

Abstract
Axon degeneration is an active, evolutionarily conserved self-destruction program by which compromised axons fragment in response to varied insults. Unlike programmed cell death, axon degeneration is poorly understood. We have combined robotic liquid handling with automated microscopy and image analysis to create a robust screening platform to measure axon degeneration in mammalian primary neuronal cultures. Using this assay, we performed an unbiased screen of 480 bioactive compounds, identifying 11 that reproducibly delay fragmentation of severed axons in vitro, including two inhibitors of glycogen synthase kinase 3 and two inhibitors of IκB kinase. Knockdown of each of these targets by shRNA lentivirus also delays axon degeneration in vitro, further supporting their role in the axon degeneration program.
Keywords: Axon, High Throughput Screening (HTS), Neurodegeneration, Neurons, Signal Transduction
Introduction
The axon is a uniquely neuronal structure whose specialized architecture facilitates the rapid transmission of information across long distances. Just as compromised cells undergo programmed cell death, damaged axons undergo an active self-destruct process that involves cytoskeletal disassembly, swelling, and eventual fragmentation with no concomitant cell death (1, 2). This process has been broadly termed axon degeneration and is intrinsic to the neuron. Despite their resemblance, axon degeneration and apoptosis are largely mechanistically distinct (1, 3); however, axon and dendrite breakdown in the contexts of developmental pruning or trophic withdrawal does involve caspases and other apoptotic machinery (5, 6, 21). Components of the axon degeneration cascade include calpain proteases and the ubiquitin-proteasome system, and calcium influx is also an important step (7–9). Axon degeneration is thought to be an early, important step in the pathologic progression and morbidity in diseases of the peripheral and central nervous systems including diabetic neuropathy, Parkinson disease, and multiple sclerosis (1, 2). Clearly, understanding the mechanisms that promote axon degeneration could have therapeutic value, yet little is known regarding the mechanisms by which axons commit to and execute this program of self-destruction.
Unbiased high-content screening is a powerful approach for uncovering biological signaling cascades. Here, we report an image-based screening assay that combines automated microscopy, liquid handling, and image analysis of primary cultured neurons to screen for experimental conditions that delay or suppress axon degeneration following axotomy. Using this assay, we identified 11 compounds from a library of 480 that delay degeneration of severed axons in vitro in a dose-dependent manner. Among these, several have biologic targets with previously reported involvement in axon degeneration, however, our findings also reveal that inhibitors of IκB kinase (IKK)2 and glycogen synthase kinase 3 (GSK3) suppress axotomy-induced axon degeneration. Finally, we demonstrate through shRNA-mediated knockdown that IKK and GSK3 are required to promote rapid axon degeneration.
EXPERIMENTAL PROCEDURES
DRG Neuron Culture
Pregnant female mice at 12 days post-coitus (Charles River Laboratories) were anesthetized prior to cervical dislocation. The dorsal root ganglia (DRG) were isolated from embryos in DMEM (Sigma), dissociated by incubation in 0.25% trypsin, and resuspended (100 μl/embryo) in complete DRG medium: neurobasal medium (Invitrogen) containing 2% B27 supplement (Invitrogen), 50 ng/ml of nerve growth factor (Harlan Laboratories), 1 μm 5-fluoro-2′-deoxyuridine (Sigma), and 1 μm uridine (Sigma). A single 0.5-μl droplet of this concentrated cell suspension containing ∼200 neurons was delivered to the dry surface of each well in 96-well coated cell culture plates (poly-d-lysine, Sigma; Laminin, Invitrogen) at a 1.5-mm offset from the well center using a Biomek FX Liquid Handler (Beckman Coulter). After 15 min incubation at 37 °C, after all neurons adhered to a 1-mm diameter region of the plate surface, 80 μl of complete DRG media was added to each well. All experiments were performed at 7 days in vitro, allowing axons to grow across the plate surface (Fig. 1B).
FIGURE 1.

In vitro axotomy model. A, DRGs were dissected from E12.5 mouse embryos and dissociated in trypsin (5 × 105 neurons/ml). Cell suspensions were delivered as single 0.5-μl droplets to the dry laminin/PDL-coated surface of each well in a 96-well microtiter plate with a liquid handling machine. Medium was then added after cells had adhered to a 1–2-mm portion of the well. B, montage of DRG spot culture at 7 days in vitro. Boxes indicate imaging regions (a, distal/injured; b, proximal/uninjured); red = β-tubulin (Tuj1 antibody), blue = DAPI. Dashed white line indicates where axons are cut (axotomy). Well diameter = 7 mm. C, screening time line: after a 30-min preincubation with compound, axons were severed with a blade, and axon integrity was quantified from brightfield images of axons taken at 0, 6, and 24 h post-axotomy. Axon fragmentation was quantified from each image (see supplemental Fig. S1). Representative images are pseudo-colored by particle circularity (see “Experimental Procedures”).
Compound Addition and Axonal Injury
The ICCB Known Bioactives Library (Enzo Life Sciences) comprises 480 compounds dissolved in DMSO arrayed in 96-well plates. In the primary screen, to screen each drug at below toxic concentrations, each compound was delivered to the cells at three separate dilutions from library stock (1:5 × 104, 1:5 × 105, and 1:5 × 106; note: the initial stock concentrations vary by compound). The compound was diluted in neurobasal media and delivered to cell culture plates using the Biomek FX 30 min prior to mechanical injury. Following preincubation, axons in each plate were manually severed with a flat surgical blade, visually guided under a light microscope. This process required 10–12 min per assay plate.
NGF withdrawal was performed in DRG cultures at 7 days in vitro by washing 3 times with media lacking NGF prior to addition of media containing anti-NGF antibody (3). Images were acquired 24 h post-insult.
Image Acquisition and Data Analysis
At three time points following axotomy (0, 6, and 24 h), axons distal to the site of transection (Fig. 1B, panel a) were visualized by automated brightfield microscopy using an InCell Analyzer 1000 fitted with a ×20 objective (GE Healthcare). From each of the 9 images of distal axon segments acquired per well, axon fragmentation was quantified using an ImageJ script modified from that described previously (10). Briefly, following background subtraction, each image was binarized based on pixel intensity to create a map of axon area. Each particle of contiguous pixels was judged either intact or fragmented based on its circularity, defined as circularity = (area)/(π × radius2), where area and radius are measured in pixels. Particles with circularity of more than 0.2 were designated fragmented. The total pixel area of fragmented particles was divided by the total axon area to give the degeneration index (DI) (supplemental Fig. S1) using,
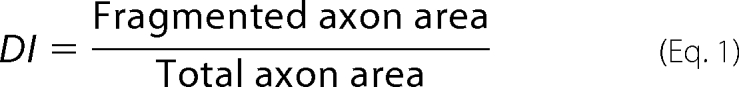 |
DI values of distal axons were averaged for each well. To facilitate comparisons between treatments, we assigned a normalized protective activity to each compound for both 6 and 24 h post-axotomy,
where ΔDI is the change in degeneration index from baseline (e.g. ΔDI24 h = DI24 h − DI0 h) and DImedian is the median DI for each assay plate.
In addition to the images of injured axons acquired at three time points, two images of uninjured axons proximal to the site of injury in each well was acquired at 24 h post-axotomy to monitor toxicity of each compound (Fig. 1B, panel b). Finally, following imaging at 24 h post-axotomy, cells were incubated with 20 μm ethidium homodimer (Botium Inc.), and fluorescent and brightfield images of cell bodies were acquired to monitor cell death as a second marker of toxicity for each compound. For each compound, the screening activity of the highest non-toxic dose was used for candidate hit selection.
Three controls were employed to monitor assay performance: 1) vehicle control (0.2% DMSO); 2) uninjured control; and 3) cytosol-localized nicotinamide mononucleotide adenyltransferase 1 (cytNmnat1) lentivirus. A minimum of four replicates of each control was monitored for each set of assay plates derived from the same cell suspension (i.e. each independent cell culture event).
Criteria for exclusion of images were established to limit the effects of unwanted variables. Images with very low axon density or with abnormally large opaque objects were excluded. All wells in which the degeneration index at 0 h post-axotomy was above 0.3 (30 wells; 2% of those screened), usually resulting from rapid drug toxicity, were excluded from further analysis.
Candidate Hit Selection and Validation
To choose compounds for validation testing, we employed criteria based on both absolute (degeneration index) and relative (protective activity) metrics: all compounds exhibiting a degeneration index below 0.7 and a protective activity greater than 2.5 median absolute deviations above the median protective activity at either time point were selected. The resulting list included 25 compounds. For each candidate hit, we performed duplicate dose-response curves. Starting with a maximum concentration 5-fold above the initial screening dose, compounds were 2-fold serially diluted in DMSO (total of 8 concentrations), prior to addition to DRG cultures. Axons were injured as in the primary screen and axon degeneration was quantified at 8 and 24 h post-axotomy as above. Degeneration indices were plotted as a function of compound concentration and sigmoid curves with Equation 3,
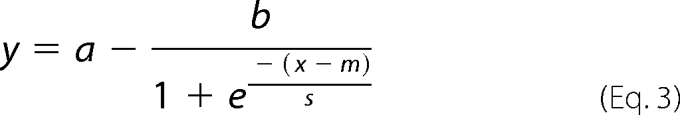 |
which were fit using the Matlab curve fitting tool. High concentrations were excluded from each fit if toxicity was observed in proximal axons.
Lentiviral Transduction
A small hairpin RNA (shRNA) interference vector targeting murine GSK3β was produced by annealing oligonucleotides containing the targeting sequence (5′-GAACCGAGAGCTCCAGATC-3′) (11) and a loop sequence into a lentiviral vector with a U6 promoter and followed by a SV40 promoter-Venus fluorescent protein cassette (3). Human GSK3β-CA (12) and GSK3β-KD mammalian expression vectors from the laboratory of Dr. Jim Woodgett were obtained through Addgene and subcloned into a lentiviral expression vector containing a ubiquitin promoter and GFP marker (13). Dominant-negative IκBα (14) was also subcloned into this vector. IKKβ shRNA viral vectors from the RNAi Consortium collection (15) were acquired from the Washington University RNAi Core (targeting sequences: shIKKβ-1, GCTGCACATTTGAATCTGTAA; shIKKβ-2, GCTCTTAGATACCTTCACGAA). GSK3 shRNA, luciferase shRNA control, cytNmnat1 (16), GSK3β-CA, GSK3β-KD, and Bcl-XL (3) viral expression vectors were used to produce lentiviral particles as described (16). Lentiviral transduction was accomplished by adding 5–10 μl of each lentivirus-containing media (∼103 particles/μl; final volume 85–100 μl/well) directly to DRG neurons at 2–3 days in vitro.
Quantitative RT-PCR
Knockdown efficiency of shRNA constructs was tested using DRG neuronal cultures grown in 24-well plates (∼104 cells/well). Cells were infected using 30 μl of lentivirus-containing supernatant (104-105 infectious particles) after 2 days in vitro. RNA was harvested after 7 days in vitro and purified by phenol-chloroform extraction using Ribozol reagent (Amresco). Transcript levels relative to GAPDH were determined by qPCR (standard curve method) using an ABI Prism 7900HT sequence detection instrument (Applied Biosystems).
Immunocytochemistry
DRG neurons were fixed in 4% paraformaldehyde, treated with 0.1% Triton X-100 in PBS with 5
BSA, and incubated with polyclonal β3-tubulin antibody (Covance; 1:1000) and monoclonal 2H3 antibody (DSHB; 1:200) overnight at 4 °C. The bound antibodies were visualized using anti-mouse Cy3 and anti-rabbit Alexa 488 antibodies (1:500, 25 °C, 1 h).
NFκB Reporter Assay
NFκB activity was measured using pκB5-FLuc, which has 5 κB motifs controlling expression of firefly luciferase (17). A Renilla luciferase plasmid (pRLuc) was employed as a transfection control. These plasmids were cotransfected into HEK 293T cells with either a dominant-negative IκBα expression construct (14) or empty vector (plasmid ratio: pRLuc/pκB5-FLuc/vector = 0.2/1/1) using the FuGENE 6 reagent (Roche Applied Science). After 2 days, cells were treated with 0, 8, or 128 ng/ml of recombinant mouse TNFα (Prospec) and after 24 h cells were harvested and assayed for FLuc and RLuc activity using the Dual Luciferase Reporter Assay System (Promega).
Statistical Analyses
All computational analyses including Student's t test, Z′ factor calculation, sigmoid curve fitting, assignment of protective activity, and plotting of data were performed using Matlab software. One-tailed Student's t tests were performed adjusting for a familywise type 1 error rate of 0.001 by the Holm-Bonferroni method. The assay Z′ factor was calculated from pooled positive (cytNmnat1) and negative (DMSO) control degeneration index values from our primary screen. Each time point was analyzed separately. Four wells each of cytNmnat1-treated and DMSO-treated neurons from six independent cell cultures were pooled and analyzed and Z′ score was calculated,
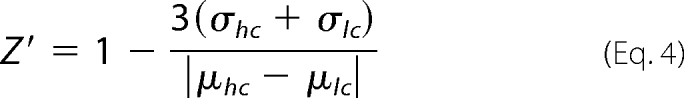 |
where hc and lc are high and low controls (DMSO, cytNmnat1), respectively (18).
RESULTS
Cell Culture Model
To develop a screening model for axon degeneration, we began by miniaturizing an existing model of in vitro axon degeneration using murine DRG sensory neurons cultured such that their cell bodies are sequestered in a cluster from which their axons grow radially (10). This arrangement permits experimental transection of the axons and imaging of the distal and proximal segments. We used liquid handling robotics to scale this model such that it could be performed in 96-well microtiter plates. Because DRG neurons grow very long axons, as few as 200 neurons are required per well, allowing the production of one 96-well assay plate from a single mouse embryo (Fig. 1A).
Quantification of Axon Degeneration
Axonal degeneration is visualized as a series of sequential morphological changes that begin with swelling and blebbing and culminate in fragmentation that is complete by 24 h in vitro. To quantify axon degeneration dynamically at multiple time points, we employed automated microscopy and image analysis. The extent of axon fragmentation can be quantified from brightfield or phase-contrast images of axons using a previously described image analysis algorithm that distinguishes fragmented from intact axonal segments (10). We acquired images of axons both distal and proximal to the site of transection (Fig. 1B, a and b boxes, respectively). Sampling the stable proximal axon segment allowed for detection of axonal toxicity caused by experimental treatments (e.g. drugs). From these images, axonal fragmentation was quantified and reported as: degeneration index − the ratio of fragmented axon area to total axon area in each field (supplemental Fig. S1). Based on our preliminary studies, we chose to quantify axon degeneration at three time points post-axotomy (Fig. 1C). Images were acquired: 1) immediately after axotomy to assure baseline axon integrity; 2) 6 h post-axotomy; and 3) 24 h post-axotomy. These time points were chosen based on mechanistic considerations; axon swelling and bleb formation occurs by 6 h in most axons. Importantly, the image analysis method employed in this screen is sensitive to axon swellings and blebs as these membrane changes disrupt axon uniformity and result in isolated round particles upon image segmentation. We reasoned that some compounds may affect early axonal changes such as swelling without blocking their eventual fragmentation. By 24 h post-axotomy, conversely, axon fragmentation is complete. Screening at this time point was performed to identify treatments that might only affect steps downstream of swelling such as cytoskeletal breakdown and to assess overall axonal protection.
Compound Library and Screen
We screened the ICCB Known Bioactives Library of 480 compounds (Enzo) for those that delay axon degeneration following axotomy. To facilitate comparisons between compounds, we developed a normalized metric called the “protective activity” of each compound that shows the extent each compound preserved axon morphology relative to median values for each assay plate (note that the protective activity of 1 is complete preservation and 0 is no effect, whereas negative values simply indicate an extent of fragmentation above the median at the indicated time point) (see Fig. 2A and “Experimental Procedures”). The distribution of protective activities of all compounds at both 6 and 24 h are plotted in Fig. 2A. Representative images from the primary screen that demonstrate high protective activity at 6 or 24 h, or both time points, are presented in Fig. 2E. To monitor assay performance throughout our screen, each assay plate contained controls including cytosol-localized Nmnat1 (cyt-Nmnat1) lentivirus, which blocked axon degeneration, and 0.2% DMSO (Fig. 2B). Based on these controls, the Z′ scores for our assay at 6 and 24 h were 0.16 and 0.66, respectively.
FIGURE 2.

Primary screen results and validation. A, normalized protective activity of all compounds tested (n = 480). The activity of the highest nontoxic dose of each compound is shown. Protective activity is calculated as: 1 = (ΔDIcompound)/(ΔDImedian), where ΔDI is the change in DI from baseline (see text). Validated hits (see text and panel C) are depicted as filled red circles and labeled. See Table 1 for compound activities. B, histograms showing distribution of degeneration indices (before normalization) for controls and compound-treated wells at each time point. Injured controls were treated with DMSO (0.2%), whereas Nmnat1 controls were treated with cytNmnat1 lentivirus. C, dose-response validation curves for representative compounds at 8 h post-axotomy. To monitor toxicity at each concentration, images of proximal/uninjured axons were acquired within each well. D, axon fragmentation time course with selected compounds. For 5 selected compounds, axon degeneration was measured at 7 time points post-axotomy in the presence of a predetermined optimal dose. All compounds tested suppressed axon degeneration at early time points, but after 22 h only some treatments resulted in reduced fragmentation. Nifedipine = calcium channel blocker, 10 μm; indirubin 3′-monoxime = GSK3 inhibitor, 5 μm; Bay 11-7082 = IKK inhibitor, 4 μm; gliotoxin = proteasome inhibitor, 2 μm; TPCK = protease inhibitor, 28 μm. E, heterogeneity in axon protection phenotype. Calcium channel inhibitor nifedipine (panels d, e, and f) protected axons from swelling at 6 h but did not block fragmentation at 24 h. Calpain inhibitor MDL-28170 did not block axon swellings at 6 h (h) but blocked the majority of axon fragmentation at 24 h (i). Proteasome inhibitor gliotoxin suppressed both swelling and fragmentation at 6 (k) and 24 (l) h post-injury. Scale bar = 100 μm.
In addition to screening compounds for their protective activity, images of proximal axons were obtained to assess which compounds in the library caused axon degeneration directly. Cell death was similarly monitored for each compound by imaging cell bodies following ethidium homodimer administration at the assay end point (24 h) (supplemental Fig. S2).
Validation of Hits by Dose-dependent Axon Protection
From our initial screen, we selected 25 compounds for subsequent analysis based on 6 and 24 h protective activity (see “Experimental Procedures”). Further validation of these compounds was sought by performing an extended dose-response curve using the same experimental platform. Eleven of those tested exhibited a dose-dependent decrease in axon degeneration at 8 or 24 h (see Fig. 2C and Table 1). Using the optimal compound concentrations obtained from these curves, we assessed the duration of axon protection for select compounds with validated activity at 8 h post-axotomy. Quantifying axon degeneration at various time points post-axotomy, we found varied durations of axon protection among different compounds (Fig. 2D).
TABLE 1.
Validated hits and reported activity
| Compound name | ICCB: reported compound activity |
|---|---|
| Gliotoxin | Inhibitor of 20S-proteasome chymotrypsin activity |
| Bay 11-7082 | Inhibits IKK kinase activation |
| Indirubin-3′-monoxime | GSK3 inhibitor |
| MDL-28170 | Calpain inhibitor |
| Nifedipine | L-type voltage-gated calcium channel inhibitor |
| Ala-Ala-Phe-CMK | Tripeptidyl peptidase II inhibitor |
| Tosyl-Phe-CMK (TPCK) | Serine protease inhibitor |
| Ac-Leu-Leu-Nle-CHO | Calpain inhibitor |
| MG-132 | Proteasome inhibitor |
| SB-415286 | GSK3 inhibitor |
| Parthenolide | IKK inhibitor |
Identification of Hits with Previously Reported Activity
The veracity of our screening methodology is highlighted by the identification of several compounds whose targets have been previously implicated in axon degeneration. For example, we identified proteasome inhibitors including MG132, which was previously shown to delay axon degeneration (8), and Gliotoxin, which provided a very robust axon-protective effect. The protease inhibitor TPCK was strongly protective at high concentrations (Fig. 2D) and is structurally related to tosyl-l-lysine chloromethyl ketone, a compound reported to delay axon degeneration (19). The calcium channel blocker nifedipine and calpain inhibitors (MDL-28170, Ac-Leu-Leu-Nle-CHO) are also well characterized inhibitors of axon degeneration (9) identified in this screen. In addition to identification of compounds consistent with previous reports, our screen identified additional axonal protective agents including the protease inhibitor Ala-Ala-Phe-CMK, two inhibitors of IKK (Bay 11-7082 and parthenolide), and two inhibitors of GSK3 (indirubin 3′-monoxime and SB-415286).
IKK and GSK3 Promote Axon Degeneration
Because GSK3 and IKK have not been implicated in axon degeneration, we sought to further validate these targets genetically. Using lentiviral shRNA vectors targeting murine IKKβ that each reduced IKKβ transcript levels in DRG neurons (shIKKβ1 70%; shIKKβ2 65%), we found that knockdown delayed axon degeneration following axotomy compared with an off-target control (shLuc; Fig. 3, A and D). To assess whether IKK-mediated activation of NFκB was required for rapid axon degeneration, we infected DRG neurons with a dominant-negative IκBα expression vector that blocks IKK-mediated NFκB activation (14). We found that this construct did not delay axon degeneration (Fig. 3, A and D), although it did potently suppress both basal and TNFα-induced NFκB reporter activity in 293T cells (Fig. 3B). This finding suggests that IKK promotes axon degeneration via an alternate target.
FIGURE 3.

IKK and GSK3 are required for rapid axon degeneration. A, knockdown of murine IKKβ by shRNA (shIKKβ 1, 2) suppressed axon degeneration measured at 9 h post-axotomy. Expression of dominant-negative IκBα had no effect. B, dominant-negative IκBα suppresses basal and TNFα-induced NFκB activity in HEK 293T cells. C, knockdown of murine GSK3α and GSK3β (shGsk3) by shRNA suppresses axon degeneration at 24 h post-axotomy, whereas expression of constitutively active (CA), but not kinase-dead (KD) human GSK3β (h-GSK3β) restored normal axon degeneration in this setting. D, representative images from panel A. E, representative images from panel C. F, protective effects of the indicated compounds when added at different times post-axotomy. Addition of compounds (indirubin 3′-monoxime, 5 μm; Bay 11-7082, 4 μm; gliotoxin, 2 μm; nifedipine, 10 μm; TPCK, 28 μm) 1 h prior (a) or minutes after axotomy (b) suppressed axon degeneration measured 9 h post-axotomy (each compound compared with DMSO added at the same time), whereas neither compound suppressed axon degeneration when added 2 h post-axotomy (c). Error bars show S.E.; *, p < 0.001 by Student's t test; N.S., not significant (p > 0.01); 4 replicates per group.
Because both murine GSK3 isoforms can be successfully knocked down with a single shRNA (11), we used lentivirus-mediated RNAi (shGsk3) to knock down murine GSK3β and GSK3α transcript levels by 95 and 75%, respectively. As knockdown was found to be toxic to DRG neurons (data not shown), experiments were performed using DRG neurons expressing Bcl-XL, which blocks shRNA-mediated toxicity but importantly does not alter axotomy-induced axon degeneration (3). Knockdown of murine GSK3 suppressed axon degeneration at 24 h post-axotomy compared with control shRNA delivery. Normal axon degeneration was restored by expression of constitutively active (CA) human GSK3β (12), whereas a kinase dead (KD) human GSK3β had no compensatory effects (Fig. 3, C and E).
IKK and GSK3 Inhibition Must Be Administered Early After Axotomy to Achieve a Protective Effect
Axonal fragmentation in severed DRG neurons is not apparent for 4–6 h after axotomy; however, the axon-protective effects of JNK inhibitors were achieved only when the compounds were present during the first 3 h post-axotomy (20), indicating that JNK activity during this latent period might commit axons to later fragmentation. To ask whether IKK and GSK3 might play a similar early role in the degeneration cascade, we tested inhibitors of these kinases as well as other hits from the screen in 3 treatment regimens: compounds were added (a) 1 h prior to axotomy; (b) immediately after axotomy (0–3 min); or (c) 2 h post-axotomy (Fig. 3F). As with JNK inhibition, both IKK and GSK3 inhibitors effectively suppressed axon degeneration when compounds were added prior to or immediately after axotomy, but not when added 2 h post-axotomy, indicating an early role for each kinase in the axon degeneration cascade. A similar pattern was observed for the proteasome inhibitor Gliotoxin and protease inhibitor TPCK; however, calcium channel blocker Nifedipine provided protection even when added 2 h post-axotomy.
Because axon degeneration involves disassembly of a dense cytoskeleton comprised of neurofilament and microtubules (8), we asked whether these cytoskeletal elements might be preserved in injured axons treated with IKK inhibitors. We found that IKK inhibition had no effect on microtubule disassembly as indicated by a punctate β3-tubulin staining pattern at 24 h post-axotomy. Interestingly, however, we found that immunoreactivity of the neurofilament medium polypeptide (Nefm/Nf-160), a protein normally degraded in degenerating axons in a proteasome-dependent manner (8), was maintained in severed axons treated with IKK inhibitors (Fig. 4). These results indicate that IKK plays a role in the disassembly or degradation of neurofilament proteins in degenerating axons.
FIGURE 4.

IKK inhibition selectively blocks neurofilament degradation in injured axons. DRG neuronal cultures were treated with IKK inhibitors Bay 11-7082 (3 μm) or parthenolide (6 μm) for 30 min prior to axotomy. Phase and immunofluorescent images of distal axons were acquired 24 h post-axotomy. Injured control axons showed complete loss of neurofilament immunoreactivity and microtubule fragmentation. IKK inhibition led to preservation of neurofilament immunoreactivity but did not affect microtubule integrity.
Finally, to next ask whether the involvement of IKK and GSK3 in axonal degeneration is specific for mechanical injury, we tested whether each kinase is required for axon degeneration induced by trophic factor withdrawal, an insult that elicits axon degeneration through a different mechanism (3, 21). GSK3 inhibition (2 μm indirubin 3′-monoxime) potently blocked axon degeneration after 24 h NGF deprivation (Fig. 5A). Similarly, knockdown of IKK suppressed trophic withdrawal-induced degeneration (Fig. 5B). Taken together, these findings suggest both IKKβ and GSK3 play important roles in axonal degeneration in response to multiple damaging stimuli.
FIGURE 5.

GSK3 and IKK promote axon degeneration following trophic factor withdrawal. DRG neurons were deprived of NGF for 24 h in the presence or absence of GSK3 inhibitors indirubin 3′-monoxime (2 μm) (A) or IKKβ shRNA lentivirus (B). Axon degeneration was quantified from phase images for all treatment groups; n = 4; *, p = 0.001 by Student's t test; scale bar = 100 μm.
DISCUSSION
In this study we present an image-based screening method to identify new members of the axon degeneration signaling cascade. Our unbiased screen of 480 bioactive compounds successfully identified previously characterized members of this cascade as well as two novel members, IKK and GSK3, which we have validated through genetic knockdown studies. The value of the screening method we describe here is 2-fold: 1) it will facilitate the identification of biological signals promoting normal axon degeneration using drug and genome-scale shRNA libraries; and 2) it can serve as a drug-discovery platform to identify therapeutics for neuropathies and other neurologic disorders in which axon degeneration is a major component. High-content screening is a powerful tool for generating new hypotheses for the study of enigmatic pathways. Screening tools for neuronal biology, however, have remained limited as primary neurons and other post-mitotic cells are generally poor substrates for high-content analysis. Unlike dividing cells, the abundance of primary post-mitotic cells is often limited to that obtained through tedious dissection. However, relatively abundant DRG neurons, with long axons extending across a 96-well microtiter plate, are an apt substrate for high-content analysis of axon biology. Using as few as 200 cells per well (∼1 embryo per 96-well assay plate), DRG axon morphology can be readily queried from brightfield images. Although the assay described herein was developed to study axon degeneration following mechanical injury, the methods described here could be adapted for screens focused on axon degeneration in models of neuropathy and neurodegenerative disease.
A particular strength of this assay is that axon degeneration is quantified from brightfield images. This allows for rapid live imaging of wild-type neurons at multiple time points, providing a more detailed analysis of axon degeneration dynamics. For instance, we found a disparity between the protective activity of some compounds at 6 and 24 h (Fig. 2, A and E). Several compounds that suppressed axonal changes at 6 h post-axotomy had no effect at 24 h (e.g. Ca2+ channel blocker nifedipine). These compounds may have incompletely blocked their biological target or perhaps blocked one of multiple redundant signals promoting normal axon degeneration. More surprisingly, some compounds partially preserved axon morphology at 24 h but not at 6 h (e.g. calpain inhibitors MDL-28170 and Ac-Leu-Leu-Nle-CHO). Because the changes seen at 6 h are predominantly axon swellings (see Fig. 2E, panels b and h), it is likely that these compounds block axon fragmentation without affecting the earlier, more modest morphologic changes. Previous reports have linked calpains to degradation of neurofilament, the principal cytoskeletal component in axons (9). Perhaps axon swelling without concomitant cytoskeletal disassembly results in incomplete fragmentation as observed here when these treatments were tested. These observations are consistent with the idea that the activities of multiple pathways are required for the degradation of various components of the axon and its eventual fragmentation.
We have previously reported that c-Jun N-terminal kinase (JNK) and its upstream activator dual leucine kinase (DLK)/Wallenda are required to promote axon degeneration of normal dynamics following injury (20). In the current study, we show that additional stress-activated kinases IKK and GSK3 are similarly required to promote rapid axon degeneration. Taken together, these findings suggest the existence of a signaling network that promotes the fragmentation of compromised axons. Moreover, we demonstrate that, as with JNK inhibitors, IKK and GSK3 inhibitors must be present during the first 2-h post-axotomy to confer a protective effect, indicating an early role for these kinases in the axon degeneration cascade. That each compound suppresses axon degeneration even when added after axonal severing indicates that these kinases act locally within the axon to promote fragmentation. JNK, IKK, and GSK3 can all affect transcriptional programs; however, it is unlikely that transcriptional regulation is important here because the severed axon is physically uncoupled from the nucleus. Understanding the pathways regulated by these kinases in the axonal degeneration context is an important future goal, as they could represent important therapeutic targets for neuropathy and neurodegeneration.
Although IKK and GSK3 have not been implicated in axon degeneration, each of these kinases has been linked to neuronal death. For example, GSK3 is involved in neuronal apoptosis in response to trophic factor withdrawal in neural precursor cells and cortical neurons (22, 23). Furthermore, in rat sympathetic neurons, GSK3 inhibition blocked a pro-apoptotic retrograde signal originating in NGF-deprived axons in compartment cultures (24). Our finding that GSK3 mediates degeneration of severed axons independent of apoptosis therefore broadens its role in neuronal degeneration. Interestingly, an IKK-related kinase (Ik2) has been shown to mediate dendrite pruning in developing Drosophila larvae through a mechanism that involves a member of the Katanin family of microtubule severing proteins (25). Interestingly, in contrast to the dendrites, axons of these developing neurons are spared. Drosophila Ik2 is more closely related to the mammalian IKK-related kinases Tank binding kinase 1 (TBK1) and IKKϵ, suggesting that these related kinases could mediate distinct forms of neuronal process degeneration.
Our screen of the ICCB library provided useful insights into the mechanism of axon degeneration through the identification of new participants. It is worth noting, however, that none of the compounds tested in this screen provided complete protection of axons at 24 h post-axotomy. This level of protection is in contrast with that provided by expression of Wallerian degeneration slow (Wlds) protein or its active component nicotinamide mononucleotide adenyltransferase 1 (Nmnat1), which protect axons from degeneration beyond 72 h in vitro. Further screening using larger libraries of compounds or genome-scale RNAi libraries will likely identify additional, more robust protective agents and provide new clues to understanding the axonal degeneration process.
Supplementary Material
Acknowledgments
We thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes-Jewish Hospital, St. Louis, MO, for the use of the High-Throughput Screening Core. The Siteman Cancer Center is supported in part by National Institutes of Health NCI Cancer Center Support Grant P30 CA091842. The Screening Core is supported by NIH Grant P50 CA91842 and an Anheuser-Busch/Emerson challenge gift. We also thank Aaron DiAntonio and Martha Bhattacharya for help with manuscript preparation, Scott Wildman for help with compound library selection, and S. Joshua Swamidass for help with the development of methods.
This work was supported, in whole or in part, by National Institutes of Health Grants AG13730 (to J. Milbrandt) and NS065053 and NS070053 (to J. Milbrandt and A. DiAntonio), NIH Neuroscience Blueprint Center Core Grant P30 NS057105 to Washington University, the HOPE Center for Neurological Disorders, and Craig H. Neilsen Foundation Grant 124030 (to J. Milbrandt).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- IKK
- IκB kinase
- GSK3
- glycogen synthase kinase 3
- DRG
- dorsal root ganglia
- RLuc
- Renilla luciferase
- FLuc
- firefly luciferase
- TPCK
- l-1-tosylamido-2-phenylethyl chloromethyl ketone
- DMSO
- dimethyl sulfoxide
- DI
- degeneration index.
REFERENCES
- 1. Coleman M. (2005) Nat. Rev. Neurosci. 6, 889–898 [DOI] [PubMed] [Google Scholar]
- 2. Saxena S., Caroni P. (2007) Prog. Neurobiol. 83, 174–191 [DOI] [PubMed] [Google Scholar]
- 3. Vohra B. P., Sasaki Y., Miller B. R., Chang J., DiAntonio A., Milbrandt J. (2010) J. Neurosci. 30, 13729–13738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kuo C. T., Jan L. Y., Jan Y. N. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 15230–15235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schoenmann Z., Assa-Kunik E., Tiomny S., Minis A., Haklai-Topper L., Arama E., Yaron A. (2010) J. Neurosci. 30, 6375–6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rumpf S., Lee S. B., Jan L. Y., Jan Y. N. (2011) Development 138, 1153–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Watts R. J., Hoopfer E. D., Luo L. (2003) Neuron 38, 871–885 [DOI] [PubMed] [Google Scholar]
- 8. Zhai Q., Wang J., Kim A., Liu Q., Watts R., Hoopfer E., Mitchison T., Luo L., He Z. (2003) Neuron 39, 217–225 [DOI] [PubMed] [Google Scholar]
- 9. George E. B., Glass J. D., Griffin J. W. (1995) J. Neurosci. 15, 6445–6452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sasaki Y., Vohra B. P., Lund F. E., Milbrandt J. (2009) J. Neurosci. 29, 5525–5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim W. Y., Zhou F. Q., Zhou J., Yokota Y., Wang Y. M., Yoshimura T., Kaibuchi K., Woodgett J. R., Anton E. S., Snider W. D. (2006) Neuron 52, 981–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stambolic V., Woodgett J. R. (1994) Biochem. J. 303, 701–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Araki T., Sasaki Y., Milbrandt J. (2004) Science 305, 1010–1013 [DOI] [PubMed] [Google Scholar]
- 14. Sun S., Elwood J., Greene W. C. (1996) Mol. Cell. Biol. 16, 1058–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moffat J., Grueneberg D. A., Yang X., Kim S. Y., Kloepfer A. M., Hinkle G., Piqani B., Eisenhaure T. M., Luo B., Grenier J. K., Carpenter A. E., Foo S. Y., Stewart S. A., Stockwell B. R., Hacohen N., Hahn W. C., Lander E. S., Sabatini D. M., Root D. E. (2006) Cell 124, 1283–1298 [DOI] [PubMed] [Google Scholar]
- 16. Sasaki Y., Vohra B. P., Baloh R. H., Milbrandt J. (2009) J. Neurosci. 29, 6526–6534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moss B. L., Gross S., Gammon S. T., Vinjamoori A., Piwnica-Worms D. (2008) J. Biol. Chem. 283, 8687–8698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Birmingham A., Selfors L. M., Forster T., Wrobel D., Kennedy C. J., Shanks E., Santoyo-Lopez J., Dunican D. J., Long A., Kelleher D., Smith Q., Beijersbergen R. L., Ghazal P., Shamu C. E. (2009) Nat. Methods 6, 569–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ikegami K., Kato S., Koike T. (2004) Brain Res. 1030, 81–93 [DOI] [PubMed] [Google Scholar]
- 20. Miller B. R., Press C., Daniels R. W., Sasaki Y., Milbrandt J., DiAntonio A. (2009) Nat. Neurosci. 12, 387–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nikolaev A., McLaughlin T., O'Leary D. D., Tessier-Lavigne M. (2009) Nature 457, 981–989 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22. Eom T. Y., Roth K. A., Jope R. S. (2007) J. Biol. Chem. 282, 22856–22864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hetman M., Cavanaugh J. E., Kimelman D., Xia Z. (2000) J. Neurosci. 20, 2567–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mok S. A., Lund K., Campenot R. B. (2009) Cell Res. 19, 546–560 [DOI] [PubMed] [Google Scholar]
- 25. Lee H. H., Jan L. Y., Jan Y. N. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 6363–6368 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.