

The C-terminal domain of a bacteriophage P2 tail-spike protein, gpV, was crystallized and its structure was solved at 1.27 Å resolution. The refined model showed a triple β-helix structure and the presence of iron, calcium and chloride ions.
Keywords: bacteriophage P2, tail-spike proteins, gpV
Abstract
The adsorption and infection of bacteriophage P2 is mediated by tail fibres and tail spikes. The tail spikes on the tail baseplate are used to irreversibly adsorb to the host cells. Recently, a P2 phage tail-spike protein, gpV, was purified and it was shown that a C-terminal domain, Ser87–Leu211, is sufficient for the binding of gpV to host Escherichia coli membranes [Kageyama et al. (2009 ▶), Biochemistry, 48, 10129–10135]. In this paper, the crystal structure of the C-terminal domain of P2 gpV is reported. The structure is a triangular pyramid and looks like a spearhead composed of an intertwined β-sheet, a triple β-helix and a metal-binding region containing iron, calcium and chloride ions.
1. Introduction
Bacteriophage P2 has a 60 nm diameter icosahedral capsid with a double-stranded DNA genome, a 135 nm long contractile tail with a baseplate to the distal side of the head, and tail fibres (Dokland et al., 1992 ▶; Fig. 1 ▶). The baseplate and tail fibres are involved in binding of the phage to the bacterial cell. In the infection processes of some myoviruses the baseplate is able to undergo large conformational changes. These conformational changes are not only involved in host adsorption but also trigger tail contraction, leading to injection of the DNA into the host. P2-like phages are a widespread group of related temperate myoviruses that grow on γ-proteobacteria (Nilsson & Haggård-Ljungquist, 2007 ▶). Since P2-like prophages are frequently found in several bacterial genomes, it seems that the P2-like phages track horizontal gene transfer and the evolution of their hosts (Kita et al., 2003 ▶). These observations indicate that P2-like phages have a broad range of host selectivity and a flexible host-binding ability to adsorb to several host membranes. The phage receptor varies for different phages; examples include lipopolysaccharide, membrane-surface proteins and bacterial pili. It is possible to speculate that the P2 phage recognizes conserved chemical or structural features of receptor molecules to adsorb to various host bacteria, although P2 receptor molecules have not yet been identified. Tail fibres and tail spikes located beneath a baseplate contribute to host adsorption of the P2 phage. Because of its morphological similarity to those of other myoviruses, we presume that the P2 baseplate has six tail fibres and six tail spikes. It is believed that the first step of adsorption for a myovirus is reversibly introduced by its tail fibres and that the host selectivity of the phages is determined by their tail-fibre specificity (Riede et al., 1987 ▶). In the second binding step, the tail-spike proteins are used to reach the host cell membranes. Because the binding of the spike proteins is irreversible, the tail spikes are as important as the tail fibres in the infection process. The P2 tail spikes consist of a subunit that is a product of gene V (gpV; Haggård-Ljungquist et al., 1995 ▶). We have recently purified the recombinant P2 tail-spike protein gpV and shown that it irreversibly binds to the Escherichia coli membrane by using a quartz crystal microbalance technique (Kageyama et al., 2009 ▶). It forms a trimer and is attached to the baseplate by its protease-sensitive N-terminal domain, whereas a C-terminal domain is sufficient for binding to E. coli membranes. These results are related to the fact that the C-terminal region 142–207 of gpV has significant homology (45.5%) to a Mu tail-spike protein. It has been observed that bacterial strains resistant to P2 are usually also resistant to Mu and P1 but not to most other phages (Kutter, 2005 ▶). We therefore consider that this homologous region may be involved in host binding and recognition. Another interesting finding is that the N- and C-terminal domains show very different thermostabilities. Our results indicate that the T m of the N-terminal domain is approximately 323 K and that the C-terminal domain is stable with a T m of 345 K. Recombinant gpV-C (the C-terminal domain of gpV; Ser87–Leu211) shows resistance to denaturation by sodium dodecyl sulfate (SDS) and remains a trimer in SDS–PAGE treatments (Kageyama et al., 2009 ▶). We surmise that the N-terminal domain of gpV is flexible and less stable in order to allow induced fitting or a conformational change during baseplate assembly, whereas the C-terminal domain is more stable in order to retain adsorption to the cell. To investigate the structural features of the C-terminal domain of gpV, we determined the three-dimensional structure of gpV-C. The structure indicated that a triple β-helix structure and bound ions may induce the extraordinary stability of gpV-C.
Figure 1.

Schematic illustration of bacteriophage P2 based on electron-microscopic observations. The P2 tail spike was found at the side face of the baseplate using immuno-electron microscopy (Haggård-Ljungquist et al., 1995 ▶). The scale bar represents 100 nm. (a) Head, (b) tail, (c) contractile sheath, (d) baseplate, (e) tail fibre. The tail spikes are depicted as black triangles.
2. Experimental procedures
2.1. Purification and crystallization of gpV-C
The expression and purification of gpV-C were performed using a previously described method (Kageyama et al., 2009 ▶). We used a pET-15-derived vector containing a DNA fragment corresponding to histidine-tagged Ser87–Leu211 of P2 phage gpV for expression (Haggård-Ljungquist et al., 1995 ▶). Transformed E. coli BL21 (DE3) pLysS cells were grown at 310 K in Luria broth containing 100 µg ml−1 ampicillin. Expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) when the absorbance at 600 nm reached 0.8. After suspension in 10 mM Tris–HCl pH 8.0 and 1 mM EDTA and sonication of the harvested cells in the presence of 1 mM phenylmethanesulfonyl fluoride (PMSF), the supernatant was recovered by centrifugation (6000 rev min−1, 10 min) and dialyzed against 20 mM phosphate pH 7.4 and 500 mM NaCl. The gpV-C fraction was then loaded onto a nickel-affinity column (5 ml, Pharmacia) equilibrated with dialysis buffer. After the column had been washed with 20 mM phosphate pH 7.4 and 50 mM imidazole, gpV was eluted with 20 mM phosphate pH 7.4 and 500 mM imidazole. The fraction was further purified on a Sephacryl S-300 column (Bio-Rad) equilibrated with 50 mM Tris–HCl pH 7.4 and 500 mM arginine. Purified gpV-C was dialyzed into the appropriate buffer and stored at 193 K.
2.2. Crystallographic data collection and processing
Purified gpV-C at a concentration of 3 mg ml−1 in 100 mM HEPES pH 7.0 was used for crystallization. Trials at 293 K with the sitting-drop vapour-diffusion method using 20 µl protein solution and 20 µl precipitant solution equilibrated against 1 ml reservoir solution in 24-well plates were carried out using Crystal Screen and Crystal Screen 2 (Hampton Research, USA). Finally, crystals of gpV-C were grown in 100 mM HEPES pH 6.5–7.0 and 20% polyethylene glycol 20 000 and were cryoprotected by soaking them in reservoir solution supplemented with 25% polyethylene glycol 20 000 prior to flash-cryocooling. The derivative crystal for phase determination was soaked in cryoprotective solution containing 5 mM PtCl6 for 6 h.
Diffraction data sets were collected at 100 K using a Rayonix MX-225HE CCD detector supported by NSSRC on the BL44XU beamline at SPring-8 (Harima, Japan). Multi-wavelength anomalous diffraction (MAD) data using Pt-derivative crystals were obtained at three wavelengths: 1.0717 Å (peak), 1.0721 Å (edge) and 1.0539 Å (remote). The data set for structure refinement was collected from a native crystal (Native1) at a wavelength of 0.9000 Å. Two anomalous data sets to identify the Fe atom were collected from a native crystal (Native2) at two wavelengths: 1.7400 and 1.7500 Å. The diffraction data were processed by HKL-2000 (Otwinowski & Minor, 1997 ▶). The crystallographic data are shown in Table 1 ▶.
Table 1. Intensity data, refinement statistics and peak heights in anomalous difference maps.
Values in parentheses are for the highest resolution shell.
| Crystal | Native1 | Pt-MAD | Native2 | |||
|---|---|---|---|---|---|---|
| Data set | Refined | Remote | Edge | Peak | 1.74 Å | 1.75 Å |
| Unit-cell parameters | ||||||
| a (Å) | 66.9 | 66.8 | 67.1 | |||
| b (Å) | 64.5 | 64.6 | 64.5 | |||
| c (Å) | 67.2 | 67.0 | 67.6 | |||
| β (°) | 91.3 | 91.1 | 91.7 | |||
| Wavelength (Å) | 0.9000 | 1.0539 | 1.0721 | 1.0717 | 1.7400 | 1.7500 |
| Resolution (Å) | 1.27 (1.29–1.27) | 1.85 (1.88–1.85) | 1.85 (1.88–1.85) | 1.68 (1.71–1.68) | 1.98 (2.01–1.98) | 2.00 (2.03–2.00) |
| No. of reflections | ||||||
| Observed | 1097561 | 358451 | 357457 | 515207 | 849469 | 831369 |
| Unique | 148533 (6225) | 48729 (2417) | 48752 (2399) | 64556 (3043) | 40194 (1828) | 39001 (1953) |
| Multiplicity† | 7.4 (5.8) | 7.4 (6.4) | 7.3 (6.3) | 8.0 (6.1) | 21.1 (15.6) | 21.3 (17.5) |
| Completeness‡ (%) | 99.0 (82.9) | 99.9 (99.8) | 99.9 (99.1) | 99.5 (93.3) | 99.5 (91.4) | 100.0 (99.6) |
| 〈I/σ(I)〉 | 29.2 (3.5) | 21.8 (4.1) | 21.2 (3.7) | 21.7 (3.1) | 34.8 (5.6) | 36.7 (7.1) |
| Rmerge§ | 0.069 (0.350) | 0.114 (0.372) | 0.113 (0.399) | 0.110 (0.354) | 0.109 (0.354) | 0.101 (0.338) |
| Phasing | ||||||
| Resolution (Å) | 2.50 | 2.50 | 2.50 | |||
| Phasing power (isomorphous)¶ | 1.32 | 0.31 | ||||
| Phasing power (anomalous)†† | 1.24 | 1.29 | 1.16 | |||
| No. of sites | 6 | |||||
| FOM (acentric/centric)‡‡ | 0.52/0.38 | |||||
| Refinement | ||||||
| Resolution (Å) | 1.27 | |||||
| R/Rfree§§ | 0.160/0.191 | |||||
| R.m.s. deviation from ideal | ||||||
| Bond lengths (Å) | 0.03 | |||||
| Bond angles (°) | 2.54 | |||||
| Average B factor (Å2) | 13.1 | |||||
| Ramachandran plot (%) | ||||||
| Favoured | 89.5 | |||||
| Allowed | 9.2 | |||||
| Generously allowed | 1.2 | |||||
| Disallowed | 0.0 | |||||
| Ratio of peak heights (Fe/Cys82)§§ | 12.0 | 0.9 | ||||
Multiplicity is the number of observed reflections for each independent reflection.
Completeness is the percentage of independent reflections observed.
R
merge = 
 , where I
i(hkl) is the intensity value of the ith measurement of hkl and 〈I(hkl)〉 is the corresponding mean value of I(hkl) for all I measurements; the summation is over reflections with I/σ(I) larger than −3.0.
, where I
i(hkl) is the intensity value of the ith measurement of hkl and 〈I(hkl)〉 is the corresponding mean value of I(hkl) for all I measurements; the summation is over reflections with I/σ(I) larger than −3.0.
Isomorphous phasing power is r.m.s. isomorphous difference divided by r.m.s. isomorphous residual lack of closure; anomalous phasing power is r.m.s. anomalous difference divided by r.m.s. anomalous residual lack of closure.
FOM is the mean figure of merit.
R is a conventional crystallographic R factor, 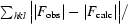
 , where F
obs and F
calc are the observed and calculated structure factors, respectively. R
free is a free R factor of the refinement evaluated for 5% of reflections that were excluded from refinement.
, where F
obs and F
calc are the observed and calculated structure factors, respectively. R
free is a free R factor of the refinement evaluated for 5% of reflections that were excluded from refinement.
The ratio of peak heights is the ratio of the Fe peak height to the S peak height of Cys82 in the anomalous difference maps at wavelengths of 1.74 and 1.75 Å.
2.3. Structure solution and refinement
Six platinum-binding sites were located using SHELX (Sheldrick, 2008 ▶) and the initial phases were calculated to 2.5 Å resolution from the MAD data using SHARP (de La Fortelle & Bricogne, 1997 ▶). Density modification and phase extension to 1.68 Å resolution were carried out with SOLOMON (Abrahams & Leslie, 1996 ▶). The initial structure was built using ARP/wARP (Langer et al., 2008 ▶). Structure refinement and rebuilding at 1.27 Å resolution were performed using REFMAC5 (Murshudov et al., 1997 ▶) and Coot (Emsley et al., 2010 ▶). The phase and refinement statistics are shown in Table 1 ▶.
2.4. Metal-ion analysis
For quantitative analysis of metals, inductively coupled plasma mass spectrometry (ICP-MS) was used. The solution containing gpV-C was dialyzed into pure water and introduced into the ICP-MS instrument (Agilent 7700s, Agilent Technologies) via a PFA nebulizer. The dialysis solution was used as a blank sample.
3. Results and discussion
In order to understand the host membrane-binding activity and thermostability of gpV-C, we investigated the structural features of gpV-C.
3.1. Overall structure of gpV-C
The protein crystallized in space group P21. The unit-cell parameters of the crystals were a = 71.0, b = 67.4, c = 72.1 Å, α = 90, β = 99.8, γ = 90°. The asymmetric unit of the crystal contained two trimers. The gpV-C trimer formed a triangular pyramid or spearhead shape. We did not find significant electron density for the histidine tag or Ser87–Ser94. A view of gpV-C is shown in Fig. 2 ▶. It was roughly divided into three regions in the structure: an intertwined β-sheet (Val95–Ile124), a triple β-helix (Lys125–Gly184) and an ion-binding region (Gly185–Leu211). Fig. 3 ▶ shows electrostatic surface potentials for a whole gpV-C trimer calculated using MolFeat v.2.6 (FiatLux, Japan).
Figure 2.

(a) Side view of the gpV-C trimer. One monomer subunit is illustrated in green in order to make it easy to trace the folding. (i) Intertwined β-sheet (Val95–Ile124), (ii) triple β-helix (Lys125–Gly184), (iii) ion-binding region (Gly185–Leu211). (b) Cα stereoscopic drawing of the gpV-C trimer.
Figure 3.

Electrostatic surface potentials calculated for the gpV-C trimer using MolFeat v.2.6 (FiatLux, Japan). The surface is coloured according to electrostatic potential, with red and blue corresponding to negative and positive, respectively.
3.2. Intertwined β-sheet
Three strands form an antiparallel β-sheet from Val95 to Ile124. The three gpV-C chains in this region were assembled together and formed an intertwined prism shape (Fig. 4 ▶). Its interior was very hydrophobic and the side chains of Leu100, Ile102, Phe104, Ile110, Tyr112, Leu119 and Val121 formed a hydrophobic cluster. In contrast, we found hydrophilic side chains on an external face and a negatively charged cluster composed of Asp98, Glu111 and Glu113.
Figure 4.

View from above of the intertwined β-sheet region [Val95–Ile124; (i) in Fig. 2 ▶ a]. One monomer subunit is illustrated in blue. The colour scheme white for carbon, red for nitrogen and blue for oxygen is used to illustrate the other subunit.
3.3. Triple β-helix structure
A triple β-helix structure was found from Lys125 to Gly184. In a triple β-helix three polypeptide chains wind around a threefold symmetry axis, conferring extraordinary stability. The interior was occupied by hydrophobic side chains (Fig. 5 ▶ a). Although each prism face was made up of hexapeptide units, the similarity of the hexapeptide sequence repeats was not so clear (Fig. 5 ▶ b). This triple β-helix has been identified in various virus proteins and has been found to be responsible for host adsorption (Mitraki et al., 2002 ▶). For example, the triple β-helix of gpV-C is very similar to part of a T4 phage short tail fibre, gp12 (Gly298–Ile334; Thomassen et al., 2003 ▶). In contrast, in T4 phage gp5, which is another T4 baseplate subunit, the internal sequence repeat and triple β-helix are composed of octapeptide units (Kanamaru et al., 2002 ▶). The prism faces of gp5 are therefore two residues wider than those of gpV-C.
Figure 5.

(a) View from above of the triple β-helix region [Lys125–Gly184; (ii) in Fig. 2 ▶ a]. The colour scheme white for carbon, red for nitrogen and blue for oxygen is used. (b) Hexapeptide sequence repeats that make up the triple β-helix. Hydrophobic residues that face the interior are labelled in bold and underlined.
The triple β-helix structures of virus tail spikes are often mediated by specific chaperones. The T4 phage short tail fibre and long tail fibre require a specific molecular chaperone, gp57A, that is encoded in the T4 phage genome itself (Herrmann & Wood, 1981 ▶). It has also been reported that several tail-spike proteins require a C-terminal chaperone domain for correct folding. These intramolecular chaperone domains are released by themselves in an autoproteolytic reaction after protein folding (Schulz et al., 2010 ▶). In contrast, gpV and gpV-C do not require a specific chaperone encoded in the P2 phage itself for folding and do not have a C-terminal chaperone domain.
3.4. Ion-binding region
The anomalous difference Fourier map at a wavelength of 0.9 Å unexpectedly revealed anomalous difference electron density in the most C-terminal region, from Gly185 to Leu211, of the gpV-C trimer. The anomalous difference electron density at the site had the highest peak at a wavelength of 1.74 Å and a lower peak than that at the sulfur site of Cys82 at a wavelength of 1.75 Å (Table 1 ▶). Since the absorption edge between 1.74 and 1.75 Å can only be an Fe atom, the anomalous density changes identified an Fe atom. The iron was coordinated by six histidine residues (His197 and His199 from each monomer in a trimer) in an octahedral conformation (Fig. 5 ▶). We also found another two ions at the point of gpV-C. To identify these ions, we performed analytical ICP-MS measurements. The results indicated that gpV-C contains Na, K, Ca, Fe, Ni and Zn ions. From the anomalous difference electron density at 1.75 Å we could eliminate the possibility that these ions were nickel or zinc. We supposed that the nickel and zinc ions identified by ICP-MS would bind to the histidine-tag sequence, which did not show any electron density in our X-ray crystallographic study. Finally, we concluded from the ligands of each ion that these ions would be calcium and chloride ions. The side chains of Asp202 and Ser203 were found to contribute to the calcium binding and the main-chain amide of Asp202 coordinated to the chloride ion (Fig. 6 ▶).
Figure 6.

(a) Side view of the gpV-C monomer polypeptide and ions. (b) Close-up view of an iron ion and calcium ions with coordinated residues in the ion-binding region. His197 and His199 contribute to the binding of an iron ion (red) in an octahedral conformation and Asp202 and Ser203 are bound to a calcium ion (grey) and a chloride ion (yellow), respectively.
3.5. The relationship between the properties of gpV-C and its structure
One of the features of gpV-C is its extraordinary stability, with a T m of 345 K (Kageyama et al., 2009 ▶). This thermostability may be caused by the ion coordination and extensive hydrophobic cluster formed by the intertwined β-sheet and triple β-helix. Another property of gpV-C is its broad range of ability to bind several host cell membranes since the P2 phage adheres to several bacterial species as hosts. Unfortunately, we did not find any ligand-binding pockets or clefts in the gpV-C structure. This finding might be related to the lower selectivity of gpV-C binding activity. Identifying the host receptor molecule will be the next step in understanding the P2 phage infection process.
Supplementary Material
PDB reference: C-terminal domain of P2 gpV, 3aqj
Acknowledgments
The authors thank Professor Haggård-Ljungquist (University of Stockholm) for the kind gift of the P2 phage gene V. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan (to ST).
References
- Abrahams, J. P. & Leslie, A. G. W. (1996). Acta Cryst. D52, 30–42. [DOI] [PubMed]
- Dokland, T., Lindqvist, B. H. & Fuller, S. D. (1992). EMBO J. 11, 839–846. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Haggård-Ljungquist, E., Jacobsen, E., Rishovd, S., Six, E. W., Nilssen, O., Sunshine, M. G., Lindqvist, B. H., Kim, K. J., Barreiro, V. & Koonin, E. V. (1995). Virology, 213, 109–121. [DOI] [PubMed]
- Herrmann, R. & Wood, W. B. (1981). Mol. Gen. Genet. 184, 125–132. [DOI] [PubMed]
- Kageyama, Y., Murayama, M., Onodera, T., Yamada, S., Fukada, H., Kudou, M., Tsumoto, K., Toyama, Y., Kado, S., Kubota, K. & Takeda, S. (2009). Biochemistry, 48, 10129–10135. [DOI] [PubMed]
- Kanamaru, S., Leiman, P. G., Kostyuchenko, V. A., Chipman, P. R., Mesyanzhinov, V. V., Arisaka, F. & Rossmann, M. G. (2002). Nature (London), 415, 553–557. [DOI] [PubMed]
- Kita, K., Kawakami, H. & Tanaka, H. (2003). J. Bacteriol. 185, 2296–2305. [DOI] [PMC free article] [PubMed]
- Kutter, E. (2005). Bacteriophages: Biology and Applications, edited by E. Kutter & A. Sulakvelidze, pp. 165–222. Boca Raton: CRC Press.
- La Fortelle, E. de & Bricogne, G. (1997). Methods Enzymol. 276, 472–494. [DOI] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Mitraki, A., Miller, S. & van Raaij, M. J. (2002). J. Struct. Biol. 137, 236–247. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nilsson, A. S. & Haggård-Ljungquist, E. (2007). Res. Microbiol. 158, 311–317. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Riede, I., Drexler, K., Schwarz, H. & Henning, U. (1987). J. Mol. Biol. 194, 23–30. [DOI] [PubMed]
- Schulz, E. C., Dickmanns, A., Urlaub, H., Schmitt, A., Mühlenhoff, M., Stummeyer, K., Schwarzer, D., Gerardy-Schahn, R. & Ficner, R. (2010). Nature Struct. Mol. Biol. 17, 210–215. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Thomassen, E., Gielen, G., Schütz, M., Schoehn, G., Abrahams, J. P., Miller, S. & van Raaij, M. J. (2003). J. Mol. Biol. 331, 361–373. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: C-terminal domain of P2 gpV, 3aqj