

Abstract
The presynaptic protein α-synuclein (α-syn), particularly in its amyloid form, is widely recognized for its involvement in Parkinson disease (PD). Recent genetic studies reveal that mutations in the gene GBA are the most widespread genetic risk factor for parkinsonism identified to date. GBA encodes for glucocerebrosidase (GCase), the enzyme deficient in the lysosomal storage disorder, Gaucher disease (GD). In this work, we investigated the possibility of a physical linkage between α-syn and GCase, examining both wild type and the GD-related N370S mutant enzyme. Using fluorescence and nuclear magnetic resonance spectroscopy, we determined that α-syn and GCase interact selectively under lysosomal solution conditions (pH 5.5) and mapped the interaction site to the α-syn C-terminal residues, 118–137. This α-syn-GCase complex does not form at pH 7.4 and is stabilized by electrostatics, with dissociation constants ranging from 1.2 to 22 μm in the presence of 25 to 100 mm NaCl. Intriguingly, the N370S mutant form of GCase has a reduced affinity for α-syn, as does the inhibitor conduritol-β-epoxide-bound enzyme. Immunoprecipitation and immunofluorescence studies verified this interaction in human tissue and neuronal cell culture, respectively. Although our data do not preclude protein-protein interactions in other cellular milieux, we suggest that the α-syn-GCase association is favored in the lysosome, and that this noncovalent interaction provides the groundwork to explore molecular mechanisms linking PD with mutant GBA alleles.
Keywords: Fluorescence, NMR, Protein Conformation, Protein-Protein Interactions, Synuclein
Introduction
Parkinson disease (PD)4 is an age-related movement disorder that features the accumulation and deposition of amyloids, insoluble aggregates enriched in α-synuclein (α-syn), a small (14 kDa) presynaptic protein of ill-defined function (1–4). These classic PD neuropathologic aggregates, known as Lewy bodies (LBs), are also associated with other neurodegenerative disorders including dementia with LBs and multiple system atrophy (4, 5). Three missense α-syn mutations, A30P, E46K, and A53T (6–8), and gene duplications or triplications are strongly associated with early-onset PD (9, 10), although the specific pathogenic role of α-syn remains to be elucidated.
Because abnormally misfolded proteins and amyloid deposits are found in other neurodegenerative diseases such as Alzheimer disease and prion encephalopathies (11), considerable research has focused at gaining molecular insights into fibril formation (12–17) and biomolecules that stimulate/inhibit this process (18–21). For example, lipid-protein interactions are of particular interest because α-syn localizes near synaptic vesicles (2, 18) and is thought to act as a chaperone in SNARE complex assembly, which is necessary for neurotransmitter release from presynaptic vesicles (23). Potential α-syn-mediated cytotoxicity of cellular targets such as mitochondria, lysosomes, and other proteolytic machinery are being investigated (3, 24, 25). It has been proposed that misfolded α-syn conformers can overwhelm, and hence impair normal cellular protein degradation pathways (26, 27).
In the past decade, several genes, including SNCA, PRKN, PINK1, DJ-1, and LRRK2 (6, 28–31) have been identified that cause parkinsonism. Mutations in SNCA and LRRK2 result in autosomal dominant forms, pointing to gain-of-toxicity mechanisms involving α-syn and leucine-rich repeat kinase 2, the two respective encoded proteins (6, 29). Moreover, emerging data including clinical observations, neuropathologic evaluations, family studies, and genetic analyses now point to a new association between GBA, the gene encoding for glucocerebrosidase (GCase, also known as acid β-glucosidase), the enzyme deficient in the lysosomal storage disorder Gaucher disease (GD), and the development of PD and related synucleinopathies (32–37).
Early observations identified patients with GD and their heterozygous relatives who developed parkinsonism manifestations (33). Subsequently, multiple independent reports (38) and an international multicenter study of over 5000 patients and an equal number of controls, established that PD patients are over five times more likely to carry a mutation in GBA (39). Importantly, autopsy studies of Gaucher patients and carriers with synucleinopathies reveal the presence of mutant GCase in α-syn positive LBs, suggesting that a potential relationship between the two proteins may contribute to PD pathogenesis (40).
GCase is a 497-residue lysosomal hydrolase that catalyzes the metabolism of the glycolipid glucosylceramide to ceramide and glucose (41). GD results from deficient GCase, because of inactivity, misfolding, and/or failure of the enzyme to reach the lysosome, leading to the accumulation of glucosylceramide (41). Approximately 300 different GBA mutations have been identified, although several distinct mutations are more frequent (42, 43). There are three types of GD, with type 1 accounting for the majority of the affected individuals (41). Types 2 and 3 are the acute and sub-acute neuronopathic forms, respectively.
α-Syn is predominantly degraded by lysosomes, in part by chaperone-mediated autophagy (24, 26, 44). It also has been shown that protein turnover is slowed in mouse models of lysosomal storage diseases (45). Therefore, it is reasonable to question whether there could be a link between α-syn clearance and GCase levels within lysosomes. Indeed, one hypothesis suggests that when mutated, GCase may contribute to aberrant α-syn aggregation and increase intracellular levels of the protein (46, 47). Conversely, it has been proposed that impaired ceramide metabolism triggers cell death (47, 48). For example, LBs may be a cellular response to altered ceramide concentration. Though there is no current consensus as to whether and how enzyme or lipid mediates cytotoxicity, experimental evidence implicates both GCase and ceramide, prompting detailed biochemical and biophysical studies on protein-protein/lipid interactions, as well as solution conditions that impact α-syn conformation and aggregation.
Here, we explored the possibility of a physical linkage between α-syn and GCase using fluorescence and nuclear magnetic resonance (NMR) spectroscopy, as well as verification by immunoprecipitation (IP) and immunofluorescence studies. We determined that the two soluble proteins associate selectively under lysosomal solution conditions, reflecting the milieu of the acidic organelle where both proteins are found. We mapped the site of interaction specifically to the C-terminal region of α-syn.
EXPERIMENTAL PROCEDURES
Protein Expression and Sample Preparation
The WT human α-syn plasmid (pRK172) was provided by M. Goedert (Medical Council Research Laboratory of Molecular Biology, Cambridge, UK) (49). Single cysteine mutants (G7C and Y136C) were generated using the Quick-Change site-directed mutagenesis kit (Stratagene) and verified by DNA sequencing. Protein was expressed and purified as previously described (50, 51). For a 1-liter culture, isotopically (13C/15N) enriched WT α-syn was produced by growing Escherichia coli BL21(DE3)pLysS in M9 medium supplemented with [13C]glucose (2 g) and 15NH4Cl (1 g). Imiglucerase, purified recombinant GCase, was obtained from Genzyme Corp. and N370S GCase was a generous gift from Dr. Timothy Edmonds (Genzyme Corp.). All samples were exchanged and concentrated into appropriate buffer (50 mm MES, 100 mm NaCl, pH 5.5 or 50 mm sodium phosphate, 100 mm NaCl, pH 7.4) before fluorescence and NMR experiments.
α-Syn Labeling
Dns-labeled proteins were prepared and purified as previously described (17). After dithiothreitol (DTT) reduction, Cys-containing α-syn was reacted with a 1.5-molar excess of the Dns-precursor (5-((((2-iodoacetyl)amino)ethyl)amino) naphthalene-1-sulfonic acid in dimethyl sulfoxide (DMSO, volume ≤ 2% (v/v)), Invitrogen) in 50 mm 4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid (HEPES) pH 8.0 buffer containing 4 m guanidinium HCl (99% pure grade, USB Corporation) and gently stirred in the dark at room temperature for 4 h. To stop the labeling reaction, DTT (20 mm) was added. Dns-α-syn was purified by anionic-exchange chromatography (MonoQ column, GE Healthcare). Protein molecular weights were confirmed by ESI-MS (Biochemistry Core Facility, NHLBI). Protein concentrations were determined using a molar absorptivity ϵ(336 nm) = 5700 m−1 cm−1 (Dns).
Fluorescence Spectroscopy
Fluorescence spectra were measured using Fluorolog-3 spectrofluorimeter (HORIBA Jobin Yvon Inc.) and conducted at 25 °C using a temperature controlled cuvette holder. GCase titrations were performed in pH 5.5 buffer (50 mm MES with increasing concentrations of 25, 75, or 100 mm NaCl). Concentrated GCase stocks were serially diluted (from 41 to 1 μm) while maintaining [Dns136-α-syn] = 1.3 μm. Dns-α-syn was excited at 340 nm and emission was monitored from 405 to 650 nm.
A stock solution of conduritol-β-epoxide (CBE, Biomol Research Labs, 50 mm dissolved in DMSO) was added to 46 μm GCase solution (50 mm MES, 100 mm NaCl, pH 5.5) to a final concentration of 46 μm. The final DMSO concentration in solution is ≤ 0.1% (v/v). The complex (CBE-GCase) was incubated at room temperature for 1 h prior to fluorescence measurements.
Intrinsic GCase Trp fluorescence was excited at 295 nm and monitored from 300 to 600 nm for Förster energy transfer measurements. Concentrated Dns136-α-syn stocks were serially diluted (3–0.5 μm) in the presence of GCase (3 μm in 50 mm MES, 100 mm NaCl, pH 5.5).
Data Analysis
Mean spectral wavelength, <λ>, was calculated according to Equation 1,
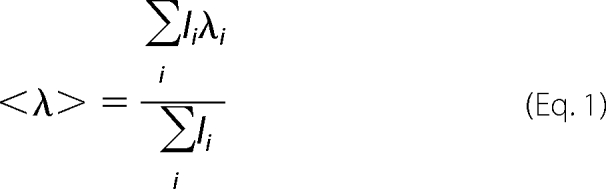 |
where Ii and λi are the emission intensity and wavelength, respectively, for i = 405–650 nm. To normalize the data, we calculated the fractional change, Δ<λ>, according to Equation 2,
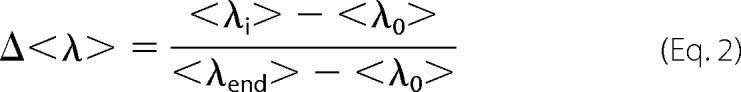 |
where <λi>, <λ0>, and <λend> are mean wavelengths determined for the different [GCase], in the absence of GCase, and the saturation value (502 nm) determined for the low salt concentration <λ>(25 mm NaCl). Estimation of apparent dissociation constants (Kd(App)) were extracted for a simple two-state model (50). Whereas there are many other possible models one can consider, we choose the simplest model because more complex models (i.e. multiple binding sites and different stoichiometry) did not significantly improve the fits; though at this time, we also cannot rule them out. Because at higher salt (100 mm NaCl) and for N370S and CBE-treated GCase, weaker binding was observed, we included a nonspecific binding term (NS), and data were fit according to Equation 3,
where Δ<λ(GCase)> is the fractional change of mean wavelength in the presence of GCase, a is [α-syn]o, b is [α-syn]o+[GCase]+Kd(app) and c is [GCase]. Data fitting were performed using IGOR Pro 6.01 (Wavemetrics).
Immunoprecipitation
50–100 mg of frozen autopsy brain samples obtained from the NIH Clinical Center Department of Pathology were used, including a subject with PD without GBA mutations (WT/WT), a GD carrier with PD (N370S/WT), a subject with GD and PD (N370S/N370S), and a subject with type 2 GD (L444P/IVS2 + 1). Multiple samples (n = 3) from the same tissue were analyzed. Samples were homogenized in 5× volume lysis buffer (containing 20 mm MES, 320 mm sucrose, 7.7 mm sodium azide, 5 mm EDTA, 0.1% Tween at either pH 5.5 or 7.4) and centrifuged at 3000 × g for 20 min at 4 °C. To avoid nonspecific binding, the supernatant was preincubated with 50 μl of μMACS protein G microbeads (Miltenyu Biotec) for 2 h and loaded on a MACS separation column. The flow-through homogenate was incubated with an antibody to α-syn (catalogue No. ab21976, Abcam) for 6–8 h. 100 μl of μMACS protein G microbeads was added before loading on a MACS separation column. Following five washes using 200 μl of wash buffer (either pH 5.5 or 7.4 containing 50 mm MES, 150 mm NaCl, 0.5% CHAPS, 0.1% SDS), bound proteins were eluted using 1× LDS buffer (Invitrogen) containing 100 mm DTT, and boiled at 95 °C for 10 min. Co-immunoprecipitated proteins were separated by SDS-PAGE on 4–12% bis-tris acrylamide gels (Invitrogen). The immunoprecipitation was repeated three different times with highly consistent results.
Immunoblotting
Samples separated by SDS-PAGE were transferred to nitrocellulose membranes (iBlot PVDF, Invitrogen). Blots were blocked in phosphate-buffered saline solution containing 0.1% Tween-20 (Sigma) and 10% fat-free milk for 1 h at RT and incubated in blocking buffer containing primary antibody (α-syn 1:1000, catalogue no.610787, BD Transduction and GCase 1:15000, custom-made polyclonal antibody) overnight at 4 °C. Membranes were washed three times for 10 min, incubated in blocking buffer containing horseradish peroxidase-conjugated secondary antibody (1:3000, KPL Inc.) for 1 h at room temperature, and developed using enhanced chemiluminescence (ECL Plus, GE Healthcare).
Immunofluorescence and Laser Scanning Confocal Microscopy
The M17 neuroblastoma cell line overexpressing wild-type α-syn (52) was transfected with pTracer-SV40 (Invitrogen) vector expressing the full-length wild-type GCase cDNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer's guidelines. Selection for transfected cells was done by Zeocin (Invitrogen) treatment for 3 weeks. Cells were grown to 60% confluency in Lab-Tek 4 chamber slides (Fisher Scientific), fixed in 3% paraformaldehyde, permeabilized with 0.1% Triton-X for 10 min and blocked in PBS containing 0.1% saponin, 100 μm glycine, 0.1% BSA, and 2% donkey serum. Incubation with goat-polyclonal cathepsin D (1:50, R&D Systems), rabbit polyclonal anti-GCase (R386) (1:500), and mouse monoclonal α-syn (LB509, 1:100, Abcam) for 2 h followed at RT. Cells were washed, incubated with secondary donkey anti-goat, anti-mouse, or anti-rabbit antibodies conjugated to ALEXA-488, ALEXA-555, or ALEXA-647, respectively (Invitrogen), rinsed, and mounted in VectaShield with DAPI (Vector Laboratories). Cells were imaged with a Zeiss 510 META confocal laser-scanning microscope using an Argon ion (458, 477, 488, 514 nm, 30 mW), a HeNe (543 nm, 1 mW) and a HeNe (633 nm) laser. Images were acquired using a Plan Apochromat 63×/1.4 oil DIC objective.
NMR Spectroscopy
15N HSQC spectra were acquired on an 800 MHz Bruker spectrometer with cryoprobe at 15 °C at pH 5.5 and pH 7.4 in the same buffers used for fluorescence measurements with 100 mm NaCl. Backbone assignments of 13C/15N labeled α-syn were done by standard procedures using HNCACB and CBCACONH experiments at pH 6.4. Additional 15N HSQC spectra were acquired at pH 6.0 and 7.0, and the backbone amide assignments extrapolated to pH 5.5 and 7.4. Spectra were processed and analyzed using NMRPipe (53). While significant peak broadening occurred due to α-syn interaction with GCase, roughly doubling the 1H linewidth for affected peaks, no chemical shift changes greater than 0.02 ppm were observed.
Molecular Modeling
pKa values for GCase were predicted using PROPKA 2.0 (54). A peptide consisting of residues 115–140 of α-syn was docked interactively and minimized using Maestro/MacroModel (Schrödinger Inc.) to GCase x-ray structure (PDB ID-3GXM, chain C). The peptide was docked with its C terminus near loop 1 (GCase residues 311–319). Three conserved histidines at the GCase surface are predicted to be neutral at pH 7.4 and positively charged at pH 5.5. The N terminus of the peptide was docked near His-223 and His-273, while the middle of the peptide lies near His-328. The three histidines are present in all mammalian GCase sequences and surface-exposed (55). The catalytic glutamate Glu-235 is predicted by PROPKA 2.0 to become protonated at lysosomal pH in the majority of GCase x-ray structures, consistent with previous predictions (56). The figures were made using PyMOL and Maestro.
RESULTS
Site-specific Fluorescent Probe of α-Syn-GCase Interaction
We hypothesized that if an electrostatic interaction occurs between GCase (pI ∼7.4) and α-syn (pI ∼4.7), it would involve the acidic α-syn C-terminal region (100–140), which contains 14 carboxylate side chains. An environmentally sensitive, dansyl (Dns) fluorophore (57) was covalently attached to a single-Cys α-syn mutant (Y136C) (Fig. 1A) and binding was examined at both cytosolic (7.4) and lysosomal (5.5) pH. As α-syn is intrinsically disordered (58), the Dns fluorophore exhibits similar spectroscopic properties independent of solution pH (mean wavelength, <λ> ∼524 nm), consistent with a fully solvent-exposed probe (<λ> ∼529 nm measured for model complex, N-acetyl-Dns-cysteine). However, upon the addition of GCase at pH 5.5, the α-syn-GCase interaction dramatically alters Dns136 fluorescence, with ∼ 2-fold intensity (I) increase and spectral blue shift (∼20 nm, <λ> = 524 → 506 nm) (Fig. 1B). In contrast, insignificant changes were observed (ΔI ≤ 2%, <λ> unchanged) at pH 7.4, suggesting minimal binding (supplemental Fig. S1).
FIGURE 1.

α-Syn-GCase interactions probed by Dns fluorescence. A, primary sequence of α-syn and structure of Dns fluorophore. The membrane binding region (1–100) and Cys-labeling sites used in this study (7 and 136) are indicated. B, fluorescence spectroscopic changes of Dns136-α-syn (1.3 μm) as a function of added GCase (up to 41 μm, gray scale) in 50 mm MES, 100 mm NaCl, pH 5.5 buffer. C, α-Syn-GCase titration curves with decreasing ionic strength (100 mm NaCl (●), 75 mm (■), and 25 mm (▴)) obtained from mean spectral wavelength (<λ>) at pH 5.5. Left and right axes denote normalized (Δ<λ>) and absolute (<λ>) wavelength changes, respectively. Error bars indicate standard deviations for at least two independent measurements. Fits are shown as solid lines.
As α-syn binds to the enzyme, residue 136 is sequestered from the aqueous to a more hydrophobic surrounding, as indicated by the spectral blue shift. An estimated dissociation constant, Kd ∼ 22(2) μm (50 mm MES, 100 mm NaCl, pH 5.5, 25 °C), was obtained by fitting the titration curve from <λ> to a two-state binding model (α-syn-GCase ⇆ α-syn + GCase) (Fig. 1C). To assess the complex formation from the perspective of GCase, we exploited its twelve intrinsic Trp residues as Förster energy transfer donors and Dns136 as the acceptor. Because of the favorable spectral overlap of Trp fluorescence and Dns absorption, energy transfer should occur when the Dns fluorophore is sufficiently close (intermolecular distance ∼11–33 Å; Förster distance, Ro ∼22 Å (57)) to any of the 12 Trp. As anticipated, Trp and Dns fluorescence decreases and increases, respectively, upon protein-enzyme interaction at pH 5.5 in the presence of 100 mm NaCl (supplemental Fig. S2). Whereas the presence of multiple donors prohibited explicit distance determination, an α-syn-GCase association was confirmed.
Probing the effect of ionic strength (100 mm to 25 mm NaCl), we found the interactions were markedly enhanced (Kd decreased from 22(2) to 1.2(1) μm) as evidenced by the lower amount of GCase required to induce a spectral shift, supporting the role of electrostatics in complex formation (Fig. 1C). An N-terminal Dns7 variant established that this binding was specific to the C terminus, as negligible spectroscopic changes (ΔI ≤ 2%, <λ> unchanged) were observed under comparable solution conditions (supplemental Fig. S3).
The Effect of N370S and Inhibitor-bound GCase on Complex Formation
N370S, a common GD mutation found in up to 4% of the Ashkenazi Jewish population (47), renders the enzyme catalytically compromised. As this mutation has clearly been associated with an increased PD risk (39), we examined the effect of N370S on the α-syn-GCase complex formation. Our titration data show that α-syn has a reduced affinity for N370S GCase (Kd ∼ 45(4) compared with 22(2) μm for WT, Fig. 2). The N370S mutation alters GCase structure near the active site, as characterized by x-ray crystallography (59). Interestingly, x-ray studies have shown that GCase bound to the inhibitor, conduritol-β-epoxide (CBE) (60), adopts a similar active site conformation, and based on our N370S result, we predicted weaker protein-protein interaction in the presence of CBE. Indeed, α-syn binds to CBE-bound GCase with an affinity comparable to the N370S mutant (Kd ∼49(7) μm, Fig. 2). These results suggest that α-syn could bind near the GCase active site.
FIGURE 2.
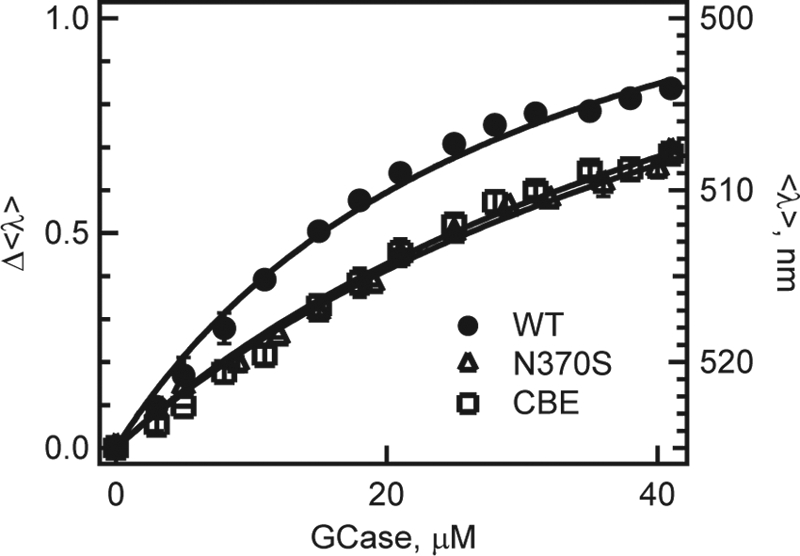
Titration curves of N370S (△) and CBE-treated GCase (□) obtained from mean spectral wavelength (<λ>) of Dns136-α-syn at pH 5.5. For comparison, WT (●) data also are shown. Left and right axis denotes normalized (Δ<λ>) and absolute (<λ>) wavelength changes, respectively. Error bars (comparable to symbol size) indicate standard deviations for at least two independent measurements. Fits are shown as lines.
NMR Identifies the α-Syn C-terminal Residues as the Site of Interaction with GCase
To map this intermolecular interaction in detail, we isotopically labeled α-syn for NMR spectroscopy. The 15N HSQC NMR spectrum provided a residue-by-residue characterization by monitoring all backbone amide hydrogen and nitrogen resonances for each non-proline residue (Fig. 3A). The backbone amide intensity was reduced over 5-fold for residues 118–137 in the presence of GCase, compared with α-syn alone (Fig. 3B). No significant reduction was observed for the N-terminal residues, in accord with the fluorescence results. Interaction of the C-terminal residues of α-syn with the 60 kDa enzyme greatly slows their effective molecular tumbling rates leading to reduced signal intensities. This region contains eight acidic residues (Asp-119, Asp-121, Glu-123, Glu-126, Glu-130, Glu-131, Asp-135, and Glu-137) as well as three tyrosines, thus providing both electrostatic and hydrophobic noncovalent contacts between the two proteins (Fig. 3B). Indeed, there are numerous positively charged side chains (17 K and 11 R) on the GCase surface that can provide electrostatic contacts upon binding. No significant interaction was seen at pH 7.4 (Fig. 3B and supplemental Fig. S4).
FIGURE 3.

15N HSQC NMR spectra of α-syn. A, overlaid 15N HSQC spectra of 13C/15N labeled α-syn (40 μm, red) and in the presence of GCase (40 μm, black) at pH 5.5. Residues undergoing significant intensity reduction are labeled. B, plot of relative spectra intensity ratio (I/I0) of the α-syn-GCase (I) and α-syn alone (I0) at neutral (7.4, gray) and acidic pH (5.5, black). The acidic C-terminal α-syn sequence is shown for residues 101–140; residues colored in black, experience the greatest reduction in intensity upon interaction (I/I0 < 0.2). Eight carboxylic acids (D119, D121, E123, E126, E130, E131, D135, and E137) and three Tyr (Y125, Y133, Y136) residues in this region are in bold and underlined, respectively.
Verification of the α-Syn-GCase Interaction in Vivo
In vivo interactions between the endogenous proteins were verified by IP of GCase from brain autopsy samples. If GCase binds to native α-syn under lysosomal solution conditions, a monoclonal α-syn antibody can be used to co-precipitate GCase from tissue homogenates at pH 5.5. We examined samples from a patient with PD (PD control, GBA genotype: WT/WT), a GD carrier with PD (N370S/WT), a PD patient with type 1 GD, (N370S/N370S), and an infant with neuronopathic GD (with minimal expression of GCase, L444P/IVS2 + 1) (Fig. 4A). IP of a tissue homogenate from a PD patient was conducted without α-syn antibody and used as a negative control.
FIGURE 4.

In vivo α-syn-GCase interaction. A, immunoprecipitation of GCase with antibody to α-syn from brain autopsy samples at pH 5.5. Shown are representative immunoblots repeated at least three times (left to right: subject with PD (GBA genotype: WT/WT); GD carrier with PD (N370S/WT); subject with GD and PD (N370S/N370S); negative control without primary antibody (WT/WT); subject with type 2 GD with very deficient GCase (L444P/IVS2 + 1)); respective lanes are total and immunoprecipitated (IP) samples; top and bottom lanes were detected with antibodies to GCase and α-syn, respectively. On Westerns probed with R386 rabbit polyclonal antibody, GCase usually appears as two bands, corresponding to different glycosylated forms. B–E, laser scanning confocal microscopy images of fixed M17 cells overexpressing GCase and α-syn. Immunofluorescence staining for GCase (B), α-syn (C), and the lysosomal marker, cathepsin-D (D). Co-localization of the three markers (E) is found in small aggregates (arrows). Scale bar, 5 μm.
GCase-positive bands were identified by Western blot analysis in the immunoprecipitated samples from the PD control and carrier with PD. As expected, GCase was not present in the type 2 patient with negligible GCase. A fainter band was seen in the sample from the PD patient with type 1 GD compared with the others, hinting that the mutation, N370S, might have an effect on α-syn binding, as observed in the recombinant proteins. In accord with the pH-dependent fluorescence data, no enzyme was detected when IP was performed at pH 7.4 (supplemental Fig. S5), suggesting that the two human proteins interact selectively at a lysosomal pH.
To explore the in vivo interaction further, we investigated the intracellular localization of GCase and α-syn in a dopaminergic human neuroblastoma BE(2)-M17 cell model where both GCase and α-syn are stably overexpressed. Cultured cells were characterized by laser scanning confocal microscopy. Cathepsin D (CatD), a mannose-6-phosphate dependent specific marker of acidic lysosomal compartments (61), was used to locate and visualize lysosomes. With immunofluorescence staining, the microscopic images revealed that GCase (Fig. 4B) co-localized with the CatD-positive lysosomes (Fig. 4D), indicating that the overexpressed GCase is indeed trafficked correctly to the lysosomal compartments.
Co-localization of GCase and α-syn (Fig. 4C) was observed in distinct cellular inclusions (Fig. 4, B, C, E), which also stained positive for CatD (Fig. 4, D, E), suggesting their close proximity in the lysosomal compartment. We excluded the possibility of antibody cross reactivity by individual staining of the three above described markers, and ascertained that they showed similar cellular localization patterns. Additionally, we performed negative controls testing the specificity of secondary antibodies in the absence of the primary antibodies. We found no background staining at the laser settings used to acquire images of the triple-labeled cells. These results suggest that GCase and α-syn can co-localize in lysosomes in a neuronal cell model.
DISCUSSION
Our study clearly shows that the lysosomal enzyme GCase interacts with the C terminus of α-syn in a pH-dependent manner. Specifically, we identified the residues 118–137 as the binding site by NMR spectroscopy (Fig. 3). The formation of this protein complex is enhanced by electrostatic interactions, and its dissociation constant ranges from 1.2 to 22 μm in the presence 25 to 100 mm NaCl, respectively (Fig. 1C). While a micromolar Kd value can be considered rather modest, the neuronal concentration of α-syn is predicted to be quite high, between 70–140 μm (62). Thus, a biologically relevant interaction with GCase at this strength is plausible.
The IP results (Fig. 4A), in accord with our data on the recombinant proteins, demonstrate that the interaction between human brain tissue derived α-syn and GCase is observed at the acidic pH, but is lost, or not measurable, under comparable experimental conditions at neutral pH typical of the cytoplasm. Moreover, immunofluorescence imaging of neuronal cell culture co-expressing the two proteins indicates co-localization in the lysosome (Fig. 4E). Whereas our data do not preclude protein-protein interactions in other cellular milieux, we suggest that the lysosome is a primary site of interaction for α-syn and GCase.
The pH and ionic strength dependence of binding affinities suggests a role for side chain protonation states at the intermolecular interface. Only one α-syn residue, His-50 with a pKa of 6.8, changes its charge from pH 7.4 to 5.5 (Glu-126 has the next closest pKa ∼4.9 (63)); because His-50 is located distal to the interacting region, we considered residues on the GCase surface as potential sites responsible for the observed pH-dependent binding behavior.
Seven surface histidines and one of the catalytic carboxylates (56) are predicted by the PROPKA 2.0 program (54) to change protonation states. Additionally, spectroscopic evidence indicates that loop 1 (residues 311–319, Fig. 5A), located near the catalytic cleft of GCase, has a pH-dependent structure (64). As determined by x-ray crystallography, loop 1 is conformationally labile at various pH (4.5–7.5), adopting a helical or extended structure where hydrogen bonding can occur between residues Trp-312 and Asn-370 (59). Taking these observations into consideration, a model structure of the α-syn-GCase interaction was generated with α-syn docked near loop 1, and near the three most highly conserved surface histidines (His-223, His-273, His-328) predicted to be charged at lysosomal pH (Fig. 5). His-223 and His-273 lie on the face of GCase opposite of loop 1, and His-328 lies in between, in the cleft between the GCase catalytic (TIM barrel) and C-terminal β-sheet domains.
FIGURE 5.

α-Syn-GCase interaction model. View of loop 1 (residues 311–319) and critical residues: A, WT: shows loop 1 in the α-turn structure (PDB code: 3GXM) and B, N370S: loop 1 in the extended structure (PDB code: 3KE0). The two catalytic glutamates, E235 and E340, are indicated. In the α-turn, D315 is buried, while in the extended conformation, D315 is partially exposed. W312 and Y313 are shown hydrogen-bonded to N370 (A) and E235 (B), respectively. Also shown is H328, a conserved surface histidine predicted to change charge at lysosomal pH. A model of the bound C-terminal residues of α-syn is depicted by the red ribbon, contacting loop 1 and H328 in the cleft region between the TIM barrel and C-terminal β-sheet domains of GCase.
In this model, the interacting C-terminal α-syn residues, 126–140, are situated near loop 1 in the groove between the GCase C terminus β-sheet domain and the TIM barrel. Lining this groove lie several charged surface residues, Lys-321, His-328, Arg-329, Lys-346, Arg-433, Lys-441, and Arg-463, that could provide electrostatic interactions with negatively charged residues, Glu-130, Glu-131, Asp-135, Glu-137, and Glu-139 of α-syn. Interestingly, the three aforementioned Arg residues are all sites of GD-related mutations (42). In the same region, GCase residues Phe-316, Leu-317, Leu-436, and Val-437 could provide hydrophobic contacts with α-syn tyrosine residues. Additionally, we note that the Dns136 fluorophore would lie near Trp-312 (∼12Å) in the model, in accord with the fluorescence energy transfer result; because GCase has 11 other Trp residues, other model structures consistent with this experimental datum are possible. Nevertheless, the model provides a possible explanation for the weaker binding observed for N370S and CBE-bound GCase. In both N370S and CBE-bound structures, loop 1 is observed in the extended structure (Fig. 5B) (59, 60) with residue Asp-315 partially surface-exposed, whereas this residue is buried in the WT structure. Thus, electrostatic repulsion between Asp-315 and the acidic α-syn C-terminal residues could lead to the observed weaker binding. While this is only one of the many potential models of an α-syn-GCase complex, it does provide molecular insights into the differences in binding affinities between N370S or CBE-bound and WT GCase.
Over 30 proteins are reported to interact with α-syn, implicating this protein in many possible functional roles (4). We suggest a new putative biological role for α-syn in the lysosome, as an auxiliary protein to GCase. Experimentally, it is known that the first 95 residues of α-syn can associate extensively with membrane surfaces, while the C-terminal residues, including those that interact with GCase, remain solvent exposed and flexible (65–68). Thus, the α-syn C terminus would be available for enzyme binding and may help recruit GCase to membranes of intra-lysosomal vesicles (Fig. 6). However, our results do not preclude α-syn interactions with other lysosomal proteins, though no interactions were found between α-syn and saposin C, a membrane-associated (69, 70) lysosomal GCase-activating protein (71) under similar solution conditions (supplemental Fig. S6).
FIGURE 6.

Schematic diagram showing a possible biological role for α-syn in the lysosome. GCase (TIM barrel motif and C-terminal β-sheet domain colored light blue and pink, respectively) is recruited by α-syn to an intra-lysosomal vesicle with gangliosides shown (lavender), a source of glucocerebroside (boxed). The membrane-bound region of α-syn is modeled as a single helix (blue, residues 1–95), though more dynamic structure is possible (68). The GCase binding residues of α-syn are colored red.
GCase is involved in the degradation pathways of gangliosides, which all have glucocerebroside as their base structure. Several studies show that α-syn preferentially interacts with membranes composed of acidic lipids (72), including those enriched with gangliosides (73–75). While this scheme clearly needs to be validated, this noncovalent interaction provides the groundwork for exploring possible mechanisms linking PD with GD mutant alleles.
Because the lysosome plays an important role in protein degradation, this newly identified interaction between α-syn and GCase can potentially influence α-syn homeostasis in neurons. Taken together with the genetic connection between GD and PD, the results imply that an altered α-syn-GCase interaction in the lysosome could perturb this equilibrium and set the stage for PD progression.
Mutations in GCase can lead to the absence of the enzyme, unstable enzyme targeted for proteasomal degradation, or stable enzyme with reduced activity (76, 77); all of which are associated with an increased risk of PD (39). Both loss- and gain-of-function theories have been proposed (22, 47). It has been postulated that the alteration in glucosylceramide/ceramide metabolism as a result of GCase deficiency may influence the sphingolipid composition of membranes, leading to the disruption of α-syn membrane binding, and hence, enhance its aggregation in the cytoplasm. However, this theory alone is insufficient to explain why GD carriers also are likely to develop PD. It has also been proposed that misfolded and accumulated mutant enzymes could contribute to pathogenesis by either impairing lysosomal function, or overwhelming the ubiquitin-proteosomal degradation pathway. In this scenario, disruption in mechanisms essential for degradation could result in the imbalance of α-syn proteostasis and consequently promote its aggregation.
Alternately, the interaction of α-syn with wild-type enzyme could have a beneficial effect by promoting lysosomal degradation of α-syn, or inhibiting adverse α-syn aggregation. Thus, mutations that decrease the amount of enzyme reaching the lysosome or weaken the interaction, as seen with the N370S mutant, could reduce this benefit and increase the probability of dysfunction. We also observed weakened interaction with GCase bound to the inhibitor CBE. A recent study showed that treatment with CBE increased α-syn levels in mice and neuroblastoma cells (46), which further supports the hypothesis that a weakened GCase interaction can result in reduced lysosomal α-syn degradation. However since only a minority of Gaucher patients and carriers develop PD, other factors mediating α-syn levels and aggregation must also be involved. Future in vivo as well as in vitro experiments, including studies of mutated GCase and of the effect of α-syn-GCase interaction on enzyme activity and α-syn amyloid formation, will further elucidate the mechanisms responsible for the increased PD risk observed, and provide insights into relevant therapeutic strategies.
Supplementary Material
Acknowledgments
We thank Timothy Edmonds (Genzyme Corp.) for the gift of N370S GCase, Nico Tjandra (Laboratory of Molecular Biophysics, NHLBI) for the use of the 800 MHz Bruker spectrometer and for the gift of saposin C, and Duck-Yeon Lee (Biochemistry Core Facility, NHBLI) for technical assistance with mass spectrometry. We also acknowledge the assistance of the National Institutes of Health Clinical Center Dept. of Pathology.
This work was supported by the Intramural Research Program at the National Institutes of Health, NHLBI and NHGRI.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6.
- PD
- Parkinson disease
- α-syn
- α-synuclein
- LBs
- Lewy bodies
- GCase
- glucocerebrosidase
- GD
- Gaucher disease
- NMR
- nuclear magnetic resonance
- IP
- immunoprecipitation
- Kd
- dissociation constant
- Dns
- dansyl
- CBE
- conduritol-β-epoxide.
REFERENCES
- 1. Lee V. M., Trojanowski J. Q. (2006) Neuron 52, 33–38 [DOI] [PubMed] [Google Scholar]
- 2. Clayton D. F., George J. M. (1998) Trends Neurosci. 21, 249–254 [DOI] [PubMed] [Google Scholar]
- 3. Cookson M. R. (2009) Mol. Neurodegener. 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Uversky V. N., Eliezer D. (2009) Curr. Protein Pept. Sci. 10, 483–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dev K. K., Hofele K., Barbieri S., Buchman V. L., van der Putten H. (2003) Neuropharmacol. 45, 14–44 [DOI] [PubMed] [Google Scholar]
- 6. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., DiIorio G., Golbe L. I., Nussbaum R. L. (1997) Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 7. Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J. T., Schöls L., Riess O. (1998) Nat. Genet. 18, 106–108 [DOI] [PubMed] [Google Scholar]
- 8. Zarranz J. J., Alegre J., Gómez-Esteban J. C., Lezcano E., Ros R., Ampuero I., Vidal L., Hoenicka J., Rodriguez O., Atares B., Llorens V., Tortosa E., del Ser T., Muñoz D. G., de Yebenes J. G. (2004) Ann. Neurol. 55, 164–173 [DOI] [PubMed] [Google Scholar]
- 9. Chartier-Harlin M. C., Kachergus J., Roumier C., Mouroux V., Douay X., Lincoln S., Levecque C., Larvor L., Andrieux J., Hulihan M., Waucquier N., Defebvre L., Amouyel P., Farrer M., Destée A. (2004) Lancet 364, 1167–1169 [DOI] [PubMed] [Google Scholar]
- 10. Singleton A. B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R., Lincoln S., Crawley A., Hanson M., Maraganore D., Adler C., Cookson M. R., Muenter M., Baptista M., Miller D., Blancato J., Hardy J., Gwinn-Hardy K. (2003) Science 302, 841–841 [DOI] [PubMed] [Google Scholar]
- 11. Chiti F., Dobson C. M. (2006) Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 12. Fink A. L. (2006) Acc. Chem. Res. 39, 628–634 [DOI] [PubMed] [Google Scholar]
- 13. Conway K. A., Harper J. D., Lansbury P. T. (1998) Nat. Med. 4, 1318–1320 [DOI] [PubMed] [Google Scholar]
- 14. Shtilerman M. D., Ding T. T., Lansbury P. T., Jr. (2002) Biochemistry 41, 3855–3860 [DOI] [PubMed] [Google Scholar]
- 15. Thirunavukkuarasu S., Jares-Erijman E. A., Jovin T. M. (2008) J. Mol. Biol. 378, 1064–1073 [DOI] [PubMed] [Google Scholar]
- 16. Nath S., Meuvis J., Hendrix J., Carl S. A., Engelborghs Y. (2010) Biophys. J. 98, 1302–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yap T. L., Pfefferkorn C. M., Lee J. C. (2011) Biochemistry 50, 1963–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maroteaux L., Campanelli J. T., Scheller R. H. (1988) J. Neurosci. 8, 2804–2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uversky V. N., Li J., Fink A. L. (2001) J. Biol. Chem. 276, 44284–44296 [DOI] [PubMed] [Google Scholar]
- 20. Zhu M., Li J., Fink A. L. (2003) J. Biol. Chem. 278, 40186–40197 [DOI] [PubMed] [Google Scholar]
- 21. Masuda M., Suzuki N., Taniguchi S., Oikawa T., Nonaka T., Iwatsubo T., Hisanaga S., Goedert M., Hasegawa M. (2006) Biochemistry 45, 6085–6094 [DOI] [PubMed] [Google Scholar]
- 22. Goldin E. (2010) Mol. Genet. Metab. 101, 307–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burré J., Sharma M., Tsetsenis T., Buchman V., Etherton M. R., Südhof T. C. (2010) Science 329, 1663–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pan T., Kondo S., Le W., Jankovic J. (2008) Brain 131, 1969–1978 [DOI] [PubMed] [Google Scholar]
- 25. Abou-Sleiman P. M., Muqit M. M., Wood N. W. (2006) Nat. Rev. Neurosci. 7, 207–219 [DOI] [PubMed] [Google Scholar]
- 26. Cuervo A. M., Stefanis L., Fredenburg R., Lansbury P. T., Sulzer D. (2004) Science 305, 1292–1295 [DOI] [PubMed] [Google Scholar]
- 27. Gruschus J. M. (2008) Amyloid-J. Protein Fold. Disord. 15, 160–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. (1998) Nature 392, 605–608 [DOI] [PubMed] [Google Scholar]
- 29. Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R. J., Calne D. B., Stoessl A. J., Pfeiffer R. F., Patenge N., Carbajal I. C., Vieregge P., Asmus F., Müller-Myhsok B., Dickson D. W., Meitinger T., Strom T. M., Wszolek Z. K., Gasser T. (2004) Neuron 44, 601–607 [DOI] [PubMed] [Google Scholar]
- 30. Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M. K., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A. R., Healy D. G., Albanese A., Nussbaum R., González-Maldonaldo R., Deller T., Salvi S., Cortelli P., Gilks W. P., Latchman D. S., Harvey R. J., Dallapiccola B., Auburger G., Wood N. W. (2004) Science 304, 1158–1160 [DOI] [PubMed] [Google Scholar]
- 31. Bonifati V., Rizzu P., van Baren M. J., Schaap O., Breedveld G. J., Krieger E., Dekker M. C. J., Squitieri F., Ibanez P., Joosse M., van Dongen J. W., Vanacore N., van Swieten J. C., Brice A., Meco G., van Duijn C. M., Oostra B. A., Heutink P. (2003) Science 299, 256–259 [DOI] [PubMed] [Google Scholar]
- 32. Goker-Alpan O., Schiffmann R., LaMarca M. E., Nussbaum R. L., McInerney-Leo A., Sidransky E. (2004) J. Med. Genet. 41, 937–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tayebi N., Walker J., Stubblefield B., Orvisky E., LaMarca M. E., Wong K., Rosenbaum H., Schiffmann R., Bembi B., Sidransky E. (2003) Mol. Genet. Metab. 79, 104–109 [DOI] [PubMed] [Google Scholar]
- 34. Aharon-Peretz J., Rosenbaum H., Gershoni-Baruch R. (2004) N. Engl. J. Med. 351, 1972–1977 [DOI] [PubMed] [Google Scholar]
- 35. Lwin A., Orvisky E., Goker-Alpan O., LaMarca M. E., Sidransky E. (2004) Mol. Genet. Metab. 81, 70–73 [DOI] [PubMed] [Google Scholar]
- 36. Goker-Alpan O., Giasson B. I., Eblan M. J., Nguyen J., Hurtig H. I., Lee V. M., Trojanowski J. Q., Sidransky E. (2006) Neurology 67, 908–910 [DOI] [PubMed] [Google Scholar]
- 37. Neudorfer O., Giladi N., Elstein D., Abrahamov A., Turezkite T., Aghai E., Reches A., Bembi B., Zimran A. (1996) QJM-Mon. J. Assoc. Physicians 89, 691–694 [DOI] [PubMed] [Google Scholar]
- 38. Hruska K. S., Goker-Alpan O., Sidransky E. (2006) J. Biomed. Biotechnol. 2006, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sidransky E., Nalls M. A., Aasly J. O., Aharon-Peretz J., Annesi G., Barbosa E. R., Bar-Shira A., Berg D., Bras J., Brice A., Chen C. M., Clark L. N., Condroyer C., De Marco E. V., Durr A., Eblan M. J., Fahn S., Farrer M. J., Fung H. C., Gan-Or Z., Gasser T., Gershoni-Baruch R., Giladi N., Griffith A., Gurevich T., Januario C., Kropp P., Lang A. E., Lee-Chen G. J., Lesage S., Marder K., Mata I. F., Mirelman A., Mitsui J., Mizuta I., Nicoletti G., Oliveira C., Ottman R., Orr-Urtreger A., Pereira L. V., Quattrone A., Rogaeva E., Rolfs A., Rosenbaum H., Rozenberg R., Samii A., Samaddar T., Schulte C., Sharma M., Singleton A., Spitz M., Tan E. K., Tayebi N., Toda T., Troiano A. R., Tsuji S., Wittstock M., Wolfsberg T. G., Wu Y. R., Zabetian C. P., Zhao Y., Ziegler S. G. (2009) N. Engl. J. Med. 361, 1651–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goker-Alpan O., Stubblefield B. K., Giasson B. I., Sidransky E. (2010) Acta Neuropathol. 120, 641–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Butters T. D. (2007) Curr. Opin. Chem. Biol. 11, 412–418 [DOI] [PubMed] [Google Scholar]
- 42. Hruska K. S., LaMarca M. E., Scott C. R., Sidransky E. (2008) Hum. Mutat. 29, 567–583 [DOI] [PubMed] [Google Scholar]
- 43. Grabowski G. A. (1997) Genet. Test. 1, 5–12 [DOI] [PubMed] [Google Scholar]
- 44. Mak S. K., McCormack A. L., Manning-Bog A. B., Cuervo A. M., Di Monte D. A. (2010) J. Biol. Chem. 285, 13621–13629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Settembre C., Fraldi A., Jahreiss L., Spampanato C., Venturi C., Medina D., de Pablo R., Tacchetti C., Rubinsztein D. C., Ballabio A. (2008) Hum. Mol. Genet. 17, 119–129 [DOI] [PubMed] [Google Scholar]
- 46. Manning-Boğ A. B., Schüle B., Langston J. W. (2009) Neurotoxicology 30, 1127–1132 [DOI] [PubMed] [Google Scholar]
- 47. Velayati A., Yu W. H., Sidransky E. (2010) Curr. Neurol. Neurosci. Rep. 10, 190–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bras J., Singleton A., Cookson M. R., Hardy J. (2008) FEBS J. 275, 5767–5773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jakes R., Spillantini M. G., Goedert M. (1994) FEBS Lett. 345, 27–32 [DOI] [PubMed] [Google Scholar]
- 50. Lee J. C., Gray H. B., Winkler J. R. (2008) J. Am. Chem. Soc. 130, 6898–6899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee J. C., Langen R., Hummel P. A., Gray H. B., Winkler J. R. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 16466–16471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Petrucelli L., O'Farrell C., Lockhart P. J., Baptista M., Kehoe K., Vink L., Choi P., Wolozin B., Farrer M., Hardy J., Cookson M. R. (2002) Neuron 36, 1007–1019 [DOI] [PubMed] [Google Scholar]
- 53. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 54. Bas D. C., Rogers D. M., Jensen J. H. (2008) Proteins 73, 765–783 [DOI] [PubMed] [Google Scholar]
- 55. Dvir H., Harel M., McCarthy A. A., Toker L., Silman I., Futerman A. H., Sussman J. L. (2003) EMBO Rep. 4, 704–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kacher Y., Brumshtein B., Boldin-Adamsky S., Toker L., Shainskaya A., Silman I., Sussman J. L., Futerman A. H. (2008) Biol. Chem. 389, 1361–1369 [DOI] [PubMed] [Google Scholar]
- 57. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd Ed., Springer, New York [Google Scholar]
- 58. Eliezer D., Kutluay E., Bussell R., Jr., Browne G. (2001) J. Mol. Biol. 307, 1061–1073 [DOI] [PubMed] [Google Scholar]
- 59. Wei R. R., Hughes H., Boucher S., Bird J. J., Guziewicz N., Van Patten S. M., Qiu H., Pan C. Q., Edmunds T. (2011) J. Biol. Chem. 286, 299–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Premkumar L., Sawkar A. R., Boldin-Adamsky S., Toker L., Silman I., Kelly J. W., Futerman A. H., Sussman J. L. (2005) J. Biol. Chem. 280, 23815–23819 [DOI] [PubMed] [Google Scholar]
- 61. Zaidi N., Maurer A., Nieke S., Kalbacher H. (2008) Biochem. Biophys. Res. Commun. 376, 5–9 [DOI] [PubMed] [Google Scholar]
- 62. van Raaij M. E., van Gestel J., Segers-Nolten I. M., de Leeuw S. W., Subramaniam V. (2008) Biophys. J. 95, 4871–7878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Croke R. L., Patil S. M., Quevreauz J., Kendall D. A., Alexandrescu A. T. (2011) Protein Sci. 20, 256–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lieberman R. L., Wustman B. A., Huertas P., Powe A. C., Jr., Pine C. W., Khanna R., Schlossmacher M. G., Ringe D., Petsko G. A. (2007) Nat. Chem. Biol. 3, 101–107 [DOI] [PubMed] [Google Scholar]
- 65. Jao C. C., Hegde B. G., Chen J., Haworth I. S., Langen R. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 19666–19671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bodner C. R., Dobson C. M., Bax A. (2009) J. Mol. Biol. 390, 775–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pfefferkorn C. M., Lee J. C. (2010) J. Phys. Chem. B 114, 4615–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Georgieva E. R., Ramlall T. F., Borbat P. P., Freed J. H., Eliezer D. (2010) J. Biol. Chem. 285, 28261–28274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. de Alba E., Weiler S., Tjandra N. (2003) Biochemistry 42, 14729–14740 [DOI] [PubMed] [Google Scholar]
- 70. Hawkins C. A., de Alba E., Tjandra N. (2005) J. Mol. Biol. 346, 1381–1392 [DOI] [PubMed] [Google Scholar]
- 71. Morimoto S., Kishimoto Y., Tomich J., Weiler S., Ohashi T., Barranger J. A., Kretz K. A., O'Brien J. S. (1990) J. Biol. Chem. 265, 1933–1937 [PubMed] [Google Scholar]
- 72. Jo E., McLaurin J., Yip C. M., St George-Hyslop P., Fraser P. E. (2000) J. Biol. Chem. 275, 34328–34334 [DOI] [PubMed] [Google Scholar]
- 73. Martinez Z., Zhu M., Han S., Fink A. L. (2007) Biochemistry 46, 1868–1877 [DOI] [PubMed] [Google Scholar]
- 74. Park J. Y., Kim K. S., Lee S. B., Ryu J. S., Chung K. C., Choo Y. K., Jou I., Kim J., Park S. M. (2009) J. Neurochem. 110, 400–411 [DOI] [PubMed] [Google Scholar]
- 75. Zabrocki P., Bastiaens I., Delay C., Barnmens T., Ghillebert R., Pellens K., De Virgilio C., Van Leuven F., Winderickx J. (2008) Biochim. Biophys. Acta, Mol. Cell Res. 1783, 1767–1780 [DOI] [PubMed] [Google Scholar]
- 76. Meivar-Levy I., Horowitz M., Futerman A. H. (1994) Biochem. J. 303, 377–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Grace M. E., Newman K. M., Scheinker V., Berg-Fussman A., Grabowski G. A. (1994) J. Biol. Chem. 269, 2283–2291 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.