

Abstract
Translation elongation in eukaryotes is mediated by the concerted actions of elongation factor 1A (eEF1A), which delivers aminoacylated tRNA to the ribosome; elongation factor 1B (eEF1B) complex, which catalyzes the exchange of GDP to GTP on eEF1A; and eEF2, which facilitates ribosomal translocation. Here we present evidence in support of a novel mode of translation regulation by hindered tRNA delivery during mitosis. A conserved consensus phosphorylation site for the mitotic cyclin-dependent kinase 1 on the catalytic delta subunit of eEF1B (termed eEF1D) is required for its posttranslational modification during mitosis, resulting in lower affinity to its substrate eEF1A. This modification is correlated with reduced availability of eEF1A·tRNA complexes, as well as reduced delivery of tRNA to and association of eEF1A with elongating ribosomes. This mode of regulation by hindered tRNA delivery, although first discovered in mitosis, may represent a more globally applicable mechanism employed under other physiological conditions that involve down-regulation of protein synthesis at the elongation level.
Keywords: Cell Cycle, Mitosis, Protein Synthesis, Translation Control, Translation Elongation Factors
Introduction
Global translation, a tightly regulated step in eukaryotic gene expression, is known to be down-regulated during mitosis (1–2). Early studies have shown that the down-regulation in mammalian cells is attributed to phosphorylation events that interfere with the formation of eIF4F3 complex (3) and eIF2·GTP·tRNAiMet ternary complex (4), both required for the initiation step. However, according to a more recent report, although mitotic polysomes become significantly less active in protein synthesis, they do not exhibit ribosomal runoff or disassembly, which is the expected outcome of inhibition at the initiation step. This work suggests that polysomes remain intact during mitosis because of simultaneous down-regulation of translation at both the initiation and postinitiation steps (5). Evidence in support of mitosis-specific translational modulation at the elongation step include: (a) the absence of stress granules, which typically form because of ribosomal runoff in response to inhibitors of translation initiation; (b) reduced ribosome disassembly in response to puromycin, which only affects actively elongating ribosomes; (c) decreased β-actin protein synthesis despite the retention of β-actin mRNA in heavy polysomes; and (d) increased ribosome transit time, indicative of a reduced elongation rate (5). The current study was designed to investigate mechanisms controlling translation elongation during mitosis.
Translation elongation in eukaryotes is mediated by the concerted actions of eukaryotic elongation factor 1A (eEF1A, the eukaryotic counterpart of the prokaryotic EF-Tu), a G-protein that binds and delivers aminoacylated tRNA (aa-tRNA) to the A-site of an elongating ribosome harboring a growing nascent peptide chain; elongation factor 1B (eEF1B), a multisubunit guanine nucleotide exchange factor (GEF) that catalyzes the exchange of GDP to GTP on eEF1A; and eukaryotic elongation factor 2 (eEF2, the eukaryotic counterpart of the prokaryotic EF-G) that facilitates ribosomal translocation following each round of peptide bond formation (reviewed in Ref. 6). In its GTP-bound form, eEF1A binds aa-tRNA (hereinafter referred to as eEF1A·tRNA complexes) with high affinity, whereas the initiator tRNA and uncharged tRNAs are presumably excluded (7). Following stable codon-anticodon binding at the ribosomal A-site, GTP is hydrolyzed and the released eEF1A·GDP is then exchanged back to eEF1A·GTP by the GEF activity of eEF1B (8). Mammalian eEF1B consists of one structural subunit, (eEF1Bγ, termed eEF1G according to NCBI Entrez Gene nomenclature) and two catalytic subunits (eEF1Bα and eEF1Bδ, termed eEF1B2 and eEF1D, respectively, according to NCBI Entrez Gene nomenclature), as well as valine-tRNA synthetase (9). eEF1B2 is highly conserved throughout eukaryotes (9) and, being the only catalytic subunit, essential for normal growth in yeast (10). However, yeast deficient in its catalytic subunit can be rescued from lethality by eEF1A overexpression (11). eEF1D, which originated from gene duplication of an eEF1B2 ancestor and fusion with a leucine zipper domain (12), is unique to higher eukaryotes, whereas a divergent catalytic subunit, termed eEF1Bβ, has developed in plants (13). Although eEF1B2 and eEF1D share a common function and highly homologous catalytic domains, they have been suggested to bind their substrate eEF1A through different sites, on the basis of the observation that binding of eEF1A to eEF1B2 results in masking of a CK2 phosphorylation site, whereas a similarly located site on eEF1D is not masked by binding to eEF1A (14). The large leucine zipper motif within eEF1D, which is absent from eEF1B2 and from plant eEF1Bβ, suggests interactions of this subunit with other proteins (15), but there is no direct interaction between eEF1B2 and eEF1D (14). The structural subunit eEF1G functions as a scaffold protein, holding together the two catalytic subunits. eEF1G does not interact directly with eEF1A or valine-tRNA synthetase. It does, however, interact directly with the two catalytic subunits, eEF1B2 and eEF1D (14, 16). It can bind both subunits simultaneously but never two subunits of the same type, suggesting the presence of two non-interchangeable binding sites. Depletion of the structural subunit in yeast is not lethal and does not result in repression of translation but instead leads to increased resistance to oxidative stress and several antibiotics (17).
In sea urchin embryos (18) as well as mammalian HeLa cells (5), the translation elongation rate decreases in synchrony with CDK1 activation during M-phase. Xenopus oocyte eEF1D was reported to undergo phosphorylation by CDK1 during metaphase at the initial stages of early development. More specifically, two CDK1 phosphorylation sites were found, Thr-131 and an unidentified serine residue that was responsible for mobility retardation on SDS-PAGE (19, 20). While the above threonine-containing motif is not present in human eEF1D, two other consensus CDK1 target sites were identified, namely Ser-133 and Thr-147. Indeed, human eEF1D was shown to be phosphorylated by CDK1 on Ser-133 in vitro (21). In vivo phosphorylation of this site was confirmed by a multitude of phosphoproteome (mass spectrometry) analyses. Still, there is lack of information regarding the implication of mitosis-specific eEF1D phosphorylation to the regulation of translational elongation. In the current study, we demonstrate that Ser-133 is essential for reduced interaction of eEF1D with its substrate eEF1A during mitosis. We show that fewer eEF1A·tRNA complexes are available for delivering charged aa-tRNA to elongating ribosomes in mitotic cells, leading to slowdown of translation elongation.
EXPERIMENTAL PROCEDURES
Cells, Synchronization, and Cell Cycle Analysis
HeLa S3 cells were grown in DMEM (Invitrogen) supplemented with 10% fetal calf serum, 2 mm l-glutamine, and antibiotics (Biological Industries). For double thymidine block (DTB), cells were treated with 2 mm thymidine (Sigma) for 16 h, released from G1/S block in fresh DMEM for 9 h, treated again with 2 mm thymidine for 16 h, released in fresh DMEM, and harvested at 9 h for mitosis. For synchronization to mitosis using nocodazole, cells were treated with 1 mm nocodazole (Sigma) for 16 h. For synchronization to mitosis using DTB-2me2, cells were treated twice with thymidine as described above, and 660 ng/ml 2me2 (Sigma) was added 4 h before harvesting. For cell cycle analysis, cells were analyzed using flow cytometry on a BD Biosciences FACSort instrument using the Cell Quest software, as described previously (22).
Generation of DNA Expression Vectors and Stable Cell Lines
Retroviral-based pQCXIP vector (Clontech) was used as the backbone for all vectors described, followed by generation of lentiviral particles. To generate pQCXIP-FLAG, FLAG1 and FLAG2 oligonucleotides were annealed and ligated into the NotI and BamHI sites of pQCXIP. These oligonucleotides also introduce an XhoI site, which is not otherwise present in pQCXIP. eEF1A and eEF1B2 were cloned from HeLa cells by PCR using eEF1A Fwd and eEF1A Rev or eEF1B2 Fwd and eEF1B2 Rev oligonucleotides, respectively. eEF1D was cloned from pCMV-eEF1D (23) by PCR using eEF1D Fwd and eEF1D Rev oligonucleotides. Amplified fragments were ligated into the XhoI and EcoRI sites of pQCXIP-FLAG. Murine eEF1G was cloned from pGFP-eEF1G (24) by digestion with HindIII (filled in) and XhoI followed by ligation into filled-in BamHI and XhoI of pQCXIP-FLAG. pQCXIP-FLAG-eEF1D(T147A), pQCXIP-FLAG-eEF1D(S133A), pQCXIP-FLAG-eEF1D(T147A;S133A), and pQCXIP-FLAG-eEF1D(S133E) were generated using primer extension according to a protocol provided by Promega using eEF1D T/A Rev, eEF1D S/A Fwd, eEF1D S/E Rev, and eEF1D S/E Fwd oligonucleotides. Transfection of all pQCXIP-based recombinant plasmids was performed into HEK293T cells using the calcium phosphate procedure. pVSVG and pGPT (Clontech) were used to generate retroviral particles for infection to generate stable cell lines. Retroviral infection of HeLa S3 cells was performed according to the protocol provided by Clontech.
DNA Primers for Cloning of PCR Fragments
FLAG1, 5′-ggccgcatggactacaaagacgatgacgacgacaagctcgagg-3′; FLAG2, 5′-gatccctcgagcttgtcgtcatcgtctttgtagtccatgc-3′; eEF1A Fwd, 5′-ggggctcgagatgggaaaggaaaagactc-3′; eEF1A Rev, 5′-gggggaattcaaacagttctgagaccgttc-3′; eEF1B2 Fwd, 5′-ggggctcgagatgggtttcggagacctgaaaagc-3′; eEF1B2 Rev, 5′-gggggaattcaatgccatgatccaggatgg-3′; eEF1D Fwd, 5′-ggggctcgagatggctacaaacttcctagc-3′; eEF1D-Rev, 5′-gggggaattcgtctttaatcgtggcagggc-3′; eEF1D T/A Rev, 5′-cctctgcaggtgcggctggctt-3′; eEF1D S/A Fwd, 5′-gcacgtagctcccatgcgcaa-3′; eEF1D S/E Rev: 5′-cagacccagcacgtagaacccatgcgcc-3′; and eEF1D S/E Fwd: 5′-cacttggcgcatgggttctacgtgctgg-3′.
Antibodies
Rabbit polyclonal antibodies against eEF1A, eEF1B2, and phospho-histone H3 were from Abcam; those against β-actin from Cell Signaling Technology, Inc.; those against eIF2α P-Ser-51 from MBL; and those against poly(A)-binding protein (PABP) from Santa Cruz Biotechnology, Inc. Mouse monoclonal antibodies against eEF1G and eEF1D were from Abcam, and those against FLAG from Sigma. Mouse monoclonal antibody against eIF2α were from E. C. Henshaw's lab (25). HRP-conjugated goat anti-mouse and anti-rabbit secondary antibodies for Western blotting were from Jackson ImmunoResearch Laboratories, Inc. Secondary antibodies for immunofluorescence were all multilabeling grade from Jackson ImmunoResearch Laboratories, Inc.
Immunoprecipitation and Immunoblot Analysis
Cells were lysed in lysis buffer (10 mm HEPES (pH 7.5), 0.5% Nonidet P-40, 100 mm NaCl, 10 mm MgCl, 1 mm sodium orthovanadate, 10 mm NaF, 20 mm β-glycerolphosphate, 1.4 μg/ml pepstatin, 2 μg/ml leupeptin, EDTA-free protease inhibitor mixture (Complete, Roche), and 0.1 μm microcystin). For immunoprecipitation, 2–5 mg of total protein extracts was incubated with anti-FLAG antibody, followed by addition of protein G-agarose beads (Santa Cruz Biotechnology, Inc.). Proteins were eluted from the beads by boiling in Laemmli sample buffer. Western blot analyses were performed following 10% SDS-PAGE according to standard procedures. Detection was by enhanced chemiluminescence (GE Healthcare). Densitometry of protein bands was performed using ImageJ software.
Immunofluorescence
Cells were grown on coverslips and fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 in PBS, blocked with 5% goat serum (Biological Industries), and primary antibodies were incubated overnight at 4 °C. Cells were then incubated with secondary antibodies (multilabeling grade, Jackson ImmunoResearch Laboratories, Inc.) in 2% BSA in PBS supplemented with 50 ng/ml Hoechst dye (Sigma) for 1 h at room temperature. Images were acquired using a Yokogawa CSU-22 confocal head and an Axiovert 200M (Zeiss) using SlideBooktm (Intelligent Imaging Innovations). Quantification of colocalization was performed by ImageJ software.
In Vitro Phosphorylation of eEF1D
eEF1D was immunoprecipitated from non-synchronized HeLa cells expressing FLAG-eEF1D as described under “Immunoprecipitation,” except that the bead-bound eEF1D was washed three times with assay buffer (20 mm MOPS (pH 7.5), 25 mm βGP, 5 mm EGTA, 1 mm DTT, 7.5 mm MgCl2, and 1 mm sodium vanadate) and incubated at 37 °C for 2 h with 100 mm ATP and 500 μg of total protein from either non-synchronized or mitotic HeLa cells homogenized in assay buffer either with or without 150 μm roscovitine (Sigma), a specific CDK1 inhibitor. Beads were then washed three times with assay buffer, boiled in sample buffer, resolved on a 10% SDS-PAGE, and subjected to immunoblot analysis with anti-FLAG antibody.
Polysomal Profile Analysis and Extraction of Proteins from Sucrose Gradients
Polysomal profiles were performed as described earlier (5). For protein extraction, each 0.5-ml fraction was diluted 1:1 with 20 mm Tris (pH 7.5) followed by incubation with 7 μl of StrataClean resin (Stratagene) overnight at 4 °C. Bound proteins were eluted by boiling in Laemmli sample buffer.
RNA Extraction and Analysis
For tRNA extraction from sucrose gradients, 0.4 ml of 8 m guanidine hydrochloride (Sigma) and 1 ml of ice-cold ethanol were added to each 0.5-ml fraction, which was then incubated for 48 h at −20 °C and centrifuged for 30 min at 20,000 × g. Next, RNA was extracted from each fraction by TRIzol reagent (Invitrogen) according to the manufacturer's instructions, except that the final precipitation was performed with 1 ml of ethanol for 24 h at −20 °C.
For tRNA extraction from FLAG-eEF1A immunoprecipitates, TRIzol reagent was used according to instructions, with the abovementioned exception concerning final precipitation. For Northern blot analysis, RNA was boiled for 2 min in loading buffer containing 49% formamide and 5 mm EDTA, separated on a 12% 8 m urea PAGE and blotted onto a Hybond-N membrane (Amersham Biosciences). A 32P-labeled DNA probe was prepared as described (26). The following DNA probes were used: tRNA-lysine, 5′-cgcccgaacagggacttgaaccctggaccctcagattaaaagtctgatgctctaccgactgagctatcc-3′; tRNA-arginine, 5′-gggccagtggcgcaatggataacgcgt-3′; and tRNA-glutamine, 5′-ttccctgaccgggaatcgaacccgggccgcggcggtgagagcgccgaatcctaaccacta-3′. Analysis of tRNA aminoacylation status was performed essentially as described (27). In brief, total RNA was extracted under acidic conditions using TRIzol reagent (Invitrogen) according to the manufacturer's instructions, except that RNA was resuspended in 10 mm sodium acetate (pH 4.0). 1 μg of total RNA was then treated with 0.2 m Tris HCl (pH 9.5) for 30 min at 37 °C, and each sample of treated or untreated RNA was mixed with sample buffer (0.1 m sodium acetate (pH 5.0) and 8 m urea) and loaded on a 14% PAGE with 8 m urea and 0.3 m sodium acetate (pH 5.0) as gel buffer and 0.3 m sodium acetate as running buffer. The gel was then stained with ethidium bromide.
RESULTS
Less tRNA Is Associated with Polysomes during Mitosis
As an initial step, we reconfirmed that polysomes remain intact in HeLa cells synchronized to mitosis by release from DTB in contrast to cells synchronized using nocodazole, a cytoskeleton-destabilizing agent often used for mitotic arrest. Indeed, polysomal profiles analyzed on sucrose gradients demonstrated that mitotic cells synchronized by DTB, but not nocodazole, contain intact heavy polysomes (Fig. 1A). We also reconfirmed that mitotic polysomes are less sensitive to inhibitors of translation initiation (supplemental Fig. 1). To better characterize the mechanism responsible for elongation slowdown during mitosis at the biochemical level, we first optimized the synchronization protocol by using a double thymidine block followed by incubation with 2-methoxyestradiol (2me2), a microtubule-stabilizing drug used to arrest cells at the prophase-metaphase boundary. Flow cytometry analysis measured 85% mitotic cells under the combined DTB/2me2 protocol, compared with 67% using the standard DTB protocol (Fig. 1B). This was also correlated with an increase in the levels of the mitotic marker phospho-histone H3 (P-H3, Fig. 1C). Polysomal profile analysis confirmed the presence of intact heavy polysomes in the DTB/2me2-arrested mitotic cells (Fig. 1D).
FIGURE 1.

Polysomes remain stable during mitosis. A, HeLa cells, either non-synchronized or synchronized to mitosis using nocodazole or release from double thymidine block (DTB release), as indicated, were analyzed for their polysomal profile. 80 S and polysomes are indicated. B, HeLa cells, either non-synchronized or synchronized to mitosis by release from DTB or by release from DTB followed by treatment with 2me2 (DTB/2me2), as described under “Experimental Procedures,” were stained for DNA content with propidium iodide followed by flow cytometry analysis and subjected to immunoblot analysis (C) using antibodies specific to the mitotic marker phospho-histone H3 (P-H3) and β-actin. D, mitotic (DTB/2me2) HeLa cells were analyzed for polysomal profile. 80 S and polysomes are indicated.
The DTB/2me2 synchronization protocol was then used to compare the amount of polysome-bound tRNA in non-synchronized and mitotic cells. To this end, cell extracts were fractionated on a sucrose gradient, followed by pooling of six consecutive fractions along the gradient. RNA was then extracted from the pooled fractions, separated by gel electrophoresis, and probed with a 32P-labeled DNA oligonucleotide specific to full-length lysyl tRNA. Equivalent loading of ribosomal RNA was confirmed by methylene blue staining (data not shown). A prominent ∼3.3-fold reduction in polysome-associated lysyl tRNA was observed in mitotic compared with non-synchronized cells (Fig. 2A). Similar results were obtained upon reprobing with 32P-labeled DNA oligonucleotide specific to arginyl tRNA and glutamyl tRNA (data not shown). Electrophoresis of tRNA samples from non-synchronized and mitotic cells under acidic conditions, which preserve aminoacylation, ruled out possible differences in the overall levels of charged aa-tRNA in mitotic cells (Fig. 2B). We speculated that the lower tRNA content in polysomal fractions of mitotic cells, i.e. a lower tRNA:rRNA ratio, may be indicative of reduced tRNA delivery to mitotic polysomes, which is in agreement with a decrease in elongation rate. This finding suggests that a reduced elongation rate may be the result of delayed cognate tRNA arrival to ribosomes because of possible regulation of the availability of eEF1A·tRNA complexes.
FIGURE 2.

Less tRNA is associated with mitotic polysomes. A, non-synchronized (NS) and mitotic (M) HeLa cells were analyzed for their polysomal profile. Free RNA, ribosomal subunits, and polysomes are indicated. RNA was extracted from six consecutive pooled fractions along the gradient and resolved on an 8 m urea-12% polyacrylamide gel followed by staining with methylene blue to verify equal loading. The RNA was then analyzed by Northern blot hybridization with 32P-labeled DNA oligonucleotide corresponding to lysyl tRNA. Shown is one representative of three independent experiments. The intensity of the lysyl tRNA signal in polysomes relative to total tRNA was quantified by densitometry. The bar graph shows the mean ± S.E. of polysome-associated tRNA in mitotic relative to non-synchronized cells. B, tRNA aminoacylation is not reduced during mitosis. RNA was extracted from NS and M HeLa cells under acidic conditions (pH 4.0), and tRNA was deacylated by base treatment (pH 9.5) for 30 min at 37 °C. Treated and untreated samples were then resolved on an acidic 8 m urea-15% polyacrylamide gel (pH 5.0) and stained with ethidium bromide. Aminoacylated tRNAs (charged tRNA) and non-aminoacylated tRNAs are indicated.
Availability of eEF1A·tRNA Complexes and Association of eEF1A with Polysomes Decrease during Mitosis
To substantiate the hypothesis that the availability of eEF1A·tRNA complexes is regulated during mitosis, we first examined the level of polysome-bound eEF1A. HeLa cells stably expressing FLAG-tagged recombinant eEF1A protein were generated with no detectable effects on the cell cycle (data not shown). Non-synchronized and mitotic cells expressing FLAG-eEF1A were analyzed for their polysomal profiles (Fig. 3A) followed by SDS-PAGE and immunoblot analysis of proteins extracted from each fraction of the gradient. This revealed a significant reduction in the proportion of polysome-bound eEF1A from 13.9% to 5.5% in non-synchronized and mitotic cells, respectively (Fig. 3B). To further confirm that the lower levels of polysome-associated tRNA and eEF1A (Figs. 2A and 3B) in mitotic cells are due to reduced availability of eEF1A·tRNA complexes, we next examined the interaction of eEF1A with tRNA. For this purpose, the amount of tRNA coimmunoprecipitated with FLAG-eEF1A was compared in lysates of mitotic and non-synchronized cells. To monitor the efficiency of RNA extraction from both samples, 100 ng of low molecular-weight RNA ladder were added to equivalent amounts of each FLAG-eEF1A immunoprecipitate (confirmed by immunoblotting using anti-FLAG antibody, Fig. 3C, top left panel) prior to the extraction procedure. Methylene blue staining of the extracted RNA marker following electrophoresis and blotting demonstrated a similar extraction efficiency and loading from both FLAG-eEF1A immunoprecipitates (Fig. 3C, middle left panel). Anti-FLAG immunoprecipitates from HeLa cells not expressing FLAG-eEF1A were used as a negative control (data not shown). The same membrane was then probed with a 32P-labeled DNA oligonucleotide specific to full-length lysyl tRNA. The lysyl tRNA signal obtained from eEF1A immunoprecipitate was considerably lower in mitotic compared with non-synchronized cells, representing a reduction of 70% in the amount of tRNA bound by eEF1A (Fig. 3C, bottom left panel and bar graph). Similar results were obtained upon reprobing with a 32P-labeled DNA oligonucleotide specific to arginyl tRNA (data not shown), confirming that the effect on eEF1A·tRNA complexes is not specific to lysyl tRNA. Given that charged tRNA levels do not change in mitosis (Fig. 2B), this observation is consistent with decreased activity of eEF1A during mitosis.
FIGURE 3.

eEF1A is depleted from polysomes and binds less tRNA during mitosis. A, non-synchronized (NS) and mitotic (M) HeLa cells stably expressing FLAG-tagged eEF1A were analyzed for polysomal profile. 80 S and polysomes are indicated. B, total protein extracted from each of 22 sucrose gradient fractions was resolved on a 10% SDS-PAGE followed by immunoblot analysis using anti-FLAG antibody. Shown in percents is the fraction of polysome-bound to total eEF1A intensity. The data represent one of two independent experiments. C, FLAG-tagged eEF1A was immunoprecipitated from NS and M HeLa cells using anti-FLAG antibody. RNA was extracted from the immunoprecipitates by TRIzol reagent spiked with a low-molecular-weight RNA ladder to monitor extraction efficiency. RNA was then resolved on an 8 m urea-12% polyacrylamide gel followed by blotting and hybridization with 32P-labeled DNA oligonucleotide corresponding to lysyl tRNA (bottom left panel). Equivalent amounts of FLAG-eEF1A immunoprecipitates were confirmed by immunoblotting with anti-FLAG antibody (top left panel). Equivalent loading of extracted RNA was confirmed by methylene blue staining (middle left panel). Shown is one representative of three independent experiments. Intensity of FLAG-eEF1A and lysyl tRNA signals was quantified by densitometry. The bar graph shows the mean ± S.E. of eEF1A-bound lysyl tRNA in mitotic relative to non-synchronized cells.
eEF1D Ser-133, a Conserved CDK1 Target Site, Is Essential for Reducing eEF1D Interaction with Its Substrate eEF1A during Mitosis
To investigate the nature of the underlying regulatory mechanism leading to reduced availability of eEF1A·tRNA complexes, we analyzed the abundance of each of the four eEF1 subunits between the G1/S boundary and M-phase. To this end, total protein from HeLa cells harvested at various time points following release from DTB was subjected to immunoblot analysis using antibodies specific to eEF1 subunits and the mitotic marker phospho-histone H3, which peaked at 9 h after block release (Fig. 4A). This experiment demonstrated that eEF1B2 and eEF1G do not exhibit any significant change in protein level or migration pattern on SDS-PAGE as the cells progress through the cell cycle and enter M-phase. eEF1A, however, showed a small and gradual increase in protein level toward M-phase. Although we are currently unable to explain this increase, it does not seem to detract from our above conclusion regarding the reduced availability of eEF1A·tRNA complexes. On the contrary; levels of polysome-bound eEF1A and eEF1A-bound tRNA are lower in mitosis despite the apparent increase in total eEF1A levels.
FIGURE 4.

Level and migration pattern of eEF1 subunits. A, HeLa cells were synchronized to the G1/S boundary using DTB and harvested at the indicated time points following release from the block. 40 μg of total protein at each time point were subjected to immunoblot analysis using antibodies specific to the indicated proteins. B, eEF1D was immunoprecipitated from non-synchronized HeLa cells expressing FLAG-tagged wild-type eEF1D. The Sepharose beads-associated FLAG-eEF1D was then incubated with buffer alone or with lysate from non-synchronized (NS) or mitotic (M) HeLa cells either in the absence or presence of roscovitine, a specific CDK1 inhibitor. The beads-associated FLAG-eEF1D was then subjected to immunoblot analysis using anti-FLAG antibody. C, total protein from HeLa cells stably expressing FLAG-tagged eEF1D wild-type and T147A, S133A, T147A;S133A, and S133E was subjected to immunoblot analysis using anti-FLAG antibody. D, total protein from non-synchronized (NS) and mitotic (DTB/2me2) HeLa cells was subjected to immunoblot analysis using anti-phospho-H3 (P-H3) and anti-eEF1D antibodies.
In contrast to other eEF1 subunits, eEF1D was detected at all time points as two distinct bands that change in intensity around M-phase (Fig. 4A). A clear shift of the lower to the upper band of eEF1D, indicative of a posttranslational modification, was observed as cells entered mitosis. A similar shift in Xenopus laevis eEF1D was attributed to phosphorylation by CDK1 during metaphase (19, 20, 28). It was therefore tempting to speculate that eEF1D activity may be directly or indirectly regulated by CDK1 during mitosis. To assess the involvement of this major mitotic kinase, we used roscovitine, a specific CDK1 inhibitor, to demonstrate that CDK1 activity in mitotic cells is necessary for the shift in migration pattern of eEF1D. In this in-lysate in vitro experiment, we immunoprecipitated eEF1D from non-synchronized cells and incubated the immunoprecipitate with lysates from non-synchronized or mitotic cells with or without roscovitine, which prevented the shift in eEF1D migration (Fig. 4B). A similar experiment cannot be performed in vivo, as the use of roscovitine would not only prevent phosphorylation of CDK1 targets but also preclude entry of cells to mitosis.
Being one of the two catalytic subunits of the eEF1B complex, we hypothesized that CDK1 phosphorylation of eEF1D may negatively affect the interaction with its substrate eEF1A, leading to down-regulation of eEF1B ability to recycle eEF1A·GDP back to its active GTP-bound form. To examine this hypothesis, we first tested whether the change in migration pattern of eEF1D on SDS-PAGE is attributed to mitosis-specific modification on Thr-147, Ser-133, or both because these two residues were identified as conserved consensus phosphorylation sites for CDK1 (29). We generated HeLa cell lines stably expressing FLAG-tagged wild-type and mutant variants of the eEF1D protein, in which Thr-147 or Ser-133 or both residues were replaced with alanine (T147A, S133A, or T147A;S133A, respectively). We also generated a HeLa cell line stably expressing a phosphomimetic mutant eEF1D variant in which Ser-133 was replaced with glutamic acid (S133E). Although the different stable cell lines expressed similar levels of the recombinant proteins, FLAG-eEF1D was detected as two bands only in cells expressing the WT and the T147A variant, whereas a single lower band was detected in cells expressing the S133A and T147TA;S133A variants (Fig. 4C), demonstrating that the observed shift in the migration of eEF1D is attributed to a posttranslational modification on Ser-133 but not on Thr-147. The variant harboring the phosphomimetic amino acid at position 133, eEF1D(S133E), migrated as a single upper band, as expected. Human eEF1D was reported previously to undergo CDK1-dependent phosphorylation both in vitro and in monkey epithelial cells on Ser-133 (21). This is supported by multiple mass spectrometry experiments showing that mammalian eEF1D is phosphorylated on Ser-133 during mitosis (30–32). Our results, taken together with these previously published reports, strongly implicate CDK1 phosphorylation on Ser-133 as the modification responsible for the shift in eEF1D mobility on SDS-PAGE, although an indirect effect downstream of CDK1 cannot be ruled out.
It is also of note that although eEF1D is detected as two bands in cells synchronized by the DTB release protocol (Fig. 4A), it is seen as a single slow-migrating band in cells synchronized by the DTB/2me2 protocol (Fig. 4D), consistent with a higher proportion of mitotic cells uniformly arrested at the end of prophase in the presence of 2me2. This also suggests that the entire population of eEF1D is modified on Ser-133 during mitosis, and thus the proportion of eEF1D bands can serve as an additional marker for mitotic enrichment and efficiency of synchronization.
To establish whether the modification of eEF1D is correlated to a change in its interactions with eEF1A during mitosis, the above HeLa cell lines stably expressing wild-type or mutated variants of FLAG-tagged eEF1D were used for coimmunoprecipitation experiments. Indeed, eEF1D modification during mitosis is correlated with a significant reduction of its binding to eEF1A, as only 52% of eEF1A was coimmunoprecipitated with FLAG-eEF1D in mitotic compared with non-synchronized cells (Fig. 5A). However, the amount of eEF1A coimmunoprecipitated with FLAG-eEF1D(T147A;S133A) was not lower in mitotic compared with non-synchronized cells (Fig. 5A). Although the FLAG-eEF1D(T147A) variant was apparently able to undergo mitosis-specific modification along with decreased binding of eEF1A, the FLAG-eEF1D(S133A) variant was apathetic to mitosis (Fig. 5B), confirming the functional significance of this putative phosphorylation on Ser-133, but not Thr-147, to eEF1D-eEF1A interactions in human cells. Furthermore, the phosphomimetic S133E variant bound less eEF1A than S133A, even in non-synchronized cells (Fig. 5C), giving further strength to the conclusion that the putative phosphorylation of eEF1D on Ser-133 may have a negative effect on eEF1D-eEF1A interactions.
FIGURE 5.

eEF1D variants and their interaction with eEF1A. A, a fraction of total protein used for immunoprecipitation (input) and the anti-FLAG immunoprecipitates (IP Flag) from non-synchronized (NS) and mitotic (M) HeLa cells expressing FLAG-tagged wild-type and T147A;S133A mutant eEF1D were immunoblotted using anti-eEF1A and anti-FLAG antibodies. B, similar to A but with HeLa cells expressing FLAG-tagged T147A or S133A mutant variants of eEF1D, respectively. C, similar to A but with HeLa cells expressing the FLAG-tagged S133E mutant variant of eEF1D. A–C represent one of three independent experiments. The intensity of bands from all experiments was quantified by densitometry. Quantitative data is represented as mean ± S.E. D, representative single-plane images (×100 magnification) of fixed non-synchronized HeLa cells stained with anti-eEF1D and anti-eEF1A taken using a spinning disc confocal microscope. Pearson's coefficients for the colocalization of eEF1D and eEF1A in 15 interphase and 15 mitotic cells is shown. Data are represented as mean ± S.E.
To determine whether the mitosis-specific reduction in eEF1A-eEF1D interactions occurs endogenously in cells that do not overexpress eEF1D, HeLa cells were subjected to double-labeling immunofluorescence experiments using antibodies specific to eEF1A and eEF1D. Representative double-labeled images of interphase and mitotic cells are shown in Fig. 5D. Pearson's coefficient of colocalization was calculated for 15 interphase and 15 mitotic cells, confirming the ∼2-fold decline in eEF1A-eEF1D association during mitosis.
Reduced Binding of eEF1A to Its GEF during Mitosis Is Specific to eEF1A-eEF1D Interaction
To test whether it is eEF1A-eEF1D interactions that are specifically interrupted during mitosis, we next analyzed the binding capacity of eEF1A to each of the two catalytic subunits, eEF1D and eEF1B2. To this end, anti-FLAG immunoprecipitates from non-synchronized and mitotic HeLa cells stably expressing FLAG-eEF1A were subjected to immunoblot analysis using antibodies specific to eEF1D and eEF1B2. As expected, less (61%) eEF1D coimmunoprecipitated with FLAG-eEF1A in mitotic compared with non-synchronized cells. However, in contrast to eEF1D, similar amounts of eEF1B2 coimmunoprecipitated with FLAG-eEF1A in non-synchronized and mitotic cells (Fig. 6A). The reciprocal experiment, analyzing anti-FLAG immunoprecipitates from HeLa cells stably expressing FLAG-eEF1B2, confirmed that similar amounts of eEF1A are bound to the eEF1B2 catalytic subunit in non-synchronized and mitotic cells (Fig. 6B). These data reveal that eEF1D-eEF1A interactions are uniquely regulated during mitosis. We then reasoned that although eEF1A is not directly associated with the structural subunit eEF1G, indirect eEF1A-eEF1G interaction should be negatively affected during mitosis as a result of the decreased association of eEF1A with eEF1D. Coimmunoprecipitation experiments using HeLa cells stably expressing FLAG-eEF1G demonstrated that only 52% eEF1A is coimmunoprecipitated with FLAG-eEF1G from lysates of mitotic compared with non-synchronized cells (Fig. 6C). This is similar to the result obtained when FLAG-eEF1D was immunoprecipitated (Fig. 5A), giving further support to the hypothesis that eEF1D-eEF1A interactions are specifically affected during mitosis. In contrast to the mitosis-specific decrease in eEF1D-mediated association of eEF1A with eEF1G, similar amounts of eEF1D and eEF1B2 were coimmunoprecipitated with FLAG-eEF1G in non-synchronized and mitotic cells (Fig. 6C). This finding seems to indicate that although eEF1A dissociates from eEF1D during mitosis, the catalytic exchange complex eEF1B, consisting of eEF1G, eEF1B2, and eEF1D, remains intact (Figs. 5 and 6). Reduced association of eEF1A with eEF1D may well lead to partial loss of eEF1B GEF activity, which is shared by the two catalytic subunits, eEF1D and eEF1B2. Such reduction may disrupt the temporal balance between eEF1A·GDP and eEF1A·GTP, leading to decreased eEF1A activity.
FIGURE 6.
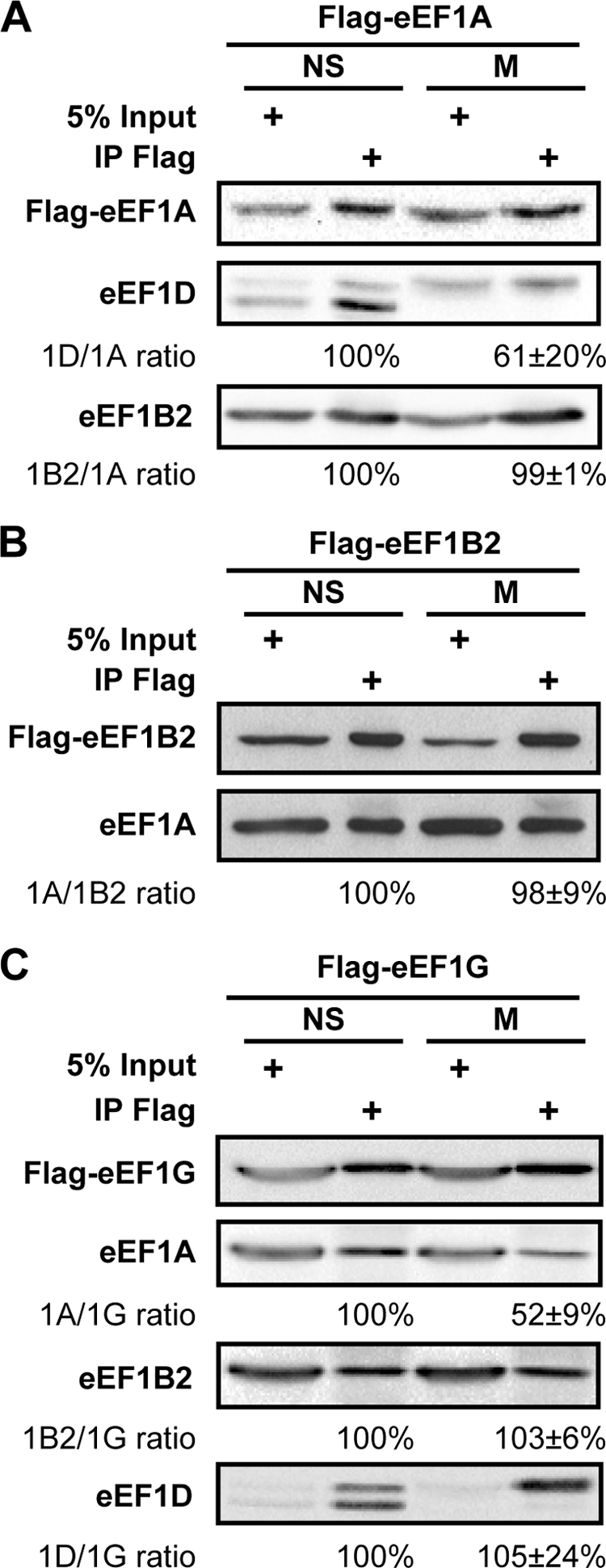
Mitosis-specific reduced binding of eEF1A to its GEF is specific to eEF1A-eEF1D interaction. A fraction of total protein used for immunoprecipitation (input) and the anti-FLAG immunoprecipitates (IP Flag) from non-synchronized (NS) and mitotic (M) HeLa cells stably expressing FLAG-eEF1A (A), FLAG-eEF1B2 (B), or FLAG-eEF1G (C) were immunoblotted for the indicated proteins. Images represent one of three or four independent experiments. The intensity of bands from all experiments was quantified by densitometry. Quantitative data is represented as mean ± S.E.
Overexpression of eEF1D(S133A) Negates the Mitosis-specific Reduction in eEF1A Activity and Destabilizes Mitotic Polysomes
On the basis of the above data, we anticipated that if Ser-133-dependent eEF1D modification is necessary for down-regulating the translation elongation rate during mitosis by reducing the availability of eEF1A·tRNA complexes, then overexpression of the phosphorylation-null eEF1D(S133A) should prevent this decrease, leading to continued elongation and subsequently destabilization or disassembly of mitotic polysomes. Indeed, we found that overexpression of eEF1D(S133A) resulted in reduced stability of mitotic polysomes, which was correlated with comparable association of eEF1A with polysomes from non-synchronized and mitotic cells (Fig. 7). This is in contrast to the dramatic decrease in eEF1A association with mitotic polysomes seen in HeLa cells not overexpressing eEF1D (Figs. 3B and 7). Therefore, this mutant may exert a dominant negative effect by abrogating the modification on Ser-133 that is responsible for reduced availability of active eEF1A. Surprisingly, a similar although smaller effect was observed with wild-type eEF1D. Compared with HeLa cells, HeLa cells overexpressing wild-type eEF1D showed some destabilization of mitotic polysomes, which was correlated with a less drastic dissociation of eEF1A from mitotic polysomes. This may be attributed to the inability of CDK1 to fully phosphorylate the excess of eEF1D in cells overexpressing the wild-type protein. This observation confirms the regulatory role of eEF1D and its putative phosphorylation on Ser-133 in managing translational elongation during mitosis.
FIGURE 7.

Overexpression of the wild-type and S133A mutant of eEF1D reverse mitotic depletion of eEF1A from polysomes. Non-synchronized (NS) and mitotic (M) HeLa cells or HeLa cells expressing either FLAG-tagged wild-type or S133A eEF1D were analyzed for their polysomal profile. Shown is one representative of two independent experiments. For each profile, the area under the curve of polysomal RNA (P) peaks and subpolysomes (SP) peaks (containing free RNA, 40 S, and 60 S, ribosomal subunits) was calculated. P/(SP+P) for NS and M cells is presented as mean ± S.E. For each profile, fractions containing polysomes (P) or subpolysomes (SP) were pooled, followed by total protein extraction from each pool. 10% of the SP samples or 100% of the P samples were resolved on a 10% SDS-PAGE followed by immunoblot analysis using anti-eEF1A and anti-PABP antibody.
DISCUSSION
Regulation of Translation Elongation during Mitosis
The macromolecular complexity of eEF1B; its multiple putative phosphorylation sites; numerous cellular partners; and accumulated data from yeast, X. laevis oocytes, and sea urchin embryos served as the basis for the hypothesis that eEF1B plays an essential role in the control of gene expression in mammalian cells, particularly during mitosis (9). In this study, we provide evidence for a regulatory role of eEF1D in translation elongation in mitotic human cells. We show for the first time that the Ser-133 residue of eEF1D, which is positioned within a conserved CDK1 phosphorylation motif, is essential for mitosis-specific reduction in eEF1D-eEF1A interaction. We also show that, during mitosis, less tRNA is bound to eEF1A and that the association of both eEF1A and tRNA with polysomes is reduced. According to our proposed model, phosphorylation of eEF1D by CDK1 leads to reduced interaction of the catalytic subunit eEF1D with its substrate eEF1A·GDP, resulting in a lower guanine nucleotide exchange rate by the eEF1B complex and, subsequently, a lower level of active eEF1A·GTP during mitosis. Consequently, less eEF1A·GTP is available for binding and delivering aa-tRNA to mitotic ribosomes, leading to translational slowdown and transient stalling of elongating ribosomes. The stability of heavy polysomes despite the well established inhibition of translation initiation during mitosis is in agreement with lack of ribosomal runoff because of ribosome stalling. We previously observed increased ribosome transit time in mitotic HeLa cells (5). This may be interpreted either as a global uniform decrease in elongation rate or as ongoing or elevated elongation by some polysomes coupled with a slowdown or complete arrest of others. The data presented in this study provide evidence for the negative effect of mitotic eEF1D modification on eEF1D-eEF1A interactions, whereas the eEF1B complex remains intact and active via the remaining eEF1B2 exchange function. This offers a basal level of eEF1A·GTP·aa-tRNA complexes that can promote translation to some extent, even during mitosis. Although first discovered in mitosis, this mechanism may also hold true for other biological conditions that require attenuation of global translation at the elongation level.
Evolution of a Phosphorylation-dependent Regulatory Mechanism
The evolutionary-conserved eEF1B2 subunit is thought to guarantee guanine nucleotide exchange on eEF1A, being the sole catalytic subunit in fungi. Therefore, the additional plant eEF1Bβ and metazoan eEF1D catalytic subunits have most likely evolved to promote specific regulatory functions. The presence of conserved phosphorylation sites on these extra subunits is attributed to the acquisition of a regulated function (20). Supporting this notion is the inability of plant eEF1Bβ subunit to complement a Saccharomyces cerevisiae mutant deleted for its single catalytic subunit, unless the sole consensus CDK1 phosphorylation site of eEF1Bβ was replaced by a non-phosphorylatable site (33). This supports our hypothesis that CDK1 phosphorylation of eEF1D down-regulates the GEF activity of the human eEF1B complex during mitosis. Additional observations supporting the regulatory role of eEF1D are the dramatic reduction in translation efficiency of cellular mRNA, but not viral mRNA, due to the interactions of eEF1D with the lentivirus protein Tat and herpes simplex virus 1 protein ICP0 (23, 34). Interestingly, in herpes simplex virus-infected cells, eEF1D is phosphorylated on Ser-133 by a viral kinase that mimics CDK1 function (21). An attractive speculation is that modulation of eEF1D activity may not only lead to inhibition of global elongation but also generate conditions that permit elongation of specific subclasses of cellular mRNAs, as it does for viral mRNAs in infected cells. Taken together with this study, we conclude that eEF1D serves as an important regulatory component of translational elongation in mammalian cells.
In human fibroblasts, eEF1B is anchored to the endoplasmic reticulum (35) by the interaction of eEF1D with kinectin (36). Disruption of eEF1D-kinectin interactions leads to specific inhibition of membrane protein expression and enhanced cytosolic protein synthesis, suggesting that eEF1B subcellular localization is important for translation regulation (37). In sea urchin embryos, just before nuclear membrane breakdown, eEF1D shifts to the nuclear envelope and concentrates as a ring around the nucleus and then as two large spheres around the mitotic spindle poles (38). Although the effects of this spatial redistribution are not yet characterized, this may be another example of translation regulation by subcellular localization of eEF1B.
tRNA molecules are transferred by eEF1A from aminoacyl tRNA synthetases to ribosomes and back to aminoacyl tRNA synthetases for aminoacylation in a closed-loop channel (39). Interestingly, aminoacyl tRNA synthetases were found to localize to the endoplasmic reticulum and cytoskeleton and to interact with eEF1 subunits (40). The reduced interaction of eEF1D with eEF1A during mitosis (this study) raises the question of whether the interaction of eEF1 with specific aminoacyl tRNA synthetases also changes in a manner dependent on eEF1D modification. Future experiments will verify the significance of CDK1-mediated eEF1D modification in mammalian cells to eEF1B subcellular localization and its role in the regulation of gene expression.
Supplementary Material
Acknowledgments
We thank David Chetrit and Marcelo Ehrlich for help with confocal microscopy, Dalia Pinchasi for technical assistance, K. T. Jeang for the pCMV-eEF1D plasmid, and P. A. Coulomb for the pGFP-eEF1G plasmid.
This work was supported by Israel Science Foundation Grant 131/07.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- elF
- eukaryotic translation initiation factor
- eEF
- eukaryotic translation elongation factor
- EF
- prokaryotic translation elongation factor
- aa-tRNA
- aminoacylated tRNA
- GEF
- guanine nucleotide exchange factor
- CDK1
- cyclin-dependent kinase 1
- DTB
- double thymidine block
- 2me2
- 2-methoxyestrdiol
- ARS
- aminoacyl tRNA synthetase
- PABP
- poly(A)-binding protein.
REFERENCES
- 1. Marash L., Liberman N., Henis-Korenblit S., Sivan G., Reem E., Elroy-Stein O., Kimchi A. (2008) Mol. Cell 30, 447–459 [DOI] [PubMed] [Google Scholar]
- 2. Sivan G., Elroy-Stein O. (2008) Cell Cycle 7, 741–744 [DOI] [PubMed] [Google Scholar]
- 3. Pyronnet S., Dostie J., Sonenberg N. (2001) Genes Dev. 15, 2083–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Datta B., Datta R., Mukherjee S., Zhang Z. (1999) Exp. Cell Res. 250, 223–230 [DOI] [PubMed] [Google Scholar]
- 5. Sivan G., Kedersha N., Elroy-Stein O. (2007) Mol. Cell. Biol. 27, 6639–6646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kapp L. D., Lorsch J. R. (2004) Annu. Rev. Biochem. 73, 657–704 [DOI] [PubMed] [Google Scholar]
- 7. Gromadski K. B., Schümmer T., Strømgaard A., Knudsen C. R., Kinzy T. G., Rodnina M. V. (2007) J. Biol. Chem. 282, 35629–35637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Merrick W. C., Nyborg J. (2000) in Translational Control of Gene Expression (Sonenberg N., Hershey J. W. B., Mathews M. B. eds) pp. 89–126, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 9. Le Sourd F., Boulben S., Le Bouffant R., Cormier P., Morales J., Belle R., Mulner-Lorillon O. (2006) Biochim. Biophys. Acta 1759, 13–31 [DOI] [PubMed] [Google Scholar]
- 10. Hiraga K., Suzuki K., Tsuchiya E., Miyakawa T. (1993) FEBS Lett. 316, 165–169 [DOI] [PubMed] [Google Scholar]
- 11. Kinzy T. G., Woolford J. L., Jr. (1995) Genetics 141, 481–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guerrucci M. A., Monnier A., Delalande C., Bellé R. (1999) Gene 233, 83–87 [DOI] [PubMed] [Google Scholar]
- 13. Le Sourd F., Cormier P., Bach S., Boulben S., Bellé R., Mulner-Lorillon O. (2006) FEBS Lett. 580, 2755–2760 [DOI] [PubMed] [Google Scholar]
- 14. Sheu G. T., Traugh J. A. (1997) J. Biol. Chem. 272, 33290–33297 [DOI] [PubMed] [Google Scholar]
- 15. Minella O., Mulner-Lorillon O., Poulhe R., Bellé R., Cormier P. (1996) Eur. J. Biochem. 237, 685–690 [DOI] [PubMed] [Google Scholar]
- 16. Mansilla F., Friis I., Jadidi M., Nielsen K. M., Clark B. F., Knudsen C. R. (2002) Biochem. J. 365, 669–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Olarewaju O., Ortiz P. A., Chowdhury W. Q., Chatterjee I., Kinzy T. G. (2004) RNA Biol. 1, 89–94 [DOI] [PubMed] [Google Scholar]
- 18. Monnier A., Morales J., Cormier P., Boulben S., Bellé R., Mulner-Lorillon O. (2001) Zygote 9, 229–236 [DOI] [PubMed] [Google Scholar]
- 19. Mulner-Lorillon O., Minella O., Cormier P., Capony J. P., Cavadore J. C., Morales J., Poulhe R., Bellé R. (1994) J. Biol. Chem. 269, 20201–20207 [PubMed] [Google Scholar]
- 20. Minella O., Cormier P., Morales J., Poulhe R., Bellé R., Mulner-Lorillon O. (1994) Cell. Mol. Biol. 40, 521–525 [PubMed] [Google Scholar]
- 21. Kawaguchi Y., Kato K., Tanaka M., Kanamori M., Nishiyama Y., Yamanashi Y. (2003) J. Virol. 77, 2359–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gerlitz G., Jagus R., Elroy-Stein O. (2002) Eur. J. Biochem. 269, 2810–2819 [DOI] [PubMed] [Google Scholar]
- 23. Xiao H., Neuveut C., Benkirane M., Jeang K. T. (1998) Biochem. Biophys. Res. Commun. 244, 384–389 [DOI] [PubMed] [Google Scholar]
- 24. Kim S., Kellner J., Lee C. H., Coulombe P. A. (2007) Nat. Struct. Mol. Biol. 14, 982–983 [DOI] [PubMed] [Google Scholar]
- 25. Scorsone K. A., Panniers R., Rowlands A. G., Henshaw E. C. (1987) J. Biol. Chem. 262, 14538–14543 [PubMed] [Google Scholar]
- 26. Brown T., Mackey K., Du T. (2004) Curr. Protoc. Mol. Biol. 4, Unit 4.9 [DOI] [PubMed] [Google Scholar]
- 27. Jester B. C., Levengood J. D., Roy H., Ibba M., Devine K. M. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 14351–14356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mulner-Lorillon O., Cormier P., Cavadore J. C., Morales J., Poulhe R., Bellé R. (1992) Exp. Cell Res. 202, 549–551 [DOI] [PubMed] [Google Scholar]
- 29. Marin O., Meggio F., Draetta G., Pinna L. A. (1992) FEBS Lett. 301, 111–114 [DOI] [PubMed] [Google Scholar]
- 30. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Daub H., Olsen J. V., Bairlein M., Gnad F., Oppermann F. S., Körner R., Greff Z., Kéri G., Stemmann O., Mann M. (2008) Mol. Cell 31, 438–448 [DOI] [PubMed] [Google Scholar]
- 32. Nagano K., Shinkawa T., Mutoh H., Kondoh O., Morimoto S., Inomata N., Ashihara M., Ishii N., Aoki Y., Haramura M. (2009) Proteomics 9, 2861–2874 [DOI] [PubMed] [Google Scholar]
- 33. Pomerening J. R., Valente L., Kinzy T. G., Jacobs T. W. (2003) Mol. Genet. Genomics 269, 776–788 [DOI] [PubMed] [Google Scholar]
- 34. Kawaguchi Y., Bruni R., Roizman B. (1997) J. Virol. 71, 1019–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanders J., Brandsma M., Janssen G. M., Dijk J., Möller W. (1996) J. Cell Sci. 109, 1113–1117 [DOI] [PubMed] [Google Scholar]
- 36. Ong L. L., Er C. P., Ho A., Aung M. T., Yu H. (2003) J. Biol. Chem. 278, 32115–32123 [DOI] [PubMed] [Google Scholar]
- 37. Ong L. L., Lin P. C., Zhang X., Chia S. M., Yu H. (2006) J. Biol. Chem. 281, 33621–33634 [DOI] [PubMed] [Google Scholar]
- 38. Boulben S., Monnier A., Le Breton M., Morales J., Cormier P., Bellé R., Mulner-Lorillon O. (2003) Cell Mol. Life Sci. 60, 2178–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hausmann C. D., Ibba M. (2008) FEMS Microbiol. Rev. 32, 705–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sang Lee J., Gyu Park S., Park H., Seol W., Lee S., Kim S. (2002) Biochem. Biophys. Res. Commun. 291, 158–164 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.