

The structure of immunoglobulin-like repeat 10 from human filamin A solved at 2.44 Å resolution suggests the potential effects of mutations correlated with otopalatodigital syndrome spectrum disorders.
Keywords: filamin, immunoglobulin-like domains, Melnick–Needles syndrome, frontometaphyseal dysplasia
Abstract
Filamin A (FlnA) plays a critical role in cytoskeletal organization, cell motility and cellular signaling. FlnA utilizes different binding sites on a series of 24 immunoglobulin-like domains (Ig repeats) to interact with diverse cytosolic proteins and with cytoplasmic portions of membrane proteins. Mutations in a specific domain, Ig10 (FlnA-Ig10), are correlated with two severe forms of the otopalatodigital syndrome spectrum disorders Melnick–Needles syndrome and frontometaphyseal dysplasia. The crystal structure of FlnA-Ig10 determined at 2.44 Å resolution provides insight into the perturbations caused by these mutations.
1. Introduction
Human filamins (Flns) are multifunctional cytoskeletal proteins that are involved in cell motility, cytoskeletal regulation and cellular signaling (Popowicz et al., 2006 ▶; Zhou et al., 2010 ▶). There are three known filamin isoforms: FlnA, FlnB and FlnC. FlnA and FlnB are expressed in many tissues and cell types, while FlnC expression is primarily restricted to muscle and cardiac tissues (van der Flier & Sonnenberg, 2001 ▶). Filamins are ∼280 kDa proteins comprised of an amino-terminal actin-binding domain and 24 immunoglobulin-like domains (Ig repeats). The 24 Ig repeats have been categorized into classes A, B, C and D (Ithychanda et al., 2009 ▶) based on sequence similarity. The FlnA-Ig10 structure presented here represents the first structure of a class D Ig repeat from FlnA to be deposited in the Protein Data Bank (PDB).
A major structural role of the filamins is to cross-link actin filaments, adding mechanical strength and flexibility to the actin cytoskeleton under load (Popowicz et al., 2006 ▶). Filamins, however, also play a direct role in linking the actin cytoskeleton to signaling and cell-surface proteins (Kim & McCulloch, 2011 ▶; Feng & Walsh, 2004 ▶). The physiological importance of the filamins is emphasized by the fact that missense mutations in FlnA and FlnB cause developmental malformations in bone, limbs and a variety of organs (Feng & Walsh, 2004 ▶). The effects of these missense mutations vary greatly and depend upon the Fln domain in which the mutation is located. Missense mutations in FlnA-Ig10 are correlated with the otopalatodigital syndrome spectrum disorders frontometaphyseal dysplasia (FMD) and Melnick–Needles syndrome (MNS). Both FMD and MNS are skeletal dysplasias found predominantly in females, as both disorders are typically embryonically or perinatally lethal to males (Robertson, 2007 ▶). MNS exhibits variable manifestations in diagnosed females ranging from mild skeletal dysplasias to significant skeletal abnormalities leading to thoracic restriction (Robertson, 2004 ▶). FMD in females is characterized by substantial skeletal dysplasia, which can include supraorbital hyperostosis, vertebral fusions and phalangeal contractures (Robertson et al., 2006 ▶). All known missense mutations correlated with MNS are located in FlnA-Ig10, while two of the three known missense mutations correlated with lethal FMD are located in FlnA-Ig10 (Robertson et al., 2006 ▶).
Here, we present the 2.44 Å resolution crystal structure of FlnA-Ig10. The structure exhibits a canonical immunoglobulin-like domain fold resembling the structures of other FlnA Ig repeats. The structure features a disulfide-linked crystal lattice, a β-mercaptoethanol thio-adduct on a single surface-exposed cysteine and an acetate ion coordinated by both side-chain-mediated and backbone-mediated hydrogen bonds. The FlnA-Ig10 crystal structure allows analysis of the potential structural consequences of missense mutations correlated with otopalatodigital syndrome spectrum disorders.
2. Materials and methods
2.1. Protein expression and purification
A pGST-parallel-1 expression vector (Sheffield et al., 1999 ▶) for the expression of GST-HsFlnA-Ig10 (residues 1158–1252) fusion protein was generously provided by Dr Sujay Ithychanda (Ithychanda et al., 2009 ▶). The vector was transformed into Escherichia coli Rosetta 2 (DE3) cells (EMD Biosciences) and the GST-HsFlnA-Ig10 fusion protein was expressed and purified as described previously (Smith et al., 2010 ▶). Purified GST-HsFlnA-Ig10 fusion protein was incubated with tobacco etch virus (TEV) protease (Kapust et al., 2002 ▶) supplemented with 5 mM β-mercaptoethanol and 1 mM EDTA. Cleaved FlnA-Ig10 was purified from the TEV cleavage reaction by size-exclusion chromatography using a Superdex 75 gel-filtration column (GE Healthcare) equilibrated with 25 mM HEPES pH 7.0, 50 mM NaCl. The purity of the Ig10 samples was confirmed by SDS–PAGE and concentrations were determined using the Pierce 660 nm protein assay (Thermo Scientific).
2.2. Crystallization and X-ray data collection
Crystals of FlnA-Ig10 were grown by hanging-drop vapor diffusion at 293 K in 2 µl drops. The hanging drops consisted of a 1:1 ratio mixture of 1 mM (10 mg ml−1) protein solution and reservoir solution. Initial screening was conducted using the sparse-matrix crystallization screens JCSG+ and ProComplex from Qiagen and Classics, Index and SaltRx from Hampton Research. Subsequent optimizations identified an optimal reservoir solution composed of 0.2 M ammonium acetate, 25%(w/v) polyethylene glycol (PEG) 3350 and 0.1 M Bis-Tris pH 5.5. Single crystals appeared after six weeks and grew for an additional two weeks. The crystals were cryoprotected by a brief transfer to reservoir solution supplemented with 20%(w/v) glycerol and flash-frozen in liquid nitrogen. Diffraction data were collected with a Cu Kα source at 1.5418 Å using a Rigaku MicroMax-007 HF generator and a Rigaku Saturn 944+ CCD detector. Data reduction was performed with d*TREK (Pflugrath, 1999 ▶). The final data set was processed to a cutoff of 2.44 Å based on significant drops in unaveraged I/σ(I) (<2.7) and completeness (<90%) in shells at higher resolutions. Data-collection and refinement statistics are given in Table 1 ▶.
Table 1. Data-collection, refinement and validation statistics for FlnA-Ig10.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| X-ray source | Rigaku MicroMax-007 HF |
| Wavelength | 1.5418 |
| Space group | P212121 |
| Unit-cell parameters (, ) | a = 36.45, b = 50.55, c = 107.17, = = = 90 |
| Resolution () | 34.512.44 (2.532.44) |
| R merge † | 0.070 (0.173) |
| R r.i.m. ‡ | 0.077 (0.194) |
| I/(I) | 13.9 (2.7) |
| Completeness (%) | 98.4 (90.7) |
| Multiplicity | 5.6 (4.4) |
| Wilson B factor (2) | 56.6 |
| No. of reflections | 42608 |
| No. of unique reflections | 7677 |
| Refinement | |
| Resolution () | 30.142.44 |
| No. of reflections for refinement | 7577 |
| R work/R free | 0.229/0.279 |
| No. of atoms | |
| Protein | 1429 |
| Water | 77 |
| Ligands | 8 |
| Average B factors (2) | |
| Protein | 38.0 |
| Water | 49.1 |
| Ligands | 42.3 |
| R.m.s. deviations | |
| Bond lengths () | 0.010 |
| Bond angles () | 1.177 |
| Ramachandran plot statistics (%) | |
| Favored regions | 100.0 |
| Allowed regions | 100.0 |
| Disallowed regions | 0.0 |
| MolProbity validation statistics | |
| Poor rotamers (%) | 0.0 |
| C deviations > 0.25 | 0 |
| MolProbity clash score | 7.09 |
| MolProbity clash percentile | 98th percentile [N = 326, 2.442 0.25] |
| MolProbity score | 1.44 |
| MolProbity score percentile | 100th percentile [N = 7752, 2.442 0.25] |
| PDB code | 3rgh |
The merging R factor is defined as R
merge = 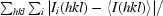
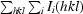 .
.
The redundancy-independent merging R factor R
r.i.m. (Weiss, 2001 ▶) is defined as 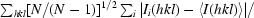 .
.
2.3. Structure solution and refinement
A molecular-replacement search model was generated by iTASSER (Roy et al., 2010 ▶) using the HsFlnA-Ig10 sequence (residues 1158–1252) as input. Phases were calculated by molecular replacement using the Phaser (McCoy et al., 2007 ▶) component of PHENIX (Adams et al., 2010 ▶). The best molecular-replacement solution was subjected to automated rebuilding in PHENIX with RESOLVE (Terwilliger, 2003 ▶), followed by iterative rounds of model building in Coot (Emsley et al., 2010 ▶) and refinement in PHENIX. The refinement protocol used isotropic atomic displacement parameters for all atoms and TLS motion analysis for protein chains (Painter & Merritt, 2006 ▶). All molecular-structure figures were generated with PyMOL (DeLano, 2002 ▶).
2.4. Validation and deposition
Stereochemical analysis of the FlnA-Ig10 structure was completed with MolProbity (Chen et al., 2010 ▶). All residues were within the favored regions of the Ramachandran plot. No poor rotamers or Cβ deviations were found. MolProbity analysis of all atom contacts calculated a clash score of 7.09, which ranks the FlnA-Ig10 structure in the 98th percentile of 326 structures deposited in the PDB that were solved at similar resolution (2.442 ± 0.25 Å). The MolProbity score, a weighted measure of stereochemical statistics, was 1.44, which ranks the FlnA-Ig10 structure in the 100th percentile of 7752 structures of similar resolution deposited in the PDB. The atomic coordinates for Ig10 have been deposited in the PDB (accession code 3rgh).
3. Results
3.1. Structural features
Human FlnA-Ig10 crystallized in the orthorhombic space group P212121 with two monomers per asymmetric unit. The r.m.s.d. between monomers, calculated over 713 equivalent atom pairs, was 0.125 Å. The structure, solved at 2.44 Å resolution, exhibits a characteristic immunoglobulin-like domain fold (Bork et al., 1994 ▶). This fold is characterized by seven β-strands that form two antiparallel β-sheets with Greek-key topology (Fig. 1 ▶ a). The FlnA-Ig10 crystal lattice is stabilized by disulfide bonds between symmetry-related copies (Fig. 1 ▶ b). Cys1225 of each chain forms a disulfide bond with Cys1225 from the opposite chain of the symmetry mate generated by x + 1/2, −y + 1/2, −z.
Figure 1.

Crystal structure of human FlnA-Ig10. (a) Ribbon diagram of FlnA-Ig10 colored as a rainbow from the N-terminus (blue) to the C-terminus (red). β-Strands A–G and loops are labeled. (b) A disulfide bond (yellow) between two symmetry-related copies of FlnA-Ig10: chain A from the asymmetric unit (magenta) and chain B of a symmetry mate (x + 1/2, −y + 1/2, −z) (cyan). (c) FlnA-Ig10 (magenta) coordinates an acetate ion using a water-mediated hydrogen-bond network utilizing the Tyr1229 hydroxyl, the Leu1224 backbone amide and a water molecule (red sphere) hydrogen bonded to the Ile1222 backbone carbonyl. (d) Cys1198 is covalently modified by a β-mercaptoethanol adduct. The β-mercaptoethanol-modified Cys1198 lies on the solvent-exposed surface of β-sheet C. 2F o − F c density (dark blue) is contoured at 1.2σ in (b)–(d).
The disulfide-linked dimer is identified by PISA (Krissinel & Henrick, 2007 ▶) as a biological multimer. However, size-exclusion chromatography during FlnA-Ig10 purification indicated that FlnA-Ig10 is monomeric in solution (data not shown). Furthermore, electron-microscopy studies on full-length filamins have identified only a single FlnA dimerization interface, which is located in the so-called dimerization domain, FlnA Ig repeat 24 (Nakamura et al., 2007 ▶). Additionally, FlnA-Ig24 dimerization occurs through β-sheet extension utilizing β-strands C and D (Seo et al., 2009 ▶; Pinotsis et al., 2009 ▶; Pudas et al., 2005 ▶; Sjekloća et al., 2007 ▶). This dimerization mode is significantly different from the disulfide-linked dimerization observed in the FlnA-Ig10 crystal, which is predominantly mediated by interactions between the EF-loops of each monomer. FlnA-Ig24 dimerization buries 18% of solvent-accessible surface area (977 Å2) per monomer (Seo et al., 2009 ▶), while the disulfide-linked FlnA-Ig10 dimer buries only 6% of the solvent-accessible surface (355 Å2) per monomer. Taken together, the data suggest that the disulfide-linked FlnA-Ig10 dimer is a crystallization artifact and not a true reflection of in vivo oligomeric status.
Each chain in the asymmetric unit binds one acetate molecule (Fig. 1 ▶ c) between loops CD and EF. The acetate-ion carboxylate is tethered by hydrogen bonds to the hydroxyl group of Tyr1229 and the backbone amide of Leu1224 and a water-mediated hydrogen-bond network with the carbonyl of Ile1222 (Fig. 1 ▶ c). In addition, the methyl group of the acetate ion participates in hydrophobic packing against the Leu1203 side chain. The electrostatic and hydrophobic interactions between FlnA-Ig10 and the acetate ion appear to stabilize the relative positioning of loops CD and EF.
Cys1198 in each chain is modified by a β-mercaptoethanol (BME) adduct (Fig. 1 ▶ d). Covalent modification of surface-exposed cysteines by BME is not uncommon; in fact, a BME thio-adduct is found in at least 150 structures in the PDB. The modification of Cys1198 is likely to be the result of local structural features that increased the reactivity of the cysteine sulfhydryl group towards BME that was present during protein purification. The side chain of Cys1198 is fully solvent-exposed, exhibiting a solvent-exposed surface area of 11.9 Å2 as calculated by WHAT IF (Vriend, 1990 ▶) and allowing facile access of BME to the sulfhydryl. Additionally, the side-chain carboxylate of Glu1196 is positioned 4.9 Å from the Cys1198 sulfhydryl. The proximity of a carboxylate is known to increase the reactivity of sulfhydryl groups. For example, a glutamate proximal to the active-site cysteine of class 2 and class 3 aldehyde dehydrogenases is critical for catalysis (Mann & Weiner, 1999 ▶). No other FlnA-Ig10 cysteine residues were modified by BME. Only Cys1198 exhibits the combination of both high solvent exposure and proximity of a negatively charged carboxylate to facilitate thio-adduct formation.
3.2. Comparison with class A and class D filamin Ig repeats
Based on sequence similarity between different Ig repeats within a given filamin isoform as well as on functional properties, the filamin Ig repeats may be categorized into four distinct groups: classes A, B, C and D (Ithychanda et al., 2009 ▶). Ig10 is a member of the class D filamin Ig repeats, a class that also includes Ig6, Ig7, Ig13 and Ig14. The FlnA-Ig10 crystal structure represents the first structure of a class D Ig repeat from FlnA. NMR structures of Ig10, Ig13 and Ig14 from the FlnB isoform have previously been solved by the RIKEN Structural Genomics Consortium. The structurally characterized class D FlnB-Ig repeats average 44% sequence identity and 60% sequence similarity to FlnA-Ig10. The structure of FlnA-Ig10 is compared with the averaged NMR structures for FlnB Ig10, Ig13 and Ig14 in Fig. 2 ▶(a). Each FlnB-Ig domain structure exhibits the canonical immunoglobulin-like domain fold and these structures superimpose on FlnA-Ig10 with 1.1 Å r.m.s.d. for backbone atoms (r.m.s.d. values were calculated over an average of 303 equivalent atom pairs per alignment).
Figure 2.

Comparison of FlnA-Ig10 to existing structures of filamin Ig repeats. (a) Overlay of structures for the class D filamin Ig repeats FlnA-Ig10 (magenta), FlnB-Ig10 (cyan; PDB entry 2dia), FlnB-Ig13 (green; PDB entry 2dj4) and FlnB-Ig14 (blue; PDB entry 2e9j) (T. Tomizawa, S. Koshiba, S. Watanabe, T. Harada, T. Kigawa & S. Yokoyama, unpublished work). (b) Sequence alignment of the CD-loop region of class D filamin Ig repeats (FlnA-Ig10, FlnB-Ig10, FlnB-Ig13 and FlnA-Ig14) and class A filamin Ig repeats (FlnA-Ig17, FlnA-Ig19, FlnA-Ig21 and FlnA-Ig23). (c) Overlay of the FlnA-Ig10 (magenta) structure with class A filamin Ig repeats FlnA-Ig17 (light pink; PDB entry 2bp3; Nakamura et al., 2006 ▶), FlnA-Ig19 (light blue; PDB entry 2j3s; Lad et al., 2008 ▶), FlnA-Ig21 (light yellow; PDB entry 3isw; Smith et al., 2010 ▶) and FlnA-Ig23 (light green; PDB entry 2k3t; Nakamura et al., 2009 ▶).
FlnA class D Ig repeats differ from class A Ig repeats, which have been identified as ligand-binding domains (Ithychanda et al., 2009 ▶; Kiema et al., 2006 ▶; Lad et al., 2008 ▶). In the class A repeats, a binding groove formed by β-strands C and D binds to unstructured motifs from binding partners. Upon binding, these motifs adopt a β-strand conformation and engage in antiparallel β-strand backbone–backbone hydrogen bonding with strand C. In addition, a highly conserved serine residue in these motifs forms a key hydrogen bond to a backbone amide group near the N-terminus of strand D (Kiema et al., 2006 ▶; Lad et al., 2008 ▶; Nakamura et al., 2006 ▶; Takala et al., 2008 ▶). Previous studies from our laboratory and others have shown that FlnA-Ig10 does not interact with class A binding ligands (Ithychanda et al., 2009 ▶; Smith et al., 2010 ▶). Using homology models, we proposed that the conformation of the CD-loop precludes the formation of the critical intermolecular hydrogen bond necessary for binding class A ligands in the groove formed by β-strands C and D (Smith et al., 2010 ▶). Sequence alignments of class A and D Ig repeats highlight a two-residue insertion in the CD-loop of class D Ig repeats (Fig. 2 ▶ b). This insertion causes the CD-loop to adopt a substantially different conformation in class D Ig repeats than in class A Ig repeats (Fig. 2 ▶ c). In combination with previous biophysical studies (Ithychanda et al., 2009 ▶; Smith et al., 2010 ▶), this key structural difference rationalizes why class D Ig repeats cannot utilize the canonical ligand-binding paradigm that has been established for class A Ig repeats.
3.3. Implications for otopalatodigital syndrome spectrum disorders
Melnick–Needles syndrome (MNS) and frontometaphyseal dysplasia (FMD) are otopalatodigital syndrome spectrum disorders correlated with specific mutations in FlnA-Ig10 (Feng & Walsh, 2004 ▶; Robertson, 2004 ▶; Robertson et al., 2006 ▶). Previous studies did not determine whether these mutations disrupt the interaction of FlnA-Ig10 with unidentified putative binding partners or whether the mutations elicit significant changes in the structure or stability of FlnA-Ig10. Analysis of the FlnA-Ig10 crystal structure suggests the potential effects of mutations correlated with MNS and FMD.
Two specific mutations correlated with MNS, A1188T and S1199L (Robertson, 2004 ▶), would result in perturbations of loops BC and CD, respectively (Fig. 3 ▶). Ala1188 has a WHAT IF-calculated side-chain solvent accessibility of 0.0 Å2. This residue acts as an end cap for the FlnA-Ig10 hydrophobic core and mutation to threonine would disrupt hydrophobic packing in the region of the BC-loop (Fig. 3 ▶ a). Specifically, all backbone-dependent A1188T rotamers (Dunbrack & Cohen, 1997 ▶) clash with the side chains of Val1163, Cys1185 and Tyr1235. The S1199L mutation would cause significant perturbations to the CD-loop region. Despite a higher side-chain solvent exposure, 4.1 Å2, no backbone-dependent S1199L rotamers could be accommodated in the CD-loop. Each leucine rotamer would result in extensive clashes with the side chains of Tyr1229, Leu1203, Ala1201 and the backbone of Glu1200 and Pro1204 (Fig. 3 ▶ b).
Figure 3.

Locations of mutations correlated with MNS. (a) Ala1188 of the BC-loop is not solvent-accessible and abuts the FlnA-Ig10 hydrophobic core. Backbone-dependent threonine rotamers for the A1188T mutation result in clashes (red disks) with neighboring residues (gray sticks). (b) Ser1199 within the CD-loop displays low side-chain solvent accessibility and all backbone-dependent leucine rotamers for the S1199L mutation lead to steric clashes (red disks) with surrounding residues (gray sticks).
Two mutations in Ig10, P1223L and V1249A, are correlated with lethal FMD, a particularly severe form of FMD (Robertson, 2004 ▶; Robertson et al., 2006 ▶). Pro1223 and Val1249 are located at the carboxy-terminal end of the FlnA-Ig10 hydrophobic core (Fig. 4 ▶ a). Both Pro1223 and Val1249 are buried, with side-chain solvent exposures of 0.0 and 0.6 Å2, respectively. All backbone-dependent leucine rotamers for the P1223L mutation would cause widespread steric clashes with neighboring residues and would perturb the FlnA-Ig10 structure in the region of loops AB and EF (Fig. 4 ▶ a). In contrast, it is less clear why the V1249A mutation is correlated with severe forms of FMD. This mutation would not result in steric clashes; instead, it may cause a hydrophobic packing defect for the carboxy-terminal end of the FlnA-Ig10 hydrophobic core.
Figure 4.

Two distinct FlnA-Ig10 regions harbor mutations correlated with FMD. (a) All preferred leucine rotamers for mutation P1223L (orange) result in significant steric clashes (red discs) with surrounding residues (gray sticks). Although mutation V1249A (purple) would not result in steric clashes, it may lead to a hydrophobic packing defect. (b) Mutations D1159A (green) and S1186L (blue) occur in surface-exposed residues. Each mutation can be accommodated by preferred rotamers without significant steric clashes (red disks); however, each mutation would preclude the formation of side-chain-mediated hydrogen bonds (black dashes).
Two additional FlnA-Ig10 mutations, D1159A and S1186L, are correlated with less severe forms of FMD (Robertson et al., 2006 ▶). In contrast to the P1223L and V1249A mutations, which cause disruptions in the hydrophobic core near the FlnA-Ig10 carboxy-terminus, the D1159A and S1186L mutations are positioned near the amino-terminus and involve residues with significant side-chain solvent exposure: 10.5 and 16.2 Å2, respectively. The D1159A mutation does not cause steric clashes and five of the nine backbone-dependent S1186L leucine rotamers are accommodated without any steric clashes (Fig. 4 ▶ b). However, while the D1159A and S1186L mutations are sterically allowed, both mutations would eliminate side-chain-mediated hydrogen bonds that appear to stabilize the conformation of the amino-terminus and BC-loop in wild-type FlnA-Ig10 (Fig. 4 ▶ b).
4. Discussion
The 2.44 Å resolution structure of FlnA-Ig10 reveals an immunoglobulin-like domain with a unique CD-loop conformation that differentiates FlnA-Ig10 from the canonical class A ligand-binding FlnA Ig repeats Ig17, Ig19, Ig21 and Ig23. This conformation precludes FlnA-Ig10 from engaging ligands in a manner similar to that characterized for class A Ig repeats. It is unclear whether FlnA-Ig10 binds ligands in an alternate manner. It has been suggested that FlnA-Ig10 mediates the interaction between FlnA and the T-cell receptor CD4 (Jiménez-Baranda et al., 2007 ▶). While this receptor does not appear to contain a class A-interacting motif, the mode of its putative interaction with FlnA-Ig10 is not defined and awaits further characterization.
The FlnA-Ig10 crystal lattice is stabilized by a disulfide bond between symmetry-related copies. However, the disulfide-linked dimer appears to be a crystal-packing artifact and not a genuine biological dimer. The β-mercaptoethanol thio-adduct on Cys1198 is a consequence of both purification strategy and local structure. The high solvent accessibility and potential activation by a neighboring glutamate side chain allowed efficient thio-adduct formation on Cys1198, while no thio-adducts were observed on either Cys1165, Cys1185 or Cys1225. Comparison of the Ig10 crystal structure with the structures of class D Ig repeats from FlnB suggests that neither the disulfide linkage between symmetry mates nor the β-mercaptoethanol thio-adduct resulted in significant perturbations of the FlnA-Ig10 structure.
Finally, the structure of FlnA-Ig10 suggests the potential structural effects of mutations correlated with severe forms of the otopalatodigital syndrome spectrum disorders MNS and FMD. Several of these mutations may alter hydrophobic packing in Ig10, possibly reducing the stability or impairing the translation of FlnA as a whole. Biophysical measurements of the stability of these FlnA-Ig10 mutants using differential thermal fluorimetry or differential scanning calorimetry should more conclusively determine whether these mutations result in reduced stability. Other mutations potentially cause loss-of-function effects by perturbing the conformations of surface loops that may be involved in protein–protein interactions or in other functions of FlnA. Our structural results provide a basis for future studies to probe the consequences of these disease-correlated mutations.
Supplementary Material
PDB reference: FlnA-Ig10, 3rgh
Acknowledgments
The authors acknowledge financial support from the US National Institutes of Health (grant RO1-GM080271 to SM). RCP was supported by American Heart Association postdoctoral fellowship 09POST2010041.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Bork, P., Holm, L. & Sander, C. (1994). J. Mol. Biol. 242, 309–320. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Dunbrack, R. L. & Cohen, F. E. (1997). Protein Sci. 6, 1661–1681. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Feng, Y. & Walsh, C. A. (2004). Nature Cell Biol. 6, 1034–1038. [DOI] [PubMed]
- Flier, A. van der & Sonnenberg, A. (2001). Biochim. Biophys. Acta, 1538, 99–117. [DOI] [PubMed]
- Ithychanda, S. S., Hsu, D., Li, H., Yan, L., Liu, D. D., Das, M., Plow, E. F. & Qin, J. (2009). J. Biol. Chem. 284, 35113–35121. [DOI] [PMC free article] [PubMed]
- Jiménez-Baranda, S., Gómez-Moutón, C., Rojas, A., Martínez-Prats, L., Mira, E., Lacalle, R. A., Valencia, A., Dimitrov, D. S., Viola, A., Delgado, R., Martínez-A. C. & Mañes, S. (2007). Nature Cell Biol. 9, 838–846. [DOI] [PubMed]
- Kapust, R. B., Tozser, J., Copeland, T. D. & Waugh, D. S. (2002). Biochem. Biophys. Res. Commun. 294, 949–955. [DOI] [PubMed]
- Kiema, T., Lad, Y., Jiang, P., Oxley, C. L., Baldassarre, M., Wegener, K. L., Campbell, I. D., Ylänne, J. & Calderwood, D. A. (2006). Mol. Cell, 21, 337–347. [DOI] [PubMed]
- Kim, H. & McCulloch, C. A. (2011). FEBS Lett. 585, 18–22. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Lad, Y., Jiang, P., Ruskamo, S., Harburger, D. S., Ylänne, J., Campbell, I. D. & Calderwood, D. A. (2008). J. Biol. Chem. 283, 35154–35163. [DOI] [PMC free article] [PubMed]
- Mann, C. J. & Weiner, H. (1999). Protein Sci. 8, 1922–1929. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Nakamura, F., Heikkinen, O., Pentikäinen, O. T., Osborn, T. M., Kasza, K. E., Weitz, D. A., Kupiainen, O., Permi, P., Kilpeläinen, I., Ylänne, J., Hartwig, J. H. & Stossel, T. P. (2009). PLoS One, 4, e4928. [DOI] [PMC free article] [PubMed]
- Nakamura, F., Osborn, T. M., Hartemink, C. A., Hartwig, J. H. & Stossel, T. P. (2007). J. Cell Biol. 179, 1011–1025. [DOI] [PMC free article] [PubMed]
- Nakamura, F., Pudas, R., Heikkinen, O., Permi, P., Kilpeläinen, I., Munday, A. D., Hartwig, J. H., Stossel, T. P. & Ylänne, J. (2006). Blood, 107, 1925–1932. [DOI] [PMC free article] [PubMed]
- Painter, J. & Merritt, E. A. (2006). Acta Cryst. D62, 439–450. [DOI] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Pinotsis, N., Abrusci, P., Djinovic Carugo, K. & Wilmanns, M. (2009). Trends Biochem. Sci. 34, 33–39. [DOI] [PubMed]
- Popowicz, G. M., Schleicher, M., Noegel, A. A. & Holak, T. A. (2006). Trends Biochem. Sci. 31, 411–419. [DOI] [PubMed]
- Pudas, R., Kiema, T. R., Butler, P. J., Stewart, M. & Ylänne, J. (2005). Structure, 13, 111–119. [DOI] [PubMed]
- Robertson, S. P. (2004). Clin. Dysmorphol. 13, 123–131. [DOI] [PubMed]
- Robertson, S. P. (2007). Eur. J. Hum. Genet. 15, 3–9. [DOI] [PubMed]
- Robertson, S. P. et al. (2006). Am. J. Med. Genet. A, 140, 1726–1736. [DOI] [PubMed]
- Roy, A., Kucukural, A. & Zhang, Y. (2010). Nature Protoc. 5, 725–738. [DOI] [PMC free article] [PubMed]
- Seo, M.-D., Seok, S.-H., Im, H., Kwon, A.-R., Lee, S. J., Kim, H.-R., Cho, Y., Park, D. & Lee, B.-J. (2009). Proteins, 75, 258–263. [DOI] [PubMed]
- Sheffield, P., Garrard, S. & Derewenda, Z. (1999). Protein Expr. Purif. 15, 34–39. [DOI] [PubMed]
- Sjekloća, L., Pudas, R., Sjoblom, B., Konarev, P., Carugo, O., Rybin, V., Kiema, T. R., Svergun, D., Ylänne, J. & Djinović Carugo, K. (2007). J. Mol. Biol. 368, 1011–1023. [DOI] [PubMed]
- Smith, L., Page, R. C., Xu, Z., Kohli, E., Litman, P., Nix, J. C., Ithychanda, S. S., Liu, J., Qin, J., Misra, S. & Liedtke, C. M. (2010). J. Biol. Chem. 285, 17166–17176. [DOI] [PMC free article] [PubMed]
- Takala, H., Nurminen, E., Nurmi, S. M., Aatonen, M., Strandin, T., Takatalo, M., Kiema, T., Gahmberg, C. G., Ylänne, J. & Fagerholm, S. C. (2008). Blood, 112, 1853–1862. [DOI] [PubMed]
- Terwilliger, T. C. (2003). Methods Enzymol. 374, 22–37. [DOI] [PubMed]
- Vriend, G. (1990). J. Mol. Graph. 8, 52–56. [DOI] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst. 34, 130–135.
- Zhou, A.-X., Hartwig, J. H. & Akyurek, L. M. (2010). Trends Cell Biol. 20, 113–123. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: FlnA-Ig10, 3rgh