with pseudoachondroplasia. Matrix Biol. 2001;20:439–450. doi: 10.1016/s0945-053x(01)00148-2. [DOI] [PubMed] [Google Scholar]
- 16.Kennedy J, Jackson GC, Barker FS, Nundlall S, Bella J, et al. Novel and recurrent mutations in the C-terminal domain of COMP cluster in two distinct regions and result in a spectrum of phenotypes within the pseudoachondroplasia—multiple epiphyseal dysplasia disease group. Hum Mutat. 2005;25:593–594. doi: 10.1002/humu.9342. [DOI] [PubMed] [Google Scholar]
- 17.Mabuchi A, Manabe N, Haga N, Kitoh H, Ikeda T, et al. Novel types of COMP mutations and genotype-phenotype association in pseudoachondroplasia and multiple epiphyseal dysplasia. Hum Genet. 2003;112:84–90. doi: 10.1007/s00439-002-0845-9. [DOI] [PubMed] [Google Scholar]
- 18.Briggs MD, Hoffman SM, King LM, Olsen AS, Mohrenweiser H, et al. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat Genet. 1995;10:330–336. doi: 10.1038/ng0795-330. [DOI] [PubMed] [Google Scholar]
- 19.Hecht JT, Nelson LD, Crowder E, Wang Y, Elder FF, et al. Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat Genet. 1995;10:325–329. doi: 10.1038/ng0795-325. [DOI] [PubMed] [Google Scholar]
- 20.Dinser R, Zaucke F, Kreppel F, Hultenby K, Kochanek S, et al. Pseudoachondroplasia is caused through both intra- and extracellular pathogenic pathways. J Clin Invest. 2002;110:505–513. doi: 10.1172/JCI14386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H, Deere M, Hecht JT, Lawler J. Cartilage oligomeric matrix protein is a calcium-binding protein, and a mutation in its type 3 repeats causes conformational changes. J Biol Chem. 2000;275:26538–26544. doi: 10.1074/jbc.M909780199. [DOI] [PubMed] [Google Scholar]
- 22.Hou J, Putkey JA, Hecht JT. Delta 469 mutation in the type 3 repeat calcium binding domain of cartilage oligomeric matrix protein (COMP) disrupts calcium binding. Cell Calcium. 2000;27:309–314. doi: 10.1054/ceca.2000.0125. [DOI] [PubMed] [Google Scholar]
- 23.Kleerekoper Q, Hecht JT, Putkey JA. Disease-causing mutations in cartilage oligomeric matrix protein cause an unstructured Ca2+ binding domain. J Biol Chem. 2002;277:10581–10589. doi: 10.1074/jbc.M109944200. [DOI] [PubMed] [Google Scholar]
- 24.Maddox BK, Mokashi A, Keene DR, Bachinger HP. A cartilage oligomeric matrix protein mutation associated with pseudoachondroplasia changes the structural and functional properties of the type 3 domain. J Biol Chem. 2000;275:11412–11417. doi: 10.1074/jbc.275.15.11412. [DOI] [PubMed] [Google Scholar]
- 25.Schmitz M, Becker A, Schmitz A, Weirich C, Paulsson M, et al. Disruption of extracellular matrix structure may cause pseudoachondroplasia phenotypes in the absence of impaired cartilage oligomeric matrix protein secretion. J Biol Chem. 2006;281:32587–32595. doi: 10.1074/jbc.M601976200. [DOI] [PubMed] [Google Scholar]
- 26.Weirich C, Keene DR, Kirsch K, Heil M, Neumann E, et al. Expression of PSACH-associated mutant COMP in tendon fibroblasts leads to increased apoptotic cell death irrespective of the secretory characteristics of mutant COMP. Matrix Biol. 2007;26:314–323. doi: 10.1016/j.matbio.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 27.Hecht JT, Hayes E, Haynes R, Cole WG. COMP mutations, chondrocyte function and cartilage matrix. Matrix Biol. 2005;23:525–533. doi: 10.1016/j.matbio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 28.Hecht JT, Makitie O, Hayes E, Haynes R, Susic M, et al. Chondrocyte cell death and intracellular distribution of COMP and type IX collagen in the pseudoachondroplasia growth plate. J Orthop Res. 2004;22:759–767. doi: 10.1016/j.orthres.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 29.Briggs MD, Mortier GR, Cole WG, King LM, Golik SS, et al. Diverse mutations in the gene for cartilage oligomeric matrix protein in the pseudoachondroplasia-multiple epiphyseal dysplasia disease spectrum. Am J Hum Genet. 1998;62:311–319. doi: 10.1086/301713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao AG, Frazier WA. Identification of a receptor candidate for the carboxyl-terminal cell binding domain of thrombospondins. J Biol Chem. 1994;269:29650–29657. [PubMed] [Google Scholar]
- 31.Gao AG, Lindberg FP, Dimitry JM, Brown EJ, Frazier WA. Thrombospondin modulates alpha v beta 3 function through integrin-associated protein. J Cell Biol. 1996;135:533–544. doi: 10.1083/jcb.135.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holden P, Keene DR, Lunstrum GP, Bachinger HP, Horton WA. Secretion of cartilage oligomeric matrix protein is affected by the signal peptide. J Biol Chem. 2005;280:17172–17179. doi: 10.1074/jbc.M411716200. [DOI] [PubMed] [Google Scholar]
- 33.Bax DV, Bernard SE, Lomas A, Morgan A, Humphries J, et al. Cell adhesion to fibrillin-1 molecules and microfibrils is mediated by alpha 5 beta 1 and alpha v beta 3 integrins. J Biol Chem. 2003;278:34605–34616. doi: 10.1074/jbc.M303159200. [DOI] [PubMed] [Google Scholar]
- 34.Adams JC, Clelland JD, Collett GD, Matsumura F, Yamashiro S, et al. Cell-matrix adhesions differentially regulate fascin phosphorylation. Mol Biol Cell. 1999;10:4177–4190. doi: 10.1091/mbc.10.12.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barazi HO, Li Z, Cashel JA, Krutzsch HC, Annis DS, et al. Regulation of integrin function by CD47 ligands. Differential effects on alpha vbeta 3 and alpha 4beta1 integrin-mediated adhesion. J Biol Chem. 2002;277:42859–42866. doi: 10.1074/jbc.M206849200. [DOI] [PubMed] [Google Scholar]
- 36.McDonald JF, Dimitry JM, Frazier WA. An amyloid-like C-terminal domain of thrombospondin-1 displays CD47 agonist activity requiring both VVM motifs. Biochemistry. 2003;42:10001–10011. doi: 10.1021/bi0341408. [DOI] [PubMed] [Google Scholar]
- 37.Lu X, Davies J, Lu D, Xia M, Wattam B, Shang D, Sun Y, Scully M, Kakkar V. The effect of the single substitution of arginine within the RGD tripeptide motif of a modified neurotoxin dendroaspin on its activity of platelet aggregation and cell adhesion. Cell Commun Adhes. 2006;13:171–183. doi: 10.1080/15419060600726183. [DOI] [PubMed] [Google Scholar]
- 38.Frazier WA, Gao AG, Dimitry J, Chung J, Brown EJ, et al. The thrombospondin receptor integrin-associated protein (CD47) functionally couples to heterotrimeric Gi. J Biol Chem. 1999;274:8554–8560. doi: 10.1074/jbc.274.13.8554. [DOI] [PubMed] [Google Scholar]
- 39.Adams JC, Lawler J. Cell-type specific adhesive interactions of skeletal myoblasts with thrombospondin-1. Mol Biol Cell. 1994;5:423–437. doi: 10.1091/mbc.5.4.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams JC, Schwartz MA. Stimulation of fascin spikes by thrombospondin-1 is mediated by the GTPases Rac and Cdc42. J Cell Biol. 2000;150:807–822. doi: 10.1083/jcb.150.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams JC. Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- 42.Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Invest. 2001;107:929–934. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kvansakul M, Adams JC, Hohenester E. Structure of a thrombospondin C-terminal fragment reveals a novel calcium core in the type 3 repeats. EMBO J. 2004;23:1223–1233. doi: 10.1038/sj.emboj.7600166. [DOI] [PMC free article] [PubMed] [Google Scholar]