

Summary
The haemostatic system is heavily involved in the host response to infection. A number of host haemostatic factors, notably plasminogen and fibrinogen have been reported to bind and interact with various bacterial proteins. This review summarises the roles of host haemostatic factors such as plasminogen, factor V and fibrinogen in host defence against group A streptococcus infection and discusses the potential of targeting the host haemostatic system for therapeutic intervention against infectious diseases.
Keywords: Group A Streptococcus, haemostatic factor, antimicrobial therapy
Introduction
Infectious diseases have been major health threats throughout human history. The discovery and broad use of antibiotics over the last century have greatly decreased the mortality from bacterial infectious diseases and increased overall life span. However, antibiotic resistance had been on the rise recently and is threatening to derail the progress made in eradicating infectious diseases [1]. Even in the 1940s, when antibiotic use was restricted to the clinic, resistance had already emerged [2]. While nearly all major pathogens have developed resistance to antibiotics, the developmental pipeline for new antibiotics is nearly dry [3–5]. A key to developing urgently needed new therapies is likely to lie in understanding the molecular mechanisms of the pathogen host interaction.
An infection begins when a bacterium gains entry into the host through one of many routes, including the skin, nasopharynx, and the gastrointestinal or urogenital tract. Bacterial pathogens produce an array of surface and secreted proteins that assist in eluding host defence mechanisms. While the role of the innate and adaptive immune systems has been well studied, the key contribution of the host haemostatic system is often overlooked. Disruption of haemostatic balance is a hallmark of infection/inflammation, and severe thrombosis and/or bleeding are common complications of bacterial infection [6,7]. In addition, a number of recent studies have demonstrated that the haemostatic system is much more than a passive bystander in the host response to bacterial invasion [7–11].
Plasminogen in the host/pathogen interaction during group A streptococcal (GAS) infection
The central mediator of the fibrinolytic system is plasminogen, a proteinase zymogen that is cleaved by plasminogen activators into its active form, plasmin, which subsequently degrades the fibrin thrombus. Host plasminogen is exploited by a number of bacterial pathogens. Borrlelia burgdorferi, the agent causing lyme disease, binds plasminogen, facilitating its conversion into plasmin by host-specific plasminogen activators [12,13]. Pla, a protease produced by Yersinia pestis is a potent human plasminogen activator. A Y. pestis pla knockout strain demonstrates markedly diminished virulence in a subcutaneous infection model in the mouse [14]. Pla is also required for primary pneumonic plaque [15] and for development of the bubonic form of the disease [16]. Staphylococcus aureus also produces a plasminogen activator, staphylokinase, which has been shown to enhance bacterial resistance to phagocytosis by interacting with HNPs (human neutrophil peptide) [17,18]. It was reported that staphylokinase could block the plasminogen activation by endogenous activators uPA and tPA, leading to further deterioration of the fibrinolytic system during infection [19].
Perhaps the best-characterised bacterial plasminogen activator is streptokinase (SK), which is produced by the common human pathogen, group A streptococcus (GAS) (Streptococcus pyogenes). S. pyogenes can cause a variety of human infections from mild conditions, such as tonsillitis, scarlet fever and impetigo to life-threatening invasive diseases, such as streptococcal toxic shock-like syndrome and necrotising fasciitis [20]. S. pyogenes is estimated to cause over 700 million cases of infection globally each year [21]. Tillett and Garner first demonstrated that lysis of a fibrin clot by an isolate from a human streptococcal infection. However, isolates from veterinary streptococcal infections failed to exhibit fibrinolytic activity against human fibrin [22]. SK was subsequently shown to be responsible for this fibrinolytic activity [23]. SK can form a complex with human plasminogen, which can hydrolytically activate other plasminogen molecules into plasmin. Furthermore, this complex is also resistant to the inhibitor α2–antiplasmin [24]. In addition, fibrinogen can also bind the streptokinase-plasminogen complex to form a trimolecular complex, which can capture and activate circulating plasminogen [25,26].
Over the years, a number of streptococci have been shown to produce streptokinases that are host-specific plasminogen actors [27]. Taken together with the observation that GAS is a strictly human pathogen, the SK/plasminogen interaction was proposed to play a role in the host-specificity of GAS infection [28]. Khil et al coinjected purified human plasminogen and GAS subcutaneously into mice, and observed a dramatic increase in mortality and skin lesion area [29]. Further insight has come from studies in a humanised mouse model for GAS infection [30]. We established a transgenic (Tg+) mouse line producing ~17% of the level human plasminogen in mouse plasma of that observed in normal human plasma. Tg+ mice demonstrated significantly increased mortality to GAS infection compared to wild type mice, suggesting that plasminogen plays a critical role in GAS pathogenicity. To further test whether the transgene expressed human plasminogen functioned in this GAS infection model through its interaction with SK, Tg+ and littermate wild type controls were infected with a GAS strain in which the SK gene had been inactivated, essentially abolishing the increased mortality observed in Tg+ mice infected with SK+ strains [30].
In addition to the plasminogen activator, SK, several GAS surface proteins have been identified as plasminogen receptors that bind plasminogen directly [31]. Plasminogen-binding group A streptococcal M-like protein (PAM), binds human plasminogen/plasmin with high affinity and is expressed from the same gene locus (emm) as M protein in the M53 GAS strain [32,33]. PAM-related protein (Prp) is another high affinity plasminogen binding M protein isolated from a severely invasive GAS infection strain (S. pyogene emm 98.1) [34]. M protein is a multifunctional virulence factor and also interacts extensively with the host haemostatic system.
Human plasminogen Tg+ mice also demonstrated markedly increased mortality compared to littermate controls following infection with a PAM-positive GAS strain, which expressed low level of SK. When these mice were infected with a PAM-negative GAS strain, which also expressed low level of SK, the increased susceptibility in Tg+ was also largely abolished [30]. In similar studies, Prp was mutated to have attenuated capacity for plasminogen binding and surface plasmin accumulation. The mutant GAS strain demonstrated a significantly decreased virulence in human plasminogen Tg+ mice in comparison to the isogenic wild type strain [35]. The ability of PAM/Prp to bind plasminogen/plasmin on the GAS surface provides another mechanism to exploit host fibrinolytic system for bacterial invasion.
In addition to PAM and Prp, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [36] and α-enolase (SEN) [37] have also been identified as plasminogen receptors. GAPDH is a multi-functional protein that binds to host plasminogen and C5a. GAPDH is involved in anti-phagocytosis likely by binding and inhibiting C5a’s chemotactic function and also mediates bacterial adhesion to host pharyngeal cells by binding receptor uPAR (urokinase plasminogen activator receptor/CD87) [38–40]. SEN is a metalloenzyme that is widely distributed in many organisms from bacteria to vertebrates. SEN is found on the surface of many eukaryotic cells such as monocytes, T cells, B cells, neuronal cells and endothelial cells [41]. GAS SEN is an octomeric molecule, which interacts with plasminogen though a combination of a C-terminal lysine residues and an internal plasminogen binding site containing lysines at amino acid position 252 and 255 [42]. The role of SEN in GAS infection is proposed to facilitate tissue invasion similar to SK [41].
Thrombosis as a host defence mechanism to contain GAS infection
The activation of the host fibrinolytic system by an invading pathogen may facilitate penetration through tissue barriers, including local thrombosis and microvessel occlusion resulting from the inflammatory response [8,43]. To test this hypothesis, we analysed mice with genetic alterations in several coagulation factors for their susceptibility to GAS infection [44].
The first set of experiments tested the effect of levels of plasma or platelet FV on host susceptibility to GAS infection. FV is distributed mainly in plasma and platelet pools. In previous studies, transgenic mice were generated to express FV at different levels in either the plasma or platelet pool [45] and were subjected to GAS infection.
Transgenic mice with low plasma FV (~15%) and deficiency in platelet FV demonstrated significantly increased mortality to GAS infection than littermate controls. Furthermore, transgenic mice with higher plasma FV (~45%) and a deficiency in platelet FV also demonstrated higher mortality to GAS infection than littermates, even though the level of plasma FV was similar to F5+/−(50%) littermates, suggesting a unique role of platelet FV in host defence against GAS infection [44]. The role of platelet FV in host defence could be due to the more procoagulant properties of platelet FV as platelet FV possesses higher prothrombinase cofactor activity and is more resistant to activated protein C (APC) [46]. In addition, it was also observed that significantly higher mortality was observed in mice with lower plasma FV level, suggesting susceptibility to GAS infection depending on quantitative level of thrombin generation. In addition, mice with low levels of platelet FV (~3%) and deficiency in plasma FV also demonstrated increased mortality to GAS infection [44]. Given that lower plasma FV level resulted in delayed and decreased thrombin generation [47], these result suggested that thrombin generation is critical for host defence against GAS and potentially other pathogens [44].
In addition to its critical role in generating fibrin, thrombin’s other functions include platelet activation, activation of the protein C anticoagulant pathway, and triggering cell signalling pathways, which could all influence the host response to infection. To distinguish the fibrin generating function of thrombin from its other potential roles in host defence, mice deficient in fibrinogen were studied for their susceptibility to GAS infection. Mice completely deficient in fibrinogen exhibited markedly increased susceptibility to GAS infection compared to wild type littermate controls [44]. Similar results were observed in wild type mice following depletion of plasma fibrinogen by Ancrod treatment [30].
In addition to providing the substance of the thrombus, fibrinogen modulates leukocyte functions through binding to the leukocyte integrin receptor, Mac-1. A genetically engineered mouse expressing a mutant form of fibrinogen lacking the αMβ2-binding motif for Mac-1 exhibits a severely compromised inflammation response, but retains normal coagulation function [48]. When challenged with GAS infection, these mice demonstrated a delayed and less severe mortality pattern compared to fibrinogen-deficient mice, suggesting that both fibrin thrombus formation and the leukocyte adhesive function of fibrinogen are important in host defence from GAS [44].
Based on these studies, we have proposed the model shown in Figure 1. When microbial pathogens such as GAS invade, the host procoagulant/inflammatory response generates local thrombosis, which serves as a physical barrier to wall off the invading bacteria and prevent systemic spread. There are multiple GAS factors that can initiate the host’s response. For example, M protein can bind and assemble factors of the host contact system (intrinsic pathway of coagulation) on the surface of the bacterium, and release bradykinin, which is a potent inflammation factor; both can lead to activation of the coagulation system [11,49]. Furthermore, it has been shown that M protein is a powerful inducer of inflammation, platelet activation and thrombosis in severe GAS infection, which contributes to various pathophysiological consequences [50,51].
Figure 1.
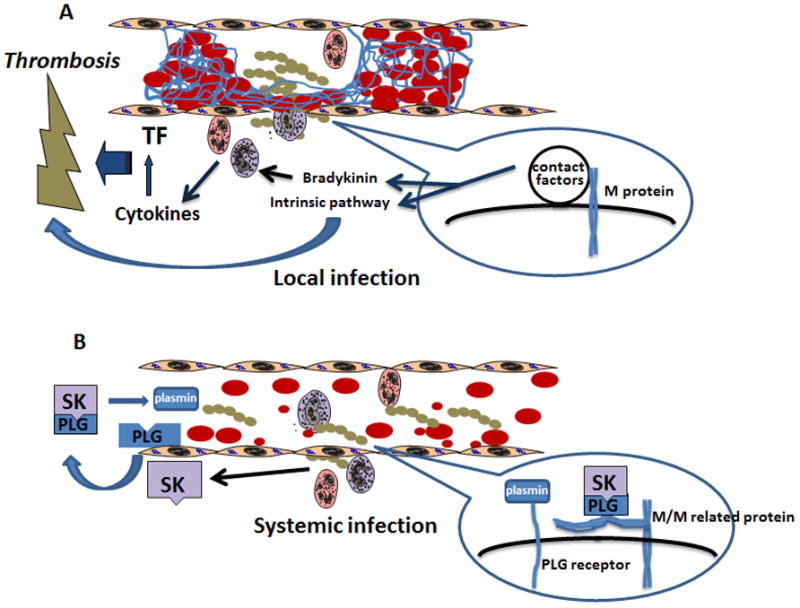
Simplified model of how GAS hijacks the host fibrinolytic system to facilitate invasion. A) At the site of GAS infection, rigorous procoagulant and inflammatory response will be initiated to release cytokines and activate coagulation system. Local thrombosis and microvascular occlusion will wall off bacteria from systemic spread. An example is given as to how a GAS virulence factor, M protein, can activate the intrinsic pathway of coagulation and also release a potent inflammation stimulant bradykinin to promote a host response. B) GAS produces the plasminogen activator streptokinase (SK) to form a SK/plasminogen complex, which will activate circulating plasminogen to plasmin. The SK/plasminogen complex is resistant to host fibrinolysis inhibitors such as α2-antiplasmin. Additionally, GAS also recruits the fibrinogen/SK/plasminogen complex by binding fibrinogen through M or M-related proteins or binding plasminogen/plasmin directly through plasminogen receptors. As a result, the bacteria are able to overcome local thrombosis to spread systemically.
An evolutionary arms race has ensued between host and pathogen, leading to the generation of pathogenicity factors such as streptokinase that hijack the host fibrinolytic system to overcome this physical barrier. In addition to generating circulating plasmin by secreting streptokinase, GAS also recruits plasminogen/plasmin to its surface by either directly binding plasminogen/plasmin through plasminogen receptors or through binding the trimolecular complex of fibrinogen-plasminogen-streptokinase. Clinical isolates from invasive human infections have been shown to assemble this trimolecular complex on the GAS surface more efficiently than isolates from uncomplicated infection [52]. Fibrinogen can bind to the GAS surface M proteins and M-related proteins to provide a binding site for streptokinase-plasminogen complex on the GAS surface to activate circulating plasminogen [53,54]. Fibrinogen fragment D has been shown to be the anchor of the surface plasminogen-activating complex [54]. The trimolecular complex formed with fibrinogen, streptokinase and plasminogen on the surface of GAS can generate plasmin [28,55]. The plasmin-coated bacteria can break the physical barrier to disseminate [8,31]. This model predicts that weakening of the host coagulation system through genetic or acquired coagulation factor deficiency will also weaken host defence.
Haemostasis as a therapeutic target for the treatment of infection related disease
Studies have shown that fibrinogen not only is involved in the host inflammatory response to infection [56], but also interacts extensively with GAS proteins. For example, GAS produces M protein to bind fibrinogen to evade phagocytosis [57,58]. It has also been shown that M protein forms a complex with fibrinogen and activates polymorphonuclear neutrophils to release heparin-binding protein, leading to vascular leakage and pulmonary damage, contributing to streptococcal toxic shock-like syndrome [59]. By binding fibrinogen, M and M-related protein can also anchor the trimolecular fibrinogen, plasminogen and streptokinase plasminogen activating complex to bacteria surface to facilitate invasion. M protein also binds other plasma proteins such as fibronectin, complement regulatory protein, immunoglobulins and albumin [60], though it is difficult to distinguish the contribution of M protein to fibronectin binding from other high affinity GAS fibronectin binding proteins [49].
It has also been shown that the host contact system can be activated by GAS proteins [11]. Severe bacterial infection may cause systemic activation of the contact system, leading to high levels of bradykinin, consumption of contact factors and inflammatory reaction [61].
A number of GAS proteins have been identified that bind human fibrinogen, fibronectin and complement regulatory protein C4BP in a protein microarray study [62]. It appears that GAS interacts with the host haemostatic system extensively to facilitate the establishment and spread of the pathogen, lead to various symptoms of infectious diseases.
The studies described above suggest that common polymorphic variations in human haemostatic factors may affect host susceptibility to infectious disease. For example, FV Leiden is a prevalent prothrombotic mutation of FV in populations of European origin that renders activated FV protein highly resistant to APC cleavage. FV Leiden was demonstrated to improve the survival rates in severe sepsis patients [63,64]. Furthermore, mice carrying one copy of FV Leiden mutation also exhibited higher survival rates in a LPS sepsis model at doses of LPS where half of the mice died from LPS [63]. However, at doses lower or higher, no significant protection was observed [65]. In a Danish population study, the FV Leiden mutation was associated with decreased risk for urinary-tract infection, increased risk for skin infection and increase mortality from sepsis [66]. No significant difference was observed in the outcome when wild type, FV Leiden heterozygote and homozygote mice were challenged with live E. coli in a sepsis peritonitis model [67]. The protective effect of FV Leiden in severe sepsis was ascribed to increased thrombin generation due to the mutation. The higher thrombin generation would lead to increased APC production, which was beneficial to survival in severe sepsis [63]. However, no increase in APC levels was detected in human FV Leiden carriers and the increase in mice was marginal comparing to wild type [65]. As a result, the mechanism of FV Leiden’s roles in severe sepsis is still unclear [68].
The above studies demonstrate the level of complexity of the effect of polymorphic variation in haemostatic factors on host susceptibility to infectious diseases. More extensive genetic and epidemiological studies on association of haemostatic factor mutations with susceptibility to infectious diseases will not only deepen our understanding of the mechanism of infectious diseases, but also lead to novel therapeutic approaches to treat and care for patients suffering from various bleeding, thrombotic and infection related diseases.
These studies suggest that the host haemostatic system could be mined for potential therapeutic targets to treat infection and related diseases. Natural anticoagulants, including antithrombin, tissue factor pathway inhibitor (TFPI) and APC have been extensively studied as potential treatments for sepsis and the overwhelming inflammatory reaction associated with severe infection [69]. Only APC has been demonstrated to improve survival in severe sepsis and is currently approved for clinical use [70]. The function of APC in the treatment of sepsis is thought to be related to its function(s) in modulating host response to infection, rather than the host/pathogen interaction. Inhibitors of the contact system have also been developed to treat sepsis [61]. An antagonist of kinin receptor B2R was used in a clinic trial with no significant effects on risk-adjusted 28 day survival rate [71].
In contrast, the interaction of bacteria with the host haemostatic system has not yet been actively targeted as a therapeutic strategy. One early experiment in this aspect was to use a synthetic peptide from domain 5 of high molecular weight kininogen (HK), which binds to the bacterial surface, to treat mice infected with invasive GAS [72]. The peptide was able to inhibit activation of the contact system and protect mice from lung damage. In addition, the peptide, in combination with an antibiotic, significantly prolonged survival time and increased the survival rate of infected mice, suggesting great potential in targeting host pathogen interactions in treating infection related diseases [72]. Such approaches could inhibit the entry and/or establishment of the pathogen in the host, mitigating disease symptoms and potentially complementing existing antibiotic therapies, given the different mechanisms of action. Combination of such novel classes of antimicrobial reagents with conventional antibiotics could potentially extend the shelf life of current antibiotics and delay the development of resistance.
Acknowledgments
The works of the author has been supported by grants (R21AI076675-01 and P01HL573461) from the National Institute of Health. I would like to thank David Ginsburg and Mark Walker for their contribution to the works cited in the review and insightful critique and discussions. I would like thank Poorna R. Karuparthi for his critique. I would also like to thank all my collaborators on the works discussed in the review. I apologise to all colleagues whose works could not be cited due to space limitations.
Footnotes
Disclosure of conflict of interests
The author states no conflict of interests
References
- 1.Alanis AJ. Resistance to antibiotics: are we in the post-antibiotic era? Arch Med Res. 2005;36:697–705. doi: 10.1016/j.arcmed.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest. 2003;111:1265–1273. doi: 10.1172/JCI18535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Norrby SR, Nord CE, Finch R. Lack of development of new antimicrobial drugs: a potential serious threat to public health. Lancet Infect Dis. 2005;5:115–119. doi: 10.1016/S1473-3099(05)01283-1. [DOI] [PubMed] [Google Scholar]
- 4.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 5.Theuretzbacher U. Future antibiotics scenarios: is the tide starting to turn? Int J Antimicrob Agents. 2009;34:15–20. doi: 10.1016/j.ijantimicag.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Levi M. Pathogenesis and treatment of disseminated intravascular coagulation in the septic patient. J Crit Care. 2001;16:167–177. doi: 10.1053/jcrc.2001.30666. [DOI] [PubMed] [Google Scholar]
- 7.Levi M, Keller TT, van Gorp E, ten Cate H. Infection and inflammation and the coagulation system. Cardiovasc Res. 2003;60:26–39. doi: 10.1016/s0008-6363(02)00857-x. [DOI] [PubMed] [Google Scholar]
- 8.Sun H. The interaction between pathogens and the host coagulation system. Physiology (Bethesda) 2006;21:281–288. doi: 10.1152/physiol.00059.2005. [DOI] [PubMed] [Google Scholar]
- 9.Bergmann S, Hammerschmidt S. Fibrinolysis and host response in bacterial infections. Thromb Haemost. 2007;98:512–520. [PubMed] [Google Scholar]
- 10.Degen JL, Bugge TH, Goguen JD. Fibrin and fibrinolysis in infection and host defense. J Thromb Haemost. 2007;5:24–31. doi: 10.1111/j.1538-7836.2007.02519.x. [DOI] [PubMed] [Google Scholar]
- 11.Frick IM, Bjorck L, Herwald H. The dual role of the contact system in bacterial infectious disease. Thromb Haemost. 2007;98:497–502. [PubMed] [Google Scholar]
- 12.Coleman JL, Gebbia JA, Piesman J, Degen JL, Bugge TH, Benach JL. Plasminogen is required for efficient dissemination of B. burgdorferi in ticks and for enhancement of spirochetemia in mice. Cell. 1997;89:1111–1119. doi: 10.1016/s0092-8674(00)80298-6. [DOI] [PubMed] [Google Scholar]
- 13.Hu LT, Perides G, Noring R, Klempner MS. Binding of human plasminogen to Borrelia burgdorferi. Infection and Immunity. 1995;63:3491–3496. doi: 10.1128/iai.63.9.3491-3496.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sodeinde OA, Subrahmanyam YV, Stark K, Quan T, Bao Y, Goguen JD. A surface protease and the invasive character of plague. Science. 1992;258:1004–1007. doi: 10.1126/science.1439793. [DOI] [PubMed] [Google Scholar]
- 15.Lathem WW, Price PA, Miller VL, Goldman WE. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science. 2007;315:509–513. doi: 10.1126/science.1137195. [DOI] [PubMed] [Google Scholar]
- 16.Sebbane F, Jarrett CO, Gardner D, Long D, Hinnebusch BJ. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc Natl Acad Sci USA. 2006;103:5526–5530. doi: 10.1073/pnas.0509544103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bokarewa MI, Jin T, Tarkowski A. Staphylococcus aureus: Staphylokinase. Int J Biochem Cell Biol. 2006;38:504–509. doi: 10.1016/j.biocel.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 18.Jin T, Bokarewa M, Foster T, Mitchell J, Higgins J, Tarkowski A. Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J Immunol. 2004;172:1169–1176. doi: 10.4049/jimmunol.172.2.1169. [DOI] [PubMed] [Google Scholar]
- 19.Jin T, Bokarewa M, Zhu Y, Tarkowski A. Staphylokinase reduces plasmin formation by endogenous plasminogen activators. Eur J Haematol. 2008;81:8–17. doi: 10.1111/j.1600-0609.2008.01066.x. [DOI] [PubMed] [Google Scholar]
- 20.Bisno AL, Stevens DL. Streptococcal infections of skin and soft tissues. N Engl J Med. 1996;334:240–245. doi: 10.1056/NEJM199601253340407. [DOI] [PubMed] [Google Scholar]
- 21.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 22.Tillett WS, Garner RL. The Fibrinolytic Activity of Hemolytic Streptococci. J Exp Med. 1933;58:485–502. doi: 10.1084/jem.58.4.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christensen LR, Macleod CM. A proteolytic enzyme of serum:characterization, activation, and reaction with inhibitors. J Gen Physiol. 1945;28:559–583. doi: 10.1085/jgp.28.6.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esmon CT, Mather T. Switching serine protease specificity. Nat Struct Biol. 1998;5:933–937. doi: 10.1038/2906. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Lottenberg R, Boyle MD. A role for fibrinogen in the streptokinase-dependent acquisition of plasmin(ogen) by group A streptococci. J Infect Dis. 1995;171:85–92. doi: 10.1093/infdis/171.1.85. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Lottenberg R, Boyle MD. Analysis of the interaction of group A streptococci with fibrinogen, streptokinase and plasminogen. Microb Pathog. 1995;18:153–166. doi: 10.1016/s0882-4010(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 27.Marcum JA, Kline DL. Species specificity of streptokinase. Comp Biochem Physiol B. 1983;75:389–394. doi: 10.1016/0305-0491(83)90345-0. [DOI] [PubMed] [Google Scholar]
- 28.Boyle MD, Lottenberg R. Plasminogen activation by invasive human pathogens. Thromb Haemost. 1997;77:1–10. [PubMed] [Google Scholar]
- 29.Khil J, Im M, Heath A, Ringdahl U, Mundada L, Cary Engleberg N, Fay WP. Plasminogen enhances virulence of group A streptococci by streptokinase-dependent and streptokinase-independent mechanisms. J Infect Dis. 2003;188:497–505. doi: 10.1086/377100. [DOI] [PubMed] [Google Scholar]
- 30.Sun H, Ringdahl U, Homeister JW, Fay WP, Engleberg NC, Yang AY, Rozek LS, Wang X, Sjobring U, Ginsburg D. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science. 2004;305:1283–1286. doi: 10.1126/science.1101245. [DOI] [PubMed] [Google Scholar]
- 31.Walker MJ, McArthur JD, McKay F, Ranson M. Is plasminogen deployed as a Streptococcus pyogenes virulence factor? Trends Microbiol. 2005;13:308–313. doi: 10.1016/j.tim.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 32.Wistedt AC, Ringdahl U, Muller-Esterl W, Sjobring U. Identification of a plasminogen-binding motif in PAM, a bacterial surface protein. Mol Microbiol. 1995;18:569–578. doi: 10.1111/j.1365-2958.1995.mmi_18030569.x. [DOI] [PubMed] [Google Scholar]
- 33.Ringdahl U, Svensson M, Wistedt AC, Renne T, Kellner R, Muller-Esterl W, Sjobring U. Molecular co-operation between protein PAM and streptokinase for plasmin acquisition by Streptococcus pyogenes. J Biol Chem. 1998;273:6424–6430. doi: 10.1074/jbc.273.11.6424. [DOI] [PubMed] [Google Scholar]
- 34.Sanderson-Smith ML, Dowton M, Ranson M, Walker MJ. The plasminogen-binding group A streptococcal M protein-related protein Prp binds plasminogen via arginine and histidine residues. J Bacteriol. 2007;189:1435–1440. doi: 10.1128/JB.01218-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanderson-Smith ML, Dinkla K, Cole JN, Cork AJ, Maamary PG, McArthur JD, Chhatwal GS, Walker MJ. M protein-mediated plasminogen binding is essential for the virulence of an invasive Streptococcus pyogenes isolate. FASEB Journal. 2008;22:2715–2722. doi: 10.1096/fj.07-105643. [DOI] [PubMed] [Google Scholar]
- 36.Winram SB, Lottenberg R. The plasmin-binding protein Plr of group A streptococci is identified as glyceraldehyde-3-phosphate dehydrogenase. Microbiology. 1996;142:2311–2320. doi: 10.1099/13500872-142-8-2311. [DOI] [PubMed] [Google Scholar]
- 37.Pancholi V, Fischetti VA. alpha-enolase, a novel strong plasmin(ogen) binding protein on the surface of pathogenic streptococci. J Biol Chem. 1998;273:14503–14515. doi: 10.1074/jbc.273.23.14503. [DOI] [PubMed] [Google Scholar]
- 38.Boel G, Jin H, Pancholi V. Inhibition of cell surface export of group A streptococcal anchorless surface dehydrogenase affects bacterial adherence and antiphagocytic properties. Infection and Immunity. 2005;73:6237–6248. doi: 10.1128/IAI.73.10.6237-6248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin H, Song YP, Boel G, Kochar J, Pancholi V. Group A streptococcal surface GAPDH, SDH, recognizes uPAR/CD87 as its receptor on the human pharyngeal cell and mediates bacterial adherence to host cells. J Mol Biol. 2005;350:27–41. doi: 10.1016/j.jmb.2005.04.063. [DOI] [PubMed] [Google Scholar]
- 40.Terao Y, Yamaguchi M, Hamada S, Kawabata S. Multifunctional glyceraldehyde-3-phosphate dehydrogenase of Streptococcus pyogenes is essential for evasion from neutrophils. J Biol Chem. 2006;281:14215–14223. doi: 10.1074/jbc.M513408200. [DOI] [PubMed] [Google Scholar]
- 41.Pancholi V. Multifunctional alpha-enolase: its role in diseases. Cell Mol Life Sci. 2001;58:902–920. doi: 10.1007/PL00000910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cork AJ, Jergic S, Hammerschmidt S, Kobe B, Pancholi V, Benesch JL, Robinson CV, Dixon NE, Aquilina JA, Walker MJ. Defining the structural basis of human plasminogen binding by streptococcal surface enolase. J Biol Chem. 2009;284:17129–17137. doi: 10.1074/jbc.M109.004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lahteenmaki K, Kuusela P, Korhonen TK. Bacterial plasminogen activators and receptors. FEMS Microbiol Rev. 2001;25:531–552. doi: 10.1111/j.1574-6976.2001.tb00590.x. [DOI] [PubMed] [Google Scholar]
- 44.Sun H, Wang X, Degen JL, Ginsburg D. Reduced thrombin generation increases host susceptibility to group A streptococcal infection. Blood. 2009;113:1358–1364. doi: 10.1182/blood-2008-07-170506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun H, Yang TL, Yang A, Wang X, Ginsburg D. The murine platelet and plasma factor V pools are biosynthetically distinct and sufficient for minimal hemostasis. Blood. 2003;102:2856–2861. doi: 10.1182/blood-2003-04-1225. [DOI] [PubMed] [Google Scholar]
- 46.Gould WR, Silveira JR, Tracy PB. Unique in vivo modifications of coagulation factor V produce a physically and functionally distinct platelet-derived cofactor: characterization of purified platelet-derived factor V/Va. J Biol Chem. 2004;279:2383–2393. doi: 10.1074/jbc.M308600200. [DOI] [PubMed] [Google Scholar]
- 47.Castoldi E, Brugge JM, Nicolaes GA, Girelli D, Tans G, Rosing J. Impaired APC cofactor activity of factor V plays a major role in the APC resistance associated with the factor V Leiden (R506Q) and R2 (H1299R) mutations. Blood. 2004;103:4173–4179. doi: 10.1182/blood-2003-10-3578. [DOI] [PubMed] [Google Scholar]
- 48.Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest. 2004;113:1596–1606. doi: 10.1172/JCI20741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oehmcke S, Shannon O, Morgelin M, Herwald H. Streptococcal M proteins and their role as virulence determinants. Clin Chim Acta. 2010;411:1172–1180. doi: 10.1016/j.cca.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 50.Pahlman LI, Morgelin M, Eckert J, Johansson L, Russell W, Riesbeck K, Soehnlein O, Lindbom L, Norrby-Teglund A, Schumann RR, Bjorck L, Herwald H. Streptococcal M protein: a multipotent and powerful inducer of inflammation. J Immunol. 2006;177:1221–1228. doi: 10.4049/jimmunol.177.2.1221. [DOI] [PubMed] [Google Scholar]
- 51.Shannon O, Hertzen E, Norrby-Teglund A, Morgelin M, Sjobring U, Bjorck L. Severe streptococcal infection is associated with M protein-induced platelet activation and thrombus formation. Mol Microbiol. 2007;65:1147–1157. doi: 10.1111/j.1365-2958.2007.05841.x. [DOI] [PubMed] [Google Scholar]
- 52.McKay FC, McArthur JD, Sanderson-Smith ML, Gardam S, Currie BJ, Sriprakash KS, Fagan PK, Towers RJ, Batzloff MR, Chhatwal GS, Ranson M, Walker MJ. Plasminogen binding by group A streptococcal isolates from a region of hyperendemicity for streptococcal skin infection and a high incidence of invasive infection. Infection and Immunity. 2004;72:364–370. doi: 10.1128/IAI.72.1.364-370.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Christner R, Li Z, Raeder R, Podbielski A, Boyle MD. Identification of key gene products required for acquisition of plasmin-like enzymatic activity by group A streptococci. J Infect Dis. 1997;175:1115–1120. doi: 10.1086/516450. [DOI] [PubMed] [Google Scholar]
- 54.Hess JL, Boyle MD. Fibrinogen fragment D is necessary and sufficient to anchor a surface plasminogen-activating complex in Streptococcus pyogenes. Proteomics. 2006;6:375–378. doi: 10.1002/pmic.200500189. [DOI] [PubMed] [Google Scholar]
- 55.D’Costa SS, Boyle MD. Interaction of a group A Streptococcus within human plasma results in assembly of a surface plasminogen activator that contributes to occupancy of surface plasmin-binding structures. Microb Pathog. 1998;24:341–349. doi: 10.1006/mpat.1998.0207. [DOI] [PubMed] [Google Scholar]
- 56.Flick MJ, Du X, Degen JL. Fibrin(ogen)-alpha M beta 2 interactions regulate leukocyte function and innate immunity in vivo. Exp Biol Med (Maywood) 2004;229:1105–1110. doi: 10.1177/153537020422901104. [DOI] [PubMed] [Google Scholar]
- 57.Ringdahl U, Svensson HG, Kotarsky H, Gustafsson M, Weineisen M, Sjobring U. A role for the fibrinogen-binding regions of streptococcal M proteins in phagocytosis resistance. Mol Microbiol. 2000;37:1318–1326. doi: 10.1046/j.1365-2958.2000.02062.x. [DOI] [PubMed] [Google Scholar]
- 58.Rivera J, Vannakambadi G, Hook M, Speziale P. Fibrinogen-binding proteins of Gram-positive bacteria. Thromb Haemost. 2007;98:503–511. [PubMed] [Google Scholar]
- 59.Herwald H, Cramer H, Morgelin M, Russell W, Sollenberg U, Norrby-Teglund A, Flodgaard H, Lindbom L, Bjorck L. M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell. 2004;116:367–379. doi: 10.1016/s0092-8674(04)00057-1. [DOI] [PubMed] [Google Scholar]
- 60.Smeesters PR, McMillan DJ, Sriprakash KS. The streptococcal M protein: a highly versatile molecule. Trends Microbiol. 2010;18:275–282. doi: 10.1016/j.tim.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 61.Oehmcke S, Herwald H. Contact system activation in severe infectious diseases. J Mol Med. 2010;88:121–126. doi: 10.1007/s00109-009-0564-y. [DOI] [PubMed] [Google Scholar]
- 62.Margarit I, Bonacci S, Pietrocola G, Rindi S, Ghezzo C, Bombaci M, Nardi-Dei V, Grifantini R, Speziale P, Grandi G. Capturing host-pathogen interactions by protein microarrays: identification of novel streptococcal proteins binding to human fibronectin, fibrinogen, and C4BP. FASEB Journal. 2009;23:3100–3112. doi: 10.1096/fj.09-131458. [DOI] [PubMed] [Google Scholar]
- 63.Kerlin BA, Yan SB, Isermann BH, Brandt JT, Sood R, Basson BR, Joyce DE, Weiler H, Dhainaut JF. Survival advantage associated with heterozygous factor V Leiden mutation in patients with severe sepsis and in mouse endotoxemia. Blood. 2003;102:3085–3092. doi: 10.1182/blood-2003-06-1789. [DOI] [PubMed] [Google Scholar]
- 64.Yan SB, Nelson DR. Effect of factor V Leiden polymorphism in severe sepsis and on treatment with recombinant human activated protein C. Crit Care Med. 2004;32:S239–S246. doi: 10.1097/01.ccm.0000126122.34119.d1. [DOI] [PubMed] [Google Scholar]
- 65.Weiler H, Kerlin B, Lytle MC. Factor V Leiden polymorphism modifies sepsis outcome: evidence from animal studies. Crit Care Med. 2004;32:S233–S238. doi: 10.1097/01.ccm.0000126126.79861.08. [DOI] [PubMed] [Google Scholar]
- 66.Benfield TL, Dahl M, Nordestgaard BG, Tybjaerg-Hansen A. Influence of the factor v leiden mutation on infectious disease susceptibility and outcome: a population-based study. J Infect Dis. 2005;192:1851–1857. doi: 10.1086/497167. [DOI] [PubMed] [Google Scholar]
- 67.Bruggemann LW, Schoenmakers SH, Groot AP, Reitsma PH, Spek CA. Role of the factor V Leiden mutation in septic peritonitis assessed in factor V Leiden transgenic mice. Crit Care Med. 2006;34:2201–2206. doi: 10.1097/01.CCM.0000228918.30931.E8. [DOI] [PubMed] [Google Scholar]
- 68.Parker RI. Factor V Leiden and sepsis: Proof positive or phenomenology? Crit Care Med. 2006;34:2254–2255. doi: 10.1097/01.CCM.0000229679.01590.4D. [DOI] [PubMed] [Google Scholar]
- 69.Esmon CT. Role of coagulation inhibitors in inflammation. Thromb Haemost. 2001;86:51–56. [PubMed] [Google Scholar]
- 70.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ., Jr Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 71.Fein AM, Bernard GR, Criner GJ, Fletcher EC, Good JT, Jr, Knaus WA, Levy H, Matuschak GM, Shanies HM, Taylor RW, Rodell TC. Treatment of severe systemic inflammatory response syndrome and sepsis with a novel bradykinin antagonist, deltibant (CP-0127). Results of a randomized, double-blind, placebo-controlled trial. CP-0127 SIRS and Sepsis Study Group. JAMA. 1997;277:482–487. [PubMed] [Google Scholar]
- 72.Oehmcke S, Shannon O, von Kockritz-Blickwede M, Morgelin M, Linder A, Olin AI, Bjorck L, Herwald H. Treatment of invasive streptococcal infection with a peptide derived from human high-molecular weight kininogen. Blood. 2009;114:444–451. doi: 10.1182/blood-2008-10-182527. [DOI] [PubMed] [Google Scholar]