

Abstract
The H3K27 histone methyltransferase, Ezh2 (enhancer of zeste 2), is a Polycomb group protein that plays important roles in many biological processes including cellular differentiation, stem cell biology, and cancer development. Up-regulation of Ezh2 is observed in various human cancers consistent with its role in cell proliferation. Thus, understanding the regulation of Ezh2 may reveal how it contributes to the cellular proliferation process. Here, we demonstrate that Ezh2 can be regulated by the cyclin-dependent kinase, CDK1, which phosphorylates Ezh2 at threonines 345 and 487. Consistent with the cell cycle phase during which CDK1 exhibits peak activity, Ezh2 phosphorylation is enriched in cells arrested in mitosis when compared with S-phase. Phosphorylation of Thr-345 and Thr-487 promotes Ezh2 ubiquitination and subsequent degradation by the proteasome. Furthermore, expression of T345A/T487A confers a proliferative disadvantage when compared with cells expressing wild-type Ezh2, which suggests that phosphorylation of Ezh2 is important for cell proliferation. Collectively, these results establish a novel function for CDK1-mediated Ezh2 phosphorylation and provide a mechanism by which Ezh2 protein levels can be regulated in cells.
Keywords: Cell Cycle, Epigenetics, Mitosis, Protein Stability, Ubiquitin, CDK1, Ezh2, Phosphorylation, Polycomb
Introduction
Ezh22 (enhancer of zeste 2) is a member of the Polycomb group (PcG) proteins, which are evolutionarily conserved from Drosophila to mammals. Originally discovered in Drosophila, PcG proteins play an important role in regulating expression of HOX genes, which encode a set of transcription factors that specify the anterior-posterior axis and segment identity (1–3). As the catalytic subunit of Polycomb repressive complex 2 (PRC2), Ezh2 has the capacity to trimethylate lysine 27 of histone H3 (H3K27me3) but requires Suz12 (suppressor of zeste) and Eed (embryonic ectoderm development) for its enzymatic function (4–8). Biologically, Ezh2 plays an important role in epigenetic gene silencing and has been linked to diverse processes including developmental patterning, X-inactivation, and stem cell biology (9–11).
In addition to the functions mentioned above, a number of studies have also implicated a role for Ezh2 in cell proliferation and oncogenesis. For example, ectopic expression of Ezh2 confers a proliferative advantage in primary cells (12). In addition, Ezh2 is often aberrantly overexpressed in a variety of human cancers when compared with normal tissues (13, 14). Consistently, RNAi-mediated knockdown of Ezh2 compromises cell proliferation (12, 14, 15).
Although PcG proteins are best known for maintaining the repression of HOX genes during development, they also play a critical role in silencing the CDKN2A (cyclin-dependent kinase inhibitor 2A) locus, which encodes for the tumor suppressor genes p16INK4A and p14ARF. In young proliferating cells, evasion of senescence requires repression of CDKN2A, which is mediated by Ezh2 and H3K27 trimethylation (16–18). Consistent with this, Ezh2 expression is high in proliferating cells but is down-regulated in stressed and senescent cells. Hence, it is believed that overexpression of Ezh2 contributes to oncogenesis by inappropriately silencing tumor suppressor genes.
The above observations illustrate the importance of maintaining proper Ezh2 levels for normal function of cells. However, limited studies have been performed with respect to how cellular levels of Ezh2 are controlled. Transcription of the Ezh2 gene is regulated by the pRB-E2F pathway and peaks at the G1- to S-phase transition (12). Degradation of Ezh2 mRNA can be targeted by miR-101, whose expression decreases during cancer progression (15), which provides one explanation as to how Ezh2 levels are deregulated in cancer. Thus, it appears that Ezh2 is regulated at the transcriptional and post-transcriptional level.
Here, we report that Ezh2 phosphorylation can occur at two highly conserved residues, threonines 345 and 487. This modification can be mediated by the cyclin-dependent kinase, CDK1. Consistent with the notion that CDK1 is a mitotic kinase, enrichment of phospho-Ezh2 is observed in cells arrested at mitosis when compared with S-phase. Interestingly, the half-life of phospho-Ezh2 is shorter when compared with total Ezh2, and phospho-Ezh2 is subject to ubiquitination. Expression of the phospho-deficient mutant T345A/T487A results in a proliferative disadvantage when compared with cells expressing wild-type Ezh2. Collectively, these studies provide the first evidence linking Ezh2 protein levels to cell cycle-regulated Ezh2 phosphorylation.
EXPERIMENTAL PROCEDURES
Cell Culture, Transfections, and Drug Treatments
HEK293T, HeLa, U2OS, and NIH3T3 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and penicillin/streptomycin. PC3 cells were cultured in DMEM/F12 (1:1) supplemented with 10% fetal bovine serum and penicillin/streptomycin. Transfections were performed using Lipofectamine 2000 (Invitrogen catalog number 11668-019) or FuGENE 6 (Roche Applied Science catalog number 11814443001). Roscovitine was purchased from Cell Signaling Technology (catalog number 9805) and used at a final concentration of 50 μm. CGP74514A (Sigma catalog number C3353) was used at a final concentration of 2 μm. Cycloheximide (Sigma catalog number C4859) was used at a final concentration of 100 μg/ml.
Antibodies
The following antibodies were used in this study: FLAG M2 mouse monoclonal (Sigma catalog number F3165), mouse phospho-serine (Chemicon catalog number AB1603), mouse phospho-threonine (Cell Signaling catalog number 9386), mouse phospho-tyrosine 4G10 (gift from Weiguo Zhang, Duke University), rabbit Ezh2 (Cell Signaling catalog number 4905), rabbit SLBP (gift from William Marzluff, University of North Carolina), mouse cyclin B1 (Santa Cruz Biotechnology), mouse α-tubulin (Sigma catalog number T6199), lamin B (Santa Cruz Biotechnology catalog number sc-6217), mouse GST (Santa Cruz Biotechnology catalog number sc-138), rabbit H3K27me3 (Millipore catalog number 07-449), rabbit H3 (Abcam catalog number ab1791-100), and rabbit Suz12 (as described in Ref. 5). Antibodies against Thr(P)-345 and Thr(P)-487 were produced by injecting keyhole limpet hemocyanin-conjugated phosphorylated peptides into rabbits using a standard protocol outlined by Pocono Rabbit Farm and Laboratory (Canadensis, PA). The sequences of the peptides used are as follows: Thr(P)-345 (CTAERIK(pT)PPKRPG-NH2) and Thr(P)-487 (Ac-CPTEDVD(pT)PPRKK) (where pT indicates Thr(P)).
Lentiviral Constructs
The lentiviral system has been described previously (19). FLAG-tagged wild-type Ezh2, T345A/T487A, and T345E/T487E were cloned into the vector pTYF under the control of the EF1α promoter. For knockdown experiments, shRNAs targeting human EZH2 as well as a control shRNAs were cloned into pTYF under the control of the U6 promoter. The targeting sequences are as follows: control knockdown (5′-GTTCAGATGTGCGGCGAGT-3′) and EZH2 knockdown (5′-GCTGCCTTAGCTTCAGGAA-3′).
Cell Extract Preparation and Immunoprecipitation
Cell pellets were resuspended with RIPA lysis buffer consisting of 50 mm Tris (pH 7.4), 1% Nonidet P-40, and 150 mm NaCl supplemented with protease and phosphatase inhibitors (Roche Applied Science Complete protease inhibitor catalog number 11697498001 and Roche Applied Science PhosSTOP catalog number 04906837001). Lysates were incubated on ice for 45 min and subjected to centrifugation at 17,000 × g for 15 min at 4 °C. The resulting supernatant was transferred to a new tube, and the protein concentration of the lysate was determined by Bradford assay (Bio-Rad Protein Assay catalog number 500-0006). For FLAG immunoprecipitations, 30 μl of α-FLAG M2 affinity gel (Sigma catalog number A2220) was added to the lysates and rotated at 4 °C overnight. Bound proteins were washed three times with RIPA lysis buffer prior to the addition of 2× SDS loading buffer. Bound proteins were then resolved by SDS-PAGE.
Metabolic Labeling
Phosphate-free medium was prepared using DMEM without sodium phosphate (Invitrogen catalog number 11971) supplemented with 10% FBS, which was dialyzed overnight against Tris-buffered saline (50 mm Tris, pH 7.4, 150 mm NaCl) using 3500 molecular weight cut-off dialysis tubing. Transfected HEK293T cells were first washed once with phosphate-free medium and then labeled with phosphate-free medium containing 1 mCi of [γ-32P]ATP (PerkinElmer Life Sciences catalog number NET155001MC) for 3.5 h at 37 °C. Cells were harvested and subjected to RIPA lysis and FLAG immunoprecipitation as described above.
Subcellular Fractionation
For HeLa cytosolic extracts, nuclear extracts, and nuclear pellet, previously prepared fractions were used (4, 20). For subcellular fractionation of NIH3T3 cells, the cell pellet was resuspended in lysis buffer A (10 mm Tris, pH 8.0, 140 mm NaCl, 1.5 mm MgCl2, and 0.5% Nonidet P-40) supplemented with protease and phosphatase inhibitors followed by incubation on ice for 5 min. Nuclei were centrifuged at 1000 × g for 3 min at 4 °C. The supernatant containing the cytosolic fraction was transferred. The pellet was resuspended in the same volume of RIPA lysis buffer containing protease and phosphatase inhibitors. After incubation on ice for 30 min, the insoluble material was centrifuged at 17,000 × g for 15 min at 4 °C. The supernatant containing the nuclear extract was recovered.
In Vitro Phosphatase and Kinase Assays
Immunoprecipitated proteins were incubated with 1 μl of PP1 (New England Biolabs catalog number P0754) or λ-PPase (New England Biolabs catalog number P0753) in the supplied reaction buffer for 1 h at 30 °C. Recombinant GST-Ezh2 was incubated with CDK1-cyclin B (New England Biolabs catalog number P6020) in the presence of 1 mm cold ATP for 30 min at 30 °C. The reaction was terminated by the addition of 5× SDS loading buffer. Reaction products were analyzed by Western blot analysis.
Cell Cycle Synchronization
HeLa cells arrested at S-phase were obtained through double thymidine block, whereas M-phase arrested cells were collected after thymidine-nocodazole block. In short, cells were first treated with 1 mm thymidine for 18 h. Cells were washed two times with PBS and released for 4 h (M-phase) or 6 h (S-phase) prior to a second block with 1 mm thymidine (S-phase) or 0.1 μg/ml nocodazole (M-phase) for 16 h.
In Vivo Ubiquitination Assay
Cell pellets were resuspended in preboiled SDS-lysis buffer (50 mm Tris, pH 7.5, 0.5 mm EDTA, 1% SDS, 1 mm DTT) and further boiled for an additional 10 min. Lysates were clarified by centrifugation at 17,000 × g for 10 min at 4 °C. The supernatant was transferred to a new tube, and protein concentration was measured by Lowry Assay (Bio-Rad DC protein assay catalog number 500-0112). For immunoprecipitations, lysates were first diluted 10-fold with RIPA lysis buffer and then incubated with 20 μl of α-FLAG M2 affinity gel (Sigma catalog number A2220) overnight at 4 °C. Bound proteins were washed three times with RIPA lysis buffer and eluted with 2× SDS loading buffer.
Cell Proliferation Assay
Cell proliferation was monitored by absorbance using the MTS assay (CellTiter 96 AQueous One Solution cell proliferation assay, Promega catalog number G3582). Approximately 5000 cells/well were seeded in triplicate in a 96-well plate. At the indicated times, 20 μl of the reagent was added to the cells and incubated at 37 °C for 1.5 h. Absorbance at 490 nm was measured in a microplate reader. Following background subtraction, the number of cells was back-calculated using an established standard curve. The number of cells at day 0 was set to 1. Additional experimental procedures can be found in supplemental methods.
RESULTS
Serine 21 Is Not a Major Ezh2 Phosphorylation Site
Ezh2 has previously been reported to be a phospho-protein (21). To confirm this finding, HEK293T cells expressing FLAG-tagged Ezh2 were metabolically labeled with [γ-32P]ATP. Following FLAG immunoprecipitation, bound proteins were treated with or without protein phosphatase 1 (PP1) or λ-protein phosphatase (λ-PPase) before being resolved by SDS-PAGE. Results shown in Fig. 1A confirm that Ezh2 is indeed phosphorylated and that treatment with either PP1 or λ-PPase diminishes the signal (lanes 2–4). Western blot analysis using FLAG antibodies verifies that FLAG-tagged Ezh2 is present at similar amounts in all three lanes. Because Ser-21 of Ezh2 was previously reported to be an Akt phosphorylation site (21), we asked whether additional phosphorylation sites other than Ser-21 exist. To this end, a parallel metabolic labeling experiment expressing a mutant FLAG-tagged Ezh2 (S21A) was performed. Results shown in Fig. 1A demonstrated that the S21A mutant Ezh2 can also be phosphorylated, roughly to the same degree as wild-type Ezh2 (lanes 5–7), indicating that Ezh2 contains other phosphorylation sites, in addition to Ser-21.
FIGURE 1.
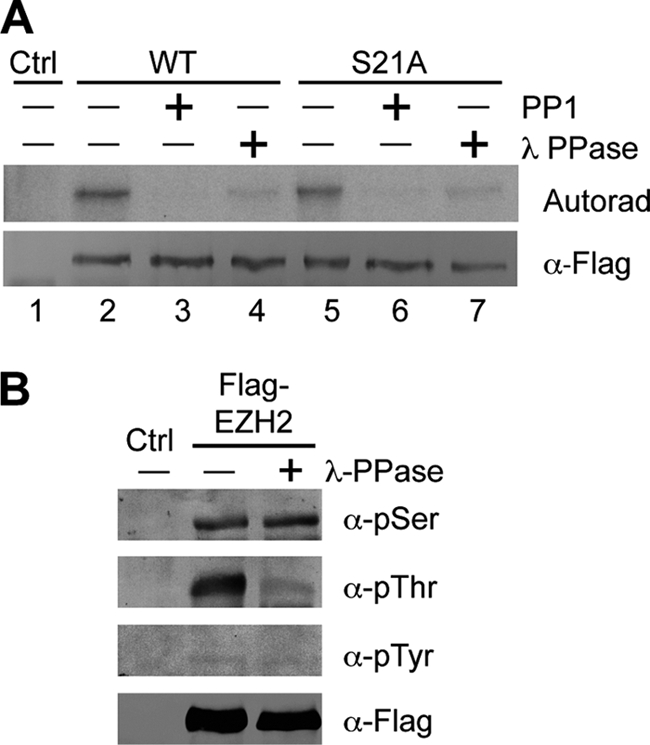
Ezh2 is threonine-phosphorylated. A, Ezh2 is phosphorylated in sites other than Ser-21. HEK293T cells exogenously expressing FLAG-tagged wild-type Ezh2 or S21A were radiolabeled with [γ-32P]ATP. Following immunoprecipitation using FLAG antibodies, bound proteins were treated with no phosphatase, with PP1, or with λ-PPase. Autoradiography (Autorad) confirms that both wild-type and S21A mutant Ezh2 are phosphorylated and that the signal diminishes upon phosphatase treatment. Bottom panel verifies that equal amounts FLAG-Ezh2 were used in the assay. Ctrl, control. B, Ezh2 is threonine-phosphorylated. FLAG-Ezh2 was transfected into HEK293T cells and immunoprecipitated using FLAG antibodies. Bound proteins were treated with or without λ-PPase and subjected to Western blot analysis using antibodies specific for phospho-serine (α-pSer), phospho-threonine (α-pThr), and phospho-tyrosine (α-pTyr).
Ezh2 Is Threonine-phosphorylated
To narrow down which amino acids of Ezh2 can be phosphorylated, we took advantage of antibodies recognizing phospho-serine, phospho-threonine, and phospho-tyrosine. HEK293T cells were transfected with FLAG-tagged Ezh2, and after immunoprecipitation using FLAG antibodies, bound proteins were treated with or without λ-PPase, which is capable of removing the phospho-group from all three residues. Western blot analysis using the residue-specific phospho-antibodies revealed that Ezh2 is threonine-phosphorylated as λ-PPase treatment greatly diminished the signal (Fig. 1B, second panel). In contrast, similar treatment did not result in significant changes in Ser or Tyr phosphorylation levels, although these results cannot rule out the possibility that Ezh2 is subjected to Ser or Tyr phosphorylation as detection of these modifications depends on both the quality of the antibody as well as the abundance of the modification. Nevertheless, the above results demonstrate that Ezh2 is subject to threonine phosphorylation.
Ezh2 Is Phosphorylated at Both Thr-345 and Thr-487 in Vivo
To determine which threonines of Ezh2 may be phosphorylated, we utilized a combination of phosphorylation site prediction programs (MotifScan and NetPhos) and published phospho-proteomic data. With this approach, two putative phosphorylation sites were of great interest, threonines 345 and 487. Under the highest stringency, both programs predicted these two sites, and in the case of MotifScan, they were the only sites that were scored within the top 1%. Previous phospho-proteomic studies also identified Thr-345 and Thr-487 of Ezh2 to be phosphorylated (22, 23). These residues are highly conserved in humans, mice, and zebrafish, whereas Thr-487 is also conserved in flies (Fig. 2A).
FIGURE 2.

Ezh2 is phosphorylated at threonines 345 and 487. A, threonines 345 and 487 are conserved across species. Human, mouse, zebrafish, and fly Ezh2 were aligned using MultAlin. Relevant regions are shown. Threonines 345 and 487 are underlined, whereas conserved amino acids are highlighted in bold. B, specificity test of the phospho-Ezh2 antibodies. Dot blot analysis was performed using 2-fold dilutions of the unmodified (Unmod.) and phosphorylated (Phos.) peptides corresponding to amino acids surrounding Thr-345 or Thr-487. C, exogenously expressed Ezh2 is phosphorylated at Thr-345 (pT345) and Thr-487 (pT487). HEK293T cells were transfected with empty vector (Ctrl) or FLAG-tagged wild-type (F-WT), T345A (F-T345A), or T487A mutant Ezh2 (F-T487A). Following FLAG immunoprecipitation, bound proteins were analyzed by Western blot analysis using the indicated antibodies. D, endogenous Ezh2 is phosphorylated at Thr-345 and Thr-487. Western blot analysis of NIH3T3 and HeLa cell extracts using the phospho-Ezh2 antibodies was performed. For NIH3T3 extracts, 100 μg of nuclear extract (NE) and the equivalent volume of cytoplasmic extract (CYT) were loaded. For HeLa extracts, 100 μg of each fraction was loaded. Recombinant PRC2 complex was used as a positive control. Subcellular fractionation was confirmed using the following controls: tubulin (cytoplasmic), Ezh2 (nuclear), and lamin B (nuclear). NP, nuclear pellet. E, knockdown (KD) of Ezh2 results in decreased levels of phospho-Ezh2. HeLa cells were infected with lentiviruses expressing either control shRNA or Ezh2 shRNA. Top panel, quantitative RT-PCR confirming Ezh2 knockdown. Bottom panel, lysates were subjected to Western blot analysis using the indicated antibodies. Error bars indicate S.D.
To gain insight into the function of their phosphorylation, we generated antibodies that specifically recognize these phosphorylation sites. After two-step affinity purification, dot blot analysis confirmed that the antibodies are specific for peptides containing the phosphorylated threonine residues (Fig. 2B). Furthermore, both phospho-specific antibodies recognize exogenously expressed wild-type Ezh2 that was immunoprecipitated, but not when the phosphorylation site was mutated to an alanine (Fig. 2C).
To determine whether endogenous Thr(P)-345 and Thr(P)-487 is present in cells, we performed Western blot analysis using subcellular protein extracts from HeLa and NIH3T3 cells. As a control, we also included recombinant PRC2 complex purified from Sf9 cells. Results shown in Fig. 2D demonstrate that the Thr(P)-345 antibody recognized a band corresponding to the size of recombinant Ezh2 as well as a slower migrating band in both the nuclear extract and the nuclear pellet fractions derived from HeLa cells as well as nuclear extracts from NIH3T3 cells (Fig. 2D, first panel). On the other hand, Western blot analysis with the Thr(P)-487 antibody detected one band corresponding to the size of Ezh2 in the NIH3T3 nuclear extract and the HeLa nuclear pellet (Fig. 2D, second panel). When more protein extract was loaded, Thr(P)-487 was also detected in HeLa nuclear extracts (data not shown). It should be noted that the Thr(P)-487 antibody was raised against the mouse sequence that contains one nonconserved amino acid when compared with the human sequence (Fig. 2A), which may account, in part, for the decreased sensitivity toward Thr(P)-487 in HeLa nuclear extracts when compared with NIH3T3 nuclear extracts. Nevertheless, the results demonstrate that the phospho-Ezh2 antibodies can detect endogenous bands corresponding to the size of recombinant Ezh2.
To further confirm that the detected bands are indeed phosphorylated Ezh2, HeLa cells stably expressing shRNAs targeting Ezh2 were generated by lentiviral infection. Following puromycin selection, we performed quantitative RT-PCR, which demonstrated roughly 80% knockdown at the mRNA level (Fig. 2E, top panel). Consistent with this observation, a dramatic decrease in Ezh2 protein levels was also detected (Fig. 2E, bottom panel). Importantly, Western blot analysis using both phospho-Ezh2 antibodies confirmed that the bands corresponding to the size of Ezh2 diminished upon Ezh2 knockdown (Fig. 2E, bottom panel). Furthermore, the slower migrating band observed in the Thr(P)-345 Western blot remained unchanged upon Ezh2 knockdown, indicating that this is a cross-reacting band, whereas the faster migrating band corresponds to Thr(P)-345 Ezh2 as this band decreases upon Ezh2 knockdown. Collectively, the above results demonstrate the specificity of our phospho-Ezh2 antibodies as well as the presence of phosphorylated Ezh2 (Thr(P)-345 and Thr(P)-487) in the cells tested.
When compared with control knockdown cells, Ezh2 knockdown HeLa cells appeared larger and rounder (supplemental Fig. S1A) and proliferated significantly more slowly (supplemental Fig. S1B). Flow cytometry analysis of DNA content indicated that there was a higher percentage of cells in G2/M-phase in the Ezh2 knockdown cells when compared with the control (supplemental Fig. S1C), consistent with previous studies in prostate cancer cells (14). Thus, these data confirm the finding that Ezh2 plays a role in cell proliferation and extend this observation to HeLa cells.
CDK1 Phosphorylates Ezh2 at both Thr-345 and Thr-487 in Vitro and in Vivo
Close inspection of the amino acid residues surrounding both Thr-345 and Thr-487 revealed the presence of a CDK1 consensus sequence ((S/T)PX(R/K)) (Fig. 2A). As a cyclin-dependent kinase, CDK1 specifically associates with cyclin B, whose level peaks during mitosis. Given that knockdown of Ezh2 results in the accumulation of cells in G2/M-phase (supplemental Fig. S1C), CDK1 was an attractive candidate for mediating Ezh2 phosphorylation at Thr-345 and Thr-487.
To explore this possibility, a cold in vitro kinase assay was performed using purified GST-tagged Ezh2 and CDK1-cyclin B followed by Western blot analysis using the phospho-Ezh2 antibodies described above. As shown in Fig. 3A, phosphorylated Ezh2 is detected only when both GST-Ezh2 and CDK1-cyclin B are present in the reaction. To gain further support that CDK1 is responsible for Thr-345 and Thr-487 phosphorylation, we used a potent CDK inhibitor, roscovitine. To this end, HEK293T cells expressing FLAG-tagged Ezh2 were treated in the presence or absence of roscovitine. Following FLAG immunoprecipitation, the phosphorylation status of Ezh2 was analyzed by Western blotting. Results shown in Fig. 3B demonstrate that phosphorylation of both Thr-345 and Thr-487 is greatly inhibited by the presence of roscovitine, indicating that CDKs (including CDK1) contribute to Ezh2 phosphorylation at both sites. Similar analysis also demonstrates that roscovitine treatment resulted in diminished endogenous phospho-Ezh2 levels in HeLa cells (Fig. 3C). Because roscovitine also inhibits CDK2 and CDK5 (24), we therefore treated HeLa cells with a CDK1-specific inhibitor, CGP74514A, for various times. Western blot analysis shown in Fig. 3D demonstrates a decrease in endogenous phospho-Ezh2 when CDK1 is specifically inhibited, indicating that CDK1 contributes to Ezh2 phosphorylation at Thr-345 and Thr-487.
FIGURE 3.

CDK1 mediates Ezh2 phosphorylation at Thr-345 and Thr-487. A, CDK1 phosphorylates Ezh2 at Thr-345 and Thr-487 (pT487) in vitro. Recombinant GST-tagged Ezh2 was subjected to a cold kinase assay using CDK1-cyclin B kinase followed by Western blot analysis using phospho-Ezh2 antibodies specific for Thr(P)-345 (α-pT345) and Thr(P)-487 (α-pT487), respectively. B, phosphorylation of Ezh2 by CDKs can be inhibited by roscovitine (Ros). HEK293T cells were transfected with empty vector (Ctrl) or FLAG-tagged Ezh2 (F-Ezh2). 24 h after transfection, cells were treated with 50 μm roscovitine for 24 h to inhibit CDKs. Following FLAG immunoprecipitation, bound proteins were subjected to Western blot analysis using the indicated antibodies. C, inhibition of CDKs results in loss of endogenous phospho-Ezh2. HeLa cells were treated with 50 μm roscovitine for the indicated times (hr Ros). Lysates were analyzed by Western blot analysis using the indicated antibodies. DMSO, dimethyl sulfoxide. D, specific inhibition of CDK1 results in loss of endogenous phospho-Ezh2. HeLa cells were treated with 2 μm CGP74514A (CGP) to specifically inhibit CDK1. Lysates were analyzed by Western blot analysis using the indicated antibodies. E, phospho-Ezh2 is enriched in cells arrested at mitosis. HeLa cells were arrested at S-phase (S) and M-phase (M) by double thymidine block and thymidine-nocodazole block, respectively. Extracts were analyzed by Western blot using the indicated antibodies. SLBP, S-phase control; cyclin B1, M-phase control; tubulin, loading control.
We then asked whether phospho-Ezh2 is enriched during mitosis, which would be expected if Ezh2 was a true CDK1 substrate. To address this question, HeLa cells were arrested at S-phase and M-phase by double thymidine block and thymidine-nocodazole block, respectively. Western blot analysis using antibodies for the cell cycle markers cyclin B1 (M-phase) and SLBP (S-phase) confirmed that the cells were properly arrested (Fig. 3E). Importantly, an enrichment of phospho-Ezh2 in the M-phase arrested cells was observed when compared with S-phase arrested cells. Thus, the above data collectively demonstrate that Ezh2 is a CDK1 substrate.
Thr-345 and Thr-487 Are Not Important for Global Levels of H3K27 Trimethylation
Phosphorylation of Ser-21 was previously reported to inhibit histone methyltransferase (HMT) activity (21). Thus, it is logical to ask whether phosphorylation of Thr-345 and Thr-487 has an effect on Ezh2 activity. There are two possible ways in which Thr-345 and Thr-487 may affect the HMT activity of Ezh2. 1) They may be crucial residues that affect the intrinsic enzymatic activity or 2) they may indirectly affect enzymatic activity through disruption of the Ezh2 interaction partners, Suz12 and Eed. It should be noted that these two possibilities are not mutually exclusive.
To address this question, we first asked whether mutation of Thr-345 and Thr-487 to alanines affected the ability of Ezh2 to interact with Suz12. NIH3T3 cells stably expressing the empty vector (control), FLAG-tagged Ezh2 (WT), or T345A/T487A (Mut-A) were generated by lentiviral infection. Immunoprecipitation using FLAG antibodies followed by Western blot analysis using Suz12 antibodies demonstrates that both WT and Mut-A bound to relatively similar amounts of Suz12 (supplemental Fig. S2A). Thus, Thr-345 and Thr-487 do not appear to be important to maintaining the integrity of the PRC2 complex.
To assess the contributions of Thr-345 and Thr-487 to Ezh2 HMT activity, a dominant negative approach was used. Given that the phosphorylation-deficient mutant Ezh2 is capable of interacting with Suz12, it is expected that overexpression of a mutant Ezh2 would compete with wild-type Ezh2 for interaction with Suz12 and Eed. If Thr-345 and Thr-487 are important residues for Ezh2 HMT activity, we would expect a global decrease of trimethylated H3K27. Using this strategy, FLAG-tagged wild-type Ezh2, T345A, or T487A were expressed in U2OS cells and were co-immunostained using antibodies against FLAG and H3K27me3. Results shown in supplemental Fig. S2B demonstrate that cells expressing wild-type or mutant Ezh2 have similar H3K27me3 levels, suggesting that mutations on these residues do not significantly alter the Ezh2 HMT activity. Consistent with this notion, Western blot analysis of histones from HeLa cells stably expressing wild-type Ezh2, T345A/T487A (Mut-A), or T345E/T487E (Mut-E) demonstrates that global levels of H3K27me3 are not significantly altered (supplemental Fig. S2C).
Phosphorylation of Ezh2 Is Important for Ubiquitin-mediated Degradation
Given that both mRNA and protein levels of Ezh2 accumulate at the G1/S-phase transition (12), we hypothesized that phosphorylation of Ezh2 during mitosis could function as a signal for degradation. To examine this possibility, we compared the half-lives of Thr(P)-345, Thr(P)-487, and total Ezh2. To this end, we transfected FLAG-tagged wild-type Ezh2 into HEK293T cells and treated cells with the protein synthesis inhibitor, cycloheximide, for different times. Western blot analysis using the FLAG antibody allowed for detection of total exogenous Ezh2, which exhibited a half-life of ∼2 h (Fig. 4A), similar to a previous study (25). In contrast, Thr(P)-345 and Thr(P)-487 Ezh2 proteins exhibited a significantly shorter half-life when compared with the total Ezh2 (Fig. 4A), although the two phosphorylated forms have similar half-lives (∼1 h).
FIGURE 4.

Phosphorylated Ezh2 is ubiquitinated and degraded via the proteasome pathway. A, phospho-Ezh2 exhibits a shorter half-life when compared with total Ezh2. Asynchronous HEK293T cells were transfected with FLAG-tagged Ezh2 and treated with 100 μg/ml cycloheximide (CHX) for the indicated times. Western blot analysis was performed using the indicated antibodies (top panel). Western blot signals were quantified using ImageJ (bottom panel). α-pT345, Thr(P)-345 antibody; α-pT487, Thr(P)-487 antibody. B, mutant Ezh2 degrades more slowly after mitosis. HeLa cells stably expressing FLAG-tagged wild-type Ezh2 (WT) or T345A/T487A (Mut-A) were synchronized at mitosis by thymidine-nocodazole block and then released into the cell cycle for the times indicated. Protein levels of Ezh2 were detected by FLAG Western blot (top panel). Cyclin B1 is a marker for mitosis, and tubulin was used as a loading control. Western blot signals were quantified using ImageJ (bottom panel). C, phospho-Ezh2 is ubiquitinated. Phospho-Ezh2 was immunoprecipitated from HeLa extracts using antibodies against Thr(P)-345 and Thr(P)-487. Total Ezh2 was immunoprecipitated using Ezh2 antibodies. Western blot analysis of bound proteins was performed using the antibodies indicated. D, ubiquitination status of Ezh2 in mitotic cells. HeLa cells stably expressing empty vector (Ctrl), FLAG-tagged wild-type Ezh2 (WT), T345A/T487A (Mut-A), and T345E/T487E (Mut-E) were generated by lentiviral infection and transfected with HA-tagged ubiquitin. Cells were synchronized and arrested at mitosis by thymidine-nocodazole block. 4 h prior to harvesting, cells were treated with or without 25 μm MG132. Lysates were prepared under denaturing conditions and subjected to immunoprecipitation (IP) using FLAG antibodies. Bound proteins were analyzed by Western blot using the indicated antibodies.
The shorter half-life of phospho-Ezh2 may be due to reduced protein stability or the actions of protein phosphatases. To assess the first possibility, we examined the protein levels of wild-type Ezh2 and T345A/T487A (Mut-A) after mitosis. HeLa cells stably expressing wild-type Ezh2 or Mut-A were mitotically arrested and released for various times. Western blot analysis demonstrates that wild-type Ezh2 is degraded more rapidly when compared with Mut-A after mitosis (Fig. 4B), suggesting that the shorter half-life of phospho-Ezh2 is due to reduced protein stability.
Because Thr(P)-345 and Thr(P)-487 appear to be less stable than total Ezh2, we investigated the possibility that phospho-Ezh2 is targeted for ubiquitination and degradation. To this end, phospho-Ezh2 and total Ezh2 were immunoprecipitated from HeLa cells with rabbit IgG as a negative control. Western blot analysis of bound proteins was performed using antibodies against Thr(P)-345, Thr(P)-487, total Ezh2, and ubiquitin. Results shown in Fig. 4C demonstrate that a ladder of bands reminiscent of polyubiquitinated proteins was observed when either Thr(P)-345 or Thr-487 is immunoprecipitated. This result supports the notion that phospho-Ezh2 may serve as a signal for ubiquitin-mediated degradation.
To further validate that Thr(P)-345 and Thr(P)-487 are important for degradation of Ezh2, in vivo ubiquitination assays were performed to assess whether mutation of these residues could affect the ubiquitination status of Ezh2. We first compared the ubiquitination status of wild-type with T345A/T487A (Mut-A) and T345E/T487E (Mut-E) mutants Ezh2 in asynchronous cells, which revealed no significant difference (supplemental Fig. S3A). Given that Ezh2 phosphorylation is enriched during mitosis (Fig. 3D) and that mitotic cells only account for ∼10–20% of the total cell population, the lack of difference between wild-type and mutants may simply be due to the low levels of phospho-Ezh2 in asynchronous cells. To circumvent this issue, we performed the in vivo ubiquitination assay using HeLa cells stably expressing Ezh2. After transfection of HA-tagged ubiquitin, cells were arrested at mitosis by thymidine-nocodazole block. Prior to harvesting, the cells were treated with or without the proteasome inhibitor, MG132. Although ubiquitinated Ezh2 was not detected in the untreated cells, likely due to its quick degradation, wild-type Ezh2 was clearly ubiquitinated when mitotic cells were treated with MG132 (Fig. 4D, right panel, lane 2). Importantly, mutation of both Thr-345 and Thr-487 to either an alanine (Mut-A) or a glutamate (Mut-E) dramatically reduced ubiquitination of Ezh2 (Fig. 4D, right panel, compare lane 2 with lanes 3 and 4). Interestingly, Mut-E was modestly ubiquitinated, suggesting that this mutation can partially act as a phospho-mimetic. Collectively, the above data demonstrate that phospho-Ezh2 is degraded via the ubiquitin pathway.
Ezh2 Thr-345 and Thr-487 Are Important for Cell Proliferation
The fact that Ezh2 is important for cell proliferation (supplemental Fig. S1B) (12) coupled with the observation that phosphorylation of Ezh2 is subject to cell cycle regulation prompted us to ask whether Thr-345 and Thr-487 of Ezh2 are important for cell proliferation. To this end, prostate cancer cells (PC3) and HeLa cells were infected with lentiviruses expressing the empty vector (control), FLAG-tagged wild-type Ezh2 (WT), or T345A/T487A mutant Ezh2 (Mut-A). Western blot analysis demonstrates that wild-type and mutant Ezh2 were expressed at relatively similar levels (Fig. 5, A and B). Cell proliferation was monitored by absorbance using the MTS assay, and the number of cells was calculated using a standard curve. Consistent with a previous study (12), overexpression of Ezh2 in both PC3 and HeLa cells results in a proliferative advantage when compared with the control (Fig. 5, C and D, compare dashed line with solid line). Interestingly, both cell lines expressing Mut-A proliferated significantly more slowly when compared with wild type (Fig. 5, C and D, compare dotted line with dashed line). These results support the importance of Thr-345 and Thr-487 of Ezh2 in regulating cell proliferation.
FIGURE 5.

Thr-345 and Thr-487 of Ezh2 are important for cell proliferation. Western blot analysis demonstrates that wild type and T345A/T487A mutant are expressed at similar levels. A and B, PC3 (A) and HeLa (B) cells were infected with lentiviruses overexpressing empty vector (Ctrl), wild-type Ezh2 (WT), or T345A/T487A (Mut-A). Exogenous Ezh2 was detected using FLAG antibodies, whereas tubulin served as a loading control. C and D, PC3 (C) and HeLa (D) cells expressing T345A/T487A proliferate more slowly when compared with wild type. Approximately 5000 cells were seeded in triplicate in a 96-well plate, and cell proliferation was monitored by absorbance using the MTS assay at the indicated times. The measured absorbance was converted to the number of cells using a standard curve. For each cell line, the number of cells at day 0 was set to 1. Error bars in panels C and D indicate S.D.
DISCUSSION
In this study, we present evidence demonstrating that Ezh2 is subject to phosphorylation at two highly conserved residues, Thr-345 and Thr-487, which are bona fide CDK1 substrates. Consistent with the cell cycle-regulated activity of CDK1, Ezh2 phosphorylation is enriched during mitosis. Phospho-Ezh2 exhibits reduced stability due to its ubiquitination and degradation by the proteasome. In addition, cells expressing Ezh2 where both threonines are mutated manifest a proliferative disadvantage in at least two cell types. Thus, our studies establish a role for Thr-345 and Thr-487 in regulating Ezh2 stability, a novel consequence of CDK1-mediated phosphorylation.
The significance of CDK1-mediated regulation of Ezh2 stems from the necessity for a cell to transmit its epigenetic marks to daughter cells during replication. The mechanisms underlying cellular memory are poorly understood, although it is evident that Ezh2 (as well as other Polycomb group proteins) plays a significant role in this process. Interestingly, the PRC2 complex can bind to H3K27me3 and was observed to co-localize with this mark during G1 as well as sites of ongoing DNA replication (26), which led to the proposal that PRC2 binds to established H3K27me3 during DNA replication and transmits the mark to the newly synthesized histones incorporated into the daughter strand. Given the amount of epigenetic information that must be transmitted to the daughter strands, one might expect to observe up-regulation of Ezh2 prior to DNA replication. Consistent with this notion, the pRB-E2F pathway up-regulates Ezh2 at both mRNA and protein levels at the G1 to S transition (12).
After the cell has successfully replicated its DNA and transmitted its epigenetic marks, excess Ezh2 accumulated during the G1 to S transition might need to be purged to restore the normal level. Thus, mitotic phosphorylation of Ezh2 and subsequent degradation by the ubiquitination pathway might serve such a purpose (Fig. 6). Because Ezh2 binds and represses the CDKN2A tumor suppressor locus (16–18), degradation of excess Ezh2 after mitosis may be important to keep cell proliferation in check.
FIGURE 6.

A model depicting the role of Ezh2 phosphorylation during mitosis. During mitosis when CDK1 kinase activity is high, Ezh2 is phosphorylated (circled P) at Thr-345 and Thr-487 (Box 1). Phosphorylation of Thr-345 facilitates Ezh2 interaction with lncRNAs including HOTAIR (Box 2), which recruits PRC2 (other components not shown) to Polycomb target genes. Initial H3K27me3 marks are placed on the newly incorporated histones of the daughter strand by PRC2 containing phospho-Ezh2 (Box 3). Phosphorylated Ezh2 is then ubiquitinated (Ub) and degraded via the proteasome, whereas additional PRC2 complexes (regardless of their Ezh2 phosphorylation status) bind to the initial trimethyl H3K27 mark and propagate the mark (Box 4).
At the time this work was nearing completion, three studies concerning CDK1-mediated phosphorylation at Thr-345 and Thr-487 were published (27–29). Chen et al. (27) provided evidence demonstrating that phosphorylation of Thr-350 (Thr-345 in mouse Ezh2) is important for the recruitment of Ezh2 and maintenance of H3K27 trimethylation at PRC2 target loci including HOXA9 and DAB2IP. This could be explained by the observation that the phospho-mimetic mutant T345E exhibits enhanced binding to the lncRNAs HOTAIR and XIST, which are known to recruit Ezh2 to target loci (28). On the other hand, Wei et al. (29) reported that phosphorylation at Thr-492 (Thr-487 in mouse Ezh2) disrupts Ezh2 binding with other PRC2 components, Suz12 and Eed, thereby attenuating Ezh2 methyltransferase activity.
Collectively, these studies show that Thr-345 and Thr-487 phosphorylation can function distinctively. Expression of human T350A, but not T492A, resulted in slower cell proliferation (27, 29), suggesting that the proliferative disadvantage we observed in cells expressing mouse T345A/T487A was due to loss of phosphorylation of Thr-345, but not Thr-487. Additionally, cancer cell migration and invasion required Thr-350 (27), but expression of the phospho-deficient mutant T492A enhanced these two processes (29). Thus, Thr(P)-350 and Thr(P)-492 appear to play opposing roles in cancer cell migration and invasion. It will be interesting to determine which of these modifications dominates given that these two phosphorylation sites can exist simultaneously within mouse Ezh2 (Fig. 4C).
A role for Thr-350 phosphorylation in regulating Ezh2 protein stability was also investigated by Chen et al. (27), but the authors concluded that it had no effect. However, the basis of their conclusion stemmed from an experiment where the half-lives of wild-type and T350A Ezh2 were compared and found to be similar. We also initially compared the half-lives of wild-type, Mut-A, and Mut-E Ezh2 and came to the same conclusion (supplemental Fig. S3B). However, phosphorylation of Ezh2 occurs during mitosis (Fig. 3E), and it is estimated that in asynchronous cells, only 1% of Ezh2 is phosphorylated at Thr-345 (28). For this reason, we compared the half-life of phospho-Ezh2 with total Ezh2 and concluded that phospho-Ezh2 has a shorter half-life when compared with the total Ezh2 (Fig. 4A). We further substantiated our conclusion by demonstrating that Mut-A is degraded less rapidly after mitosis (Fig. 4B) and that ubiquitination of Ezh2 is impaired in the mutants Mut-A and Mut-E (Fig. 4, C and D).
At first glance, it may seem contradictory that phosphorylation of Thr-345 leads to Ezh2 instability given that this modification is also important for lncRNA binding to facilitate recruitment of Ezh2 to target promoters (28). However, it should be noted that these two consequences are not necessarily mutually exclusive. Because Thr(P)-345 constitutes a small fraction of total Ezh2, Kaneko et al. (28) suggest that this form of Ezh2 may be important for the initial establishment of the H3K27me3 mark. This mark can later be propagated by other PRC2 complexes regardless of their Ezh2 phosphorylation status especially because PRC2 can bind to H3K27me3 (26). Thus, Ezh2 Thr(P)-345 may be degraded after the recruitment of PRC2 to their targets by lncRNAs and the initial establishment of H3K27me3 marks (Fig. 6). This model is particularly attractive because initial recruitment of Ezh2 may be more specific due to the use of lncRNAs. Given that we presently do not know the kinetics of Ezh2 recruitment, we cannot confirm this possibility.
In summary, the findings described in this study coupled with the studies by other groups demonstrate an important role for CDK1-mediated phosphorylation of Ezh2. Like Ezh2, CDK1 activity is positively associated with cell proliferation and cancer (30) and has emerged as a central player in controlling ES cell self-renewal and lineage specification (31). Thus, this mechanism of Ezh2 regulation may provide valuable insight into the development of therapeutic drugs as well as methods in regenerative medicine.
Supplementary Material
Acknowledgments
We thank Krzysztof Krajewski for peptide synthesis and Jake Dmochowski for technical assistance. We also thank William Marzluff for the SLBP antibody, Yue Xiong for the HA-tagged ubiquitin construct, and Weiguo Zhang for the phospho-tyrosine 4G10 antibody.
This work was supported, in whole or in part, by National Institutes of Health Grant GM68804 (to Y. Z.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental methods and Figs. S1–S3.
- Ezh2
- enhancer of zeste 2
- PcG
- Polycomb group
- CDK
- cyclin-dependent kinase
- CDKN2A
- cyclin-dependent kinase inhibitor 2A
- PRC2
- Polycomb repressive complex 2
- Suz12
- suppressor of zeste
- Eed
- embryonic ectoderm development
- RIPA
- radioimmune precipitation
- PP1
- protein phosphatase 1
- PPase
- protein phosphatase
- lncRNA
- long noncoding RNA
- SLBP
- stem-loop-binding protein
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
- HMT
- histone methyltransferase.
REFERENCES
- 1. Duncan I. M. (1982) Genetics 102, 49–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lewis E. B. (1978) Nature 276, 565–570 [DOI] [PubMed] [Google Scholar]
- 3. Struhl G. (1981) Nature 293, 36–41 [DOI] [PubMed] [Google Scholar]
- 4. Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R. S., Zhang Y. (2002) Science 298, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 5. Cao R., Zhang Y. (2004) Mol. Cell 15, 57–67 [DOI] [PubMed] [Google Scholar]
- 6. Czermin B., Melfi R., McCabe D., Seitz V., Imhof A., Pirrotta V. (2002) Cell 111, 185–196 [DOI] [PubMed] [Google Scholar]
- 7. Kuzmichev A., Nishioka K., Erdjument-Bromage H., Tempst P., Reinberg D. (2002) Genes Dev. 16, 2893–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Müller J., Hart C. M., Francis N. J., Vargas M. L., Sengupta A., Wild B., Miller E. L., O'Connor M. B., Kingston R. E., Simon J. A. (2002) Cell 111, 197–208 [DOI] [PubMed] [Google Scholar]
- 9. Cao R., Zhang Y. (2004) Curr. Opin. Genet. Dev. 14, 155–164 [DOI] [PubMed] [Google Scholar]
- 10. Lee T. I., Jenner R. G., Boyer L. A., Guenther M. G., Levine S. S., Kumar R. M., Chevalier B., Johnstone S. E., Cole M. F., Isono K., Koseki H., Fuchikami T., Abe K., Murray H. L., Zucker J. P., Yuan B., Bell G. W., Herbolsheimer E., Hannett N. M., Sun K., Odom D. T., Otte A. P., Volkert T. L., Bartel D. P., Melton D. A., Gifford D. K., Jaenisch R., Young R. A. (2006) Cell 125, 301–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Plath K., Fang J., Mlynarczyk-Evans S. K., Cao R., Worringer K. A., Wang H., de la Cruz C. C., Otte A. P., Panning B., Zhang Y. (2003) Science 300, 131–135 [DOI] [PubMed] [Google Scholar]
- 12. Bracken A. P., Pasini D., Capra M., Prosperini E., Colli E., Helin K. (2003) EMBO J. 22, 5323–5335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simon J. A., Lange C. A. (2008) Mutat. Res. 647, 21–29 [DOI] [PubMed] [Google Scholar]
- 14. Varambally S., Dhanasekaran S. M., Zhou M., Barrette T. R., Kumar-Sinha C., Sanda M. G., Ghosh D., Pienta K. J., Sewalt R. G., Otte A. P., Rubin M. A., Chinnaiyan A. M. (2002) Nature 419, 624–629 [DOI] [PubMed] [Google Scholar]
- 15. Varambally S., Cao Q., Mani R. S., Shankar S., Wang X., Ateeq B., Laxman B., Cao X., Jing X., Ramnarayanan K., Brenner J. C., Yu J., Kim J. H., Han B., Tan P., Kumar-Sinha C., Lonigro R. J., Palanisamy N., Maher C. A., Chinnaiyan A. M. (2008) Science 322, 1695–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Agherbi H., Gaussmann-Wenger A., Verthuy C., Chasson L., Serrano M., Djabali M. (2009) PLoS One 4, e5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bracken A. P., Kleine-Kohlbrecher D., Dietrich N., Pasini D., Gargiulo G., Beekman C., Theilgaard-Mönch K., Minucci S., Porse B. T., Marine J. C., Hansen K. H., Helin K. (2007) Genes Dev. 21, 525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kotake Y., Cao R., Viatour P., Sage J., Zhang Y., Xiong Y. (2007) Genes Dev. 21, 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He J., Kallin E. M., Tsukada Y., Zhang Y. (2008) Nat. Struct. Mol. Biol. 15, 1169–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang H., Wang L., Erdjument-Bromage H., Vidal M., Tempst P., Jones R. S., Zhang Y. (2004) Nature 431, 873–878 [DOI] [PubMed] [Google Scholar]
- 21. Cha T. L., Zhou B. P., Xia W., Wu Y., Yang C. C., Chen C. T., Ping B., Otte A. P., Hung M. C. (2005) Science 310, 306–310 [DOI] [PubMed] [Google Scholar]
- 22. Beausoleil S. A., Jedrychowski M., Schwartz D., Elias J. E., Villén J., Li J., Cohn M. A., Cantley L. C., Gygi S. P. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choudhary C., Olsen J. V., Brandts C., Cox J., Reddy P. N., Böhmer F. D., Gerke V., Schmidt-Arras D. E., Berdel W. E., Müller-Tidow C., Mann M., Serve H. (2009) Mol. Cell 36, 326–339 [DOI] [PubMed] [Google Scholar]
- 24. Meijer L., Borgne A., Mulner O., Chong J. P., Blow J. J., Inagaki N., Inagaki M., Delcros J. G., Moulinoux J. P. (1997) Eur. J. Biochem. 243, 527–536 [DOI] [PubMed] [Google Scholar]
- 25. Dimri M., Bommi P. V., Sahasrabuddhe A. A., Khandekar J. D., Dimri G. P. (2010) Carcinogenesis 31, 489–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hansen K. H., Bracken A. P., Pasini D., Dietrich N., Gehani S. S., Monrad A., Rappsilber J., Lerdrup M., Helin K. (2008) Nat. Cell Biol. 10, 1291–1300 [DOI] [PubMed] [Google Scholar]
- 27. Chen S., Bohrer L. R., Rai A. N., Pan Y., Gan L., Zhou X., Bagchi A., Simon J. A., Huang H. (2010) Nat. Cell Biol. 12, 1108–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaneko S., Li G., Son J., Xu C. F., Margueron R., Neubert T. A., Reinberg D. (2010) Genes Dev. 24, 2615–2620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wei Y., Chen Y. H., Li L. Y., Lang J., Yeh S. P., Shi B., Yang C. C., Yang J. Y., Lin C. Y., Lai C. C., Hung M. C. (2011) Nat. Cell Biol. 13, 87–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Malumbres M., Barbacid M. (2009) Nat. Rev. Cancer 9, 153–166 [DOI] [PubMed] [Google Scholar]
- 31. Van Hoof D., Muñoz J., Braam S. R., Pinkse M. W., Linding R., Heck A. J., Mummery C. L., Krijgsveld J. (2009) Cell Stem Cell 5, 214–226 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.