

Abstract
Purpose of review
Studies on the mechanisms of distal K+ secretion have highlighted the importance of the renal outer-medullary K+ (ROMK) and maxi-K channels. This review considers several human disorders characterized by hypo- and hyperkalemia, as well as mouse models of these disorders, and the mechanisms by which ROMK and maxi-K may be dysregulated.
Recent findings
Analysis of knockout mice lacking ROMK, a model for type II Bartter’s syndrome, has shown a role for maxi-K in distal K+ secretion. Knockout mice lacking either the α or β1 subunits of maxi-K also show deficits in flow-dependent K+ secretion. Analysis of transgenic and knock-in mouse models of pseudohypoaldsoteronism type II (PHA2), in which mutant forms of with-no-lysine kinase 4 (WNK4) are expressed, suggests ways in which ROMK and maxi-K may be dysregulated to result in hyperkalemia. Modeling studies also provide insights into the role of Na+ delivery versus flow in K+ secretion.
Summary
The importance of both ROMK and maxi-K to distal K+ secretion is now well-established, but the relative role each of these two channels plays in normal and diseased states has not been definitively established. Analysis of human and animal model data can generate hypotheses for future experiments.
Keywords: Ca2+-activated K+ channel, maxi-K, rat outer medullary K+ channel, distal K+ secretion, pseudohypoaldosteronism type II, Bartter’s, diuretics, hypokalemia, hyperkalemia
Introduction
Since the 1964 study of Malnic et al, in which the distal nephron was shown to play an essential role in the regulation of urinary K+ excretion [1], much progress has been made in defining the molecular physiology regulating distal K+ transport. Nevertheless, questions remain regarding the mechanisms by which renal K+ homeostasis is disturbed in a variety of disorders. This review will consider the role of the K+ channels ROMK (KCNJ1, Kir1.1, SK) and maxi-K (BK), in several clinical disorders, including diuretic use, Bartter’s disease and PHA2, in light of our understanding from animal and human data.
ROMK and Maxi-K: distal tubule potassium channels
ROMK is an inwardly rectifying K+ channel that has a high open probability and small conductance [2]. Maxi-K, which has a large conductance, has a low open probability at baseline, but can be activated by increases in intracellular Ca2+ and membrane depolarization [3]. Both ROMK and maxi-K are found in the apical membrane of thick ascending limb (TAL), distal convoluted tubule (DCT), connecting tubule (CNT), and cortical collecting duct (CCD). Much of the characterization of these two channels has been done in CCD due to its experimental accessibility, but increasing evidence points to the importance of late DCT and CNT [4,5]. In modeling experiments, the bulk of K+ secretion was shown to occur in CNT, with some contribution from the late DCT [6••]. In support of this, mice with a collecting duct-specific knockout of the α-subunit of the epithelial Na+ channel (ENaC) are able to increase urinary K+ excretion in response to a K+ load as well as wild-type mice [7]. Since electrogenic Na+ reabsorption by ENaC provides the driving force for K+ secretion, this suggests that DCT and CNT can account for the bulk of K+ secretion.
Flow-stimulated K+ secretion
Flow dependence of distal K+ secretion was recognized decades ago [8-11]. Although this was initially thought to occur solely through ROMK, characterization of maxi-K suggested that this channel could also contribute. Hunter et al first reported the existence of calcium-activated K+ channels in the apical membrane of CCD [12], and in subsequent studies characterized the Ca2+-sensitivity of the channels. They suggested that calcium-activated K+ channels may play a role in K+ secretion in conditions that increase intracellular Ca2+ [13]. Taniguchi and Guggino showed that membrane stretch increased the open probability of calcium-activated K+ channels in cell-attached patches [14]. Subsequent studies showed that increasing luminal flow in CCD can increase intracellular calcium [15,16], and that flow-dependent K+ secretion can be blocked by inhibitors of maxi-K [17,18]. These results established that maxi-K is responsible for flow-dependent increases in K+ secretion in isolated tubules.
Role of maxi-K in type II Bartter’s
In patients treated with loop diuretics, or those with Bartter’s syndrome, the inhibition or decreased function of the sodium-potassium-chloride cotransporter (NKCC2) in TAL results in increased distal delivery of Na+. Increased Na+ reabsorption by ENaC, with increased lumen-negative potential difference (PD), increases K+ secretion, effects which are potentiated by increased aldosterone levels due to volume contraction. Patients with type II Bartter’s are hypokalemic despite mutations in ROMK [19], suggesting that another K+ channel must be mediating K+ secretion in these patients. Similarly, knockout mice lacking ROMK, in which there is a complete absence of small conductance K+ channel activity in TAL and CCD, have increased urinary K+ excretion compared to wild-type mice [20,21]. In addition, ROMK knockout mice have increased fractional excretion of K+ in response to hydrochlorothiazide and furosemide [22•], presumably due to K+ secretion through maxi-K. This was examined by Bailey et al [23], who showed that iberiotoxin, a maxi-K+ inhibitor, decreased K+ secretion by about 50% in late distal tubules from wild-type mice, and abolished K+ secretion completely in tubules from ROMK knockout mice. This suggests that ROMK and maxi-K account for the entirety of K+ secretion in late distal tubule, and provides a mechanism by which K+ secretion can continue in the absence of ROMK. Interestingly, type II Bartter’s patients can have transient neonatal hyperkalemia [24], perhaps due to a delay in the expression of maxi-K. In animal models, neither ROMK nor maxi-K is expressed at high levels at birth, but the appearance of maxi-K is delayed compared to ROMK [25].
Maxi-K knockout mice have defects in flow-dependent K+ secretion
The functional role of maxi-K in flow-stimulated K+ secretion has been examined in two additional knockout mice. The maxi-K channel is composed of a pore-forming α–subunit, as well as accessory β–subunits, of which four have been described [3,26]. The β1 subunit, which enhances the voltage and Ca2+ sensitivity of maxi-K, is expressed in connecting tubule [27]. In maxi-K β1 knockout mice, urinary excretion of K+ after volume expansion with intravenous fluids was blunted compared to wild-type mice [28]. In addition, flow-dependent K+ excretion, as estimated by measuring urinary flow rates versus K+ excretion in volume-expanded mice, was also reduced in the knockout mice [27].
In mice lacking the maxi-K channel α-subunit, treatment with a V2-receptor antagonist increased urinary flow rate by an equal amount compared to wild-type mice. Whereas urinary K+ excretion increased in wild-type mice, it did not increase in the knockouts [29], again suggesting a role for maxi-K in flow-dependent K+ secretion. Interestingly, there was a marked upregulation of ROMK in α-subunit knockout mice fed a high K+ diet, and aldosterone levels were increased in the knockout mice on low K+, control, or high K+ diets, presumably compensatory responses for the lack of maxi-K.
Which is more important to distal K+ secretion: fluid flow or Na+ delivery?
In patients on diuretics or with Bartter’s, increased distal flow is accompanied by increased distal delivery of Na+. This raises an interesting question as to the importance of fluid flow vs Na+ delivery in flow-stimulated K+ secretion. Good and Wright showed that the flow dependence of K+ secretion is independent of luminal Na+ concentration, except at very low luminal Na+ concentrations [9,10]. In addition, flow dependence of K+ secretion is seen even in the presence of amiloride [11]. Hence, increased distal flow, even without changes in distal Na+ delivery and reabsorption via ENaC, may be sufficient to increase distal K+ secretion. This can be explained by fluid flow stimulating apical Ca2+ entry to activate maxi-K channels (Fig. 1). The results of Rieg et al [29], showing that K+ secretion was increased by fluid flow induced by V2-receptor antagonist administration, which would not be expected to increase distal Na+ delivery, further support this idea.
Figure 1. Flow-dependent increase in K+ secretion involves both ROMK and Maxi-K.
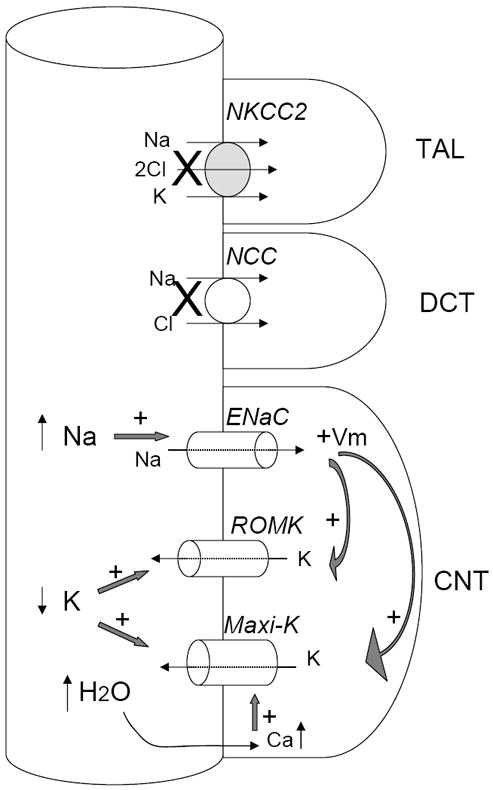
Decreases or loss of function of NKCC2 or NCC increase fluid and Na+ delivery to CNT, the key site for K+ secretion. Increased fluid flow decreases luminal K+ concentration. Increased Na+ delivery increases Na+ reabsorption via ENaC and depolarizes apical membrane potential (indicated by “+Vm”). Both factors increase K+ secretion via ROMK and maxi-K. In addition, increased fluid flow stimulates Ca2+ entry to activate maxi-K. Thus, both ROMK and maxi-K contribute to flow-dependent K+ secretion at the low flow rate. The relative contribution by maxi-K is increased at the high flow rate. Maxi-K is also activated by membrane depolarization. The contribution from membrane polarization due to increased ENaC activity to the activation of maxi-K, however, is relatively small compared to that from increases in fluid flow and the rise in intracellular Ca2+ concentration. See text for abbreviations.
Distal Na+ delivery, however, is indispensable for K+ secretion as it provides the electrical driving for K+ efflux via maxi-K or ROMK (Fig. 1). In distal micropuncture experiments, a decrease in luminal Na+ concentration sufficient to result in decreased transepithelial voltage resulted in decreased K+ secretion, even under high-flow conditions [10]. In other distal micropuncture experiments, rats treated with loop or thiazide diuretics, in which flow into both early and late distal tubule is increased, the increase in K+ secretion along the distal tubule exceeded the increase in Na+ reabsorption in this segment [30]. This suggests that both maxi-K activity stimulated by flow, as well as increased Na+ reabsorption by ENaC with generation of a more lumen-negative PD, is likely contributing to the kaliuresis in diuretic-treated animals. Type II Bartter’s patients are less hypokalemic than patients with Bartter’s due to mutations in NKCC2 or the Kb subunit of the basolateral chloride channel [31], suggesting that both K+ channels contribute to the hypokalemia seen in diuretic use (Fig. 1). Similarly, the effect of furosemide and hydrochlorothiazide on kaliuresis is blunted in ROMK knockout mice [22•].
Lessons learned from pseudohypoaldosteronism type II
Patients with the autosomal-dominant disorder pseudohypoaldosteronism type II (PHA2) invariably have hyperkalemia, and many have hypertension, though the onset of high blood pressure can be delayed [32,33]. Early studies showed that urinary K+ excretion was at or below that of daily K+ intake, despite hyperkalemia, indicating a defect in urinary K+ excretion [33-40].
PHA2 is caused by mutations in the related kinases WNK1 and WNK4 [41], and animal models have furthered our understanding of the pathophysiology of this disorder. Specifically, a transgenic mouse expressing a mutant allele of WNK4 (Q562E), corresponding to one of the mutations found in human PHA2, has been generated [42], as well as a knock-in mouse in which the wild-type WNK4 has been replaced by a mutant allele (D561A) corresponding to a second PHA2 mutation [43]. Like humans with PHA2, the mutant mice are hypertensive and hyperkalemic. The Q562E transgenic mice have a hyperplastic DCT, and the D561A knock-in mice have increased expression of NCC as well as increased phosphorylation and apical density of this transporter. These results suggested that overactivity of NCC could explain the phenotypes seen in the WNK4 mutant mice as well as in humans with corresponding mutations. Indeed, the hyperkalemia and hypertension seen in the Q562E transgenic mice was suppressed by deletion of NCC, and treatment of the D561A knock-in mice with hydrochlorothiazide also corrected hypertension, as well as increasing the urinary fractional excretion of K+, with normalization of serum K+. Patients with PHA2, at least those with mutations in WNK4, are also responsive to thiazide treatment, and it has been estimated that their sensitivity to thiazide is increased approximately six-fold compared to normal individuals [32,38].
Mechanism of hyperkalemia in PHA2: role of NCC, ROMK, and maxi-K
A proposed mechanism to explain these patients’ hyperkalemia, then, is that there is increased Na+ reabsorption in DCT due to increased NCC activity, with decreased distal delivery of Na+, less negative luminal PD, and therefore less K+ secretion in CNT (Fig. 2). Indeed, increased surface expression of ENaC, seen in the CCD of the D561A knock-in mice, likely reflects a compensatory response to this reduced fluid flow and Na+ delivery. In modeling experiments, a decrease in distal Na+ delivery from 65 mM to 25 mM, if flow rate is kept constant at 6 nl/min, would be expected to decrease K+ secretion in CNT by about 50% [6••]. However, this mechanism alone will not lead to sustained hyperkalemia unless the expected compensatory increase in the function of ROMK and maxi-K is abolished. In PHA2, disease-causing mutations in WNK1 and WNK4 result in inhibition of ROMK activity via increased endocytosis [44-46]. The apparently normal level of ROMK in the apical membrane of distal nephron in PHA2-mimicking mice compared to controls [42,43] could be interpreted as a relative deficiency in surface ROMK considering that these mice have severe hyperkalemia. Second, although there is a compensatory increase in the expression of the maxi-K α-subunit by ~3 fold [43], K+ secretion via this channel is likely below normal in the knock-in mice as a result of reduced fluid flow and Na+ delivery – that is, there is a functional deficiency in the activity of the channel (Fig. 2). However, the upregulation of maxi-K likely explains why maneuvers which increase distal flow, such as sodium sulfate infusion or treatment with hydrochlorothiazide, lead to brisk kaliuresis and improvement in hyperkalemia [34,36,38-40,43,47].
Figure 2. Three potential mechanisms for hyperkalemia in patients of PHA2 with WNK4 mutations.
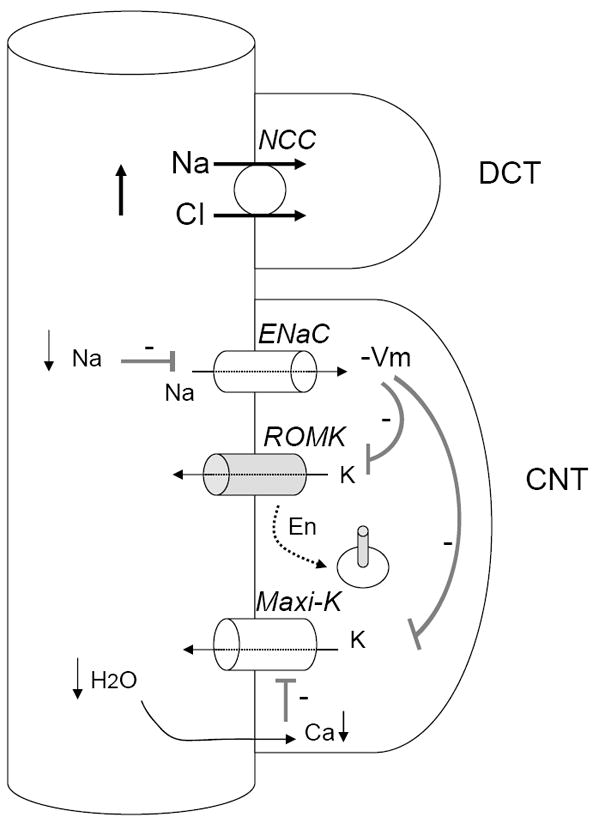
First, overactivity of NCC decreases Na+ reabsorption via ENaC, hyperpolarizes membrane potential (indicated by “-Vm”), and decreases K+ efflux via ROMK and maxi-K. Second, mutations in WNK4 that cause PHA2 inhibit ROMK by increasing its endocytosis (indicated by “En”). Third, reduced fluid flow to CNT decreases the opening of maxi-K. Because hyperkalemia causes upregulation of ROMK and maxi-K, the increase in the endocytosis of ROMK and “functional” inhibition of maxi-K (i.e., secondary to reduced Na+ and fluid delivery) are likely critical for sustained hyperkalemia.
Na+ transport in PHA2 in steady-state
The argument has been advanced that changes in Na+ delivery or distal flow could not explain differences in tubular K+ excretion in PHA2 patients because they are in steady-state. However, steady-state in a patient with disease may not recapitulate a non-diseased state. For example, assume that in a normal individual there is reabsorption of 90% of filtered Na+ in proximal tubule and TAL, 6% in DCT, and 3% in the CNT and collecting duct, with 1% excretion in the urine (Fig. 3). In a patient with PHA2, if the primary defect is increased Na+ reabsorption by NCC in DCT, such that DCT reabsorption is now 12%, there may be a compensatory decrease in proximal and thick limb reabsorption to 86%, but if the final urinary excretion of filtered Na+ is still 1%, the connecting tubule and collecting duct will see only 2% of the filtered Na+ as compared to 4% in a normal individual. This reduction in Na+ (and water) delivery could be sufficient to explain the observed defects in urinary K+ excretion. Ultimately, however, micropuncture experiments in WNK4 mutant mice are needed to resolve this issue.
Figure 3. Hypothetical model for steady-state tubular Na+ handling in PHA2 patients with WNK4 mutations.
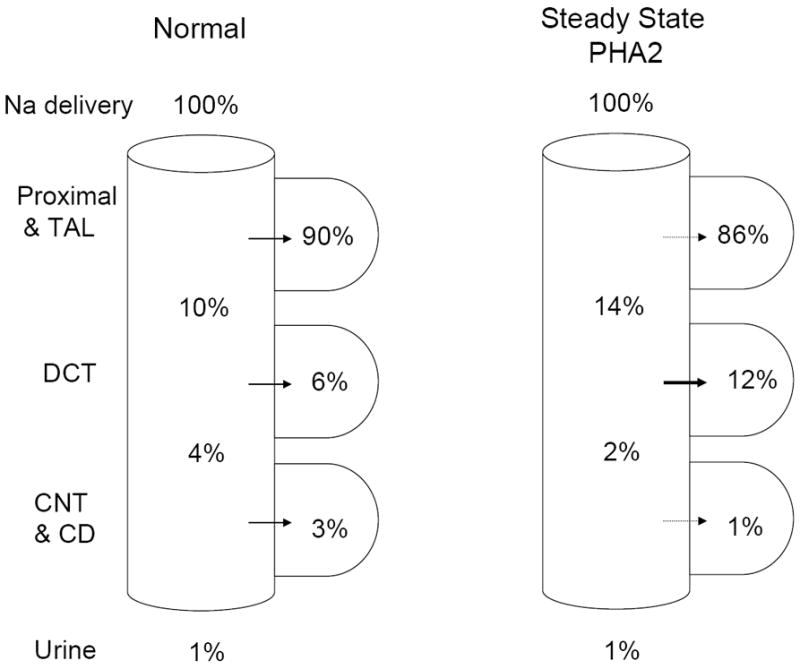
It is assumed that WNK4 mutations increase Na+ reabsorption in DCT from 6% (of filtered load) in control to 12% in the diseased state. Assuming that the compensatory decrease in Na+ reabsorption (in response to volume expansion and hypertension) occurs in the tubular segments proximal as well as distal to DCT, Na+ delivery to the CNT/CD will be reduced compared to the control (2% vs 4% in PHA2 vs control, respectively). It should be mentioned that the axial fluid profile does not parallel that for Na+ delivery because DCT lacks aquaporin-2.
Conclusions
Despite advances in our understanding of the K+ secretory channels ROMK and maxi-K, the relative roles of these two channels in regulating tubular K+ secretion in health and disease have yet to be completely elucidated. A combination of pharmacologic studies, perhaps with novel drugs allowing inhibition of maxi-K and ROMK after systemic administration, physiologic investigations including patch-clamp and micropuncture studies, and genetic studies in which the channels are manipulated in a spatially and temporally restricted fashion, could help advance our understanding of these questions.
Acknowledgments
Supported in part by a grant from NIH (DK-59530) and an EIA award from AHA (0440019N).
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
-
•
of special interest
-
••
of outstanding interest
- 1.Malnic G, Klose RM, Giebisch G. Micropuncture study of renal potassium excretion in the rat. Am J Physiol. 1964;206:674–686. doi: 10.1152/ajplegacy.1964.206.4.674. [DOI] [PubMed] [Google Scholar]
- 2.Hebert SC. An ATP-regulated, inwardly rectifying potassium channel from rat kidney (ROMK) Kidney Int. 1995;48:1010–1016. doi: 10.1038/ki.1995.383. [DOI] [PubMed] [Google Scholar]
- 3.Pluznick JL, Sansom SC. BK channels in the kidney: role in K+ secretion and localization of molecular components. Am J Physiol Renal Physiol. 2006;291:F517–F529. doi: 10.1152/ajprenal.00118.2006. [DOI] [PubMed] [Google Scholar]
- 4.Meneton P, Loffing J, Warnock DG. Sodium and potassium handling by the aldosterone-sensitive distal nephron: the pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol. 2004;287:F593–F601. doi: 10.1152/ajprenal.00454.2003. [DOI] [PubMed] [Google Scholar]
- 5.Palmer LG, Frindt G. Na+ and K+ transport by the renal connecting tubule. Curr Opin Nephrol Hypertens. 2007;16:477–483. doi: 10.1097/MNH.0b013e32820ac850. [DOI] [PubMed] [Google Scholar]
- 6••.Weinstein AM. A mathematical model of distal nephron acidification: diuretic effects. Am J Physiol Renal Physiol. 2008;295:F1353–F1364. doi: 10.1152/ajprenal.90356.2008. This paper incorporates data from prior micropuncture and microperfusion studies to quantitatively estimate proton and electrolyte fluxes under varying conditions of Na+ delivery to the distal tubule. It highlights the importance of CNT in K+ secretion and that loop and thiazide diuretics enhance K+ secretion by shifting Na+ delivery to the CNT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rubera I, Loffing J, Palmer LG, et al. Collecting duct-specific gene inactivation of α-ENaC in the mouse kidney does not impair sodium and potassium balance. J Clin Invest. 2003;112:554–565. doi: 10.1172/JCI16956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khuri RN, Wiederholt M, Strieder N, Giebisch G. Effects of flow rate and potassium intake on distal tubular potassium transfer. Am J Physiol. 1975;228:1249–1261. doi: 10.1152/ajplegacy.1975.228.4.1249. [DOI] [PubMed] [Google Scholar]
- 9.Good DW, Wright FS. Luminal influences on potassium secretion: sodium concentration and fluid flow rate. Am J Physiol. 1979;236:F192–F205. doi: 10.1152/ajprenal.1979.236.2.F192. [DOI] [PubMed] [Google Scholar]
- 10.Good DW, Velazquez H, Wright FS. Luminal influences on potassium secretion: low sodium concentration. Am J Physiol. 1984;246:F609–F619. doi: 10.1152/ajprenal.1984.246.5.F609. [DOI] [PubMed] [Google Scholar]
- 11.Malnic G, Berliner RW, Giebisch G. Flow dependence of K+ secretion in cortical distal tubules of the rat. Am J Physiol. 1989;256:F932–F941. doi: 10.1152/ajprenal.1989.256.5.F932. [DOI] [PubMed] [Google Scholar]
- 12.Hunter M, Lopes AG, Boulpaep EL, Giebisch G. Single channel recordings of calcium-activated potassium channels in the apical membrane of rabbit cortical collecting tubules. Proc Natl Acad Sci USA. 1984;81:4237–4239. doi: 10.1073/pnas.81.13.4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunter M, Lopes AG, Boulpaep E, Giebisch G. Regulation of single potassium ion channels from apical membrane of rabbit collecting tubule. Am J Physiol. 1986;251:F725–F733. doi: 10.1152/ajprenal.1986.251.4.F725. [DOI] [PubMed] [Google Scholar]
- 14.Taniguchi J, Guggino WB. Membrane stretch: a physiological stimulator of Ca2+-activated K+ channels in thick ascending limb. Am J Physiol. 1989;257:F347–F352. doi: 10.1152/ajprenal.1989.257.3.F347. [DOI] [PubMed] [Google Scholar]
- 15.Woda CB, Leite M, Jr, Rohatgi R, Satlin LM. Effects of luminal flow and nucleotides on [Ca2+]i in rabbit cortical collecting duct. Am J Physiol Renal Physiol. 2002;283:F437–F446. doi: 10.1152/ajprenal.00316.2001. [DOI] [PubMed] [Google Scholar]
- 16.Liu W, Xu S, Woda C, et al. Effect of flow and stretch on the [Ca2+]i response of principal and intercalated cells in cortical collecting duct. Am J Physiol Renal Physiol. 2003;285:F998–F1012. doi: 10.1152/ajprenal.00067.2003. [DOI] [PubMed] [Google Scholar]
- 17.Taniguchi J, Imai M. Flow-dependent activation of maxi K+ channels in apical membrane of rabbit connecting tubule. J Membrane Biol. 1998;164:35–45. doi: 10.1007/s002329900391. [DOI] [PubMed] [Google Scholar]
- 18.Woda CB, Bragin A, Kleyman TR, Satlin LM. Flow-dependent K+ secretion in the cortical collecting duct is mediated by a maxi-K channel. Am J Physiol Renal Physiol. 2001;49:F786–F793. doi: 10.1152/ajprenal.2001.280.5.F786. [DOI] [PubMed] [Google Scholar]
- 19.Simon DB, Karet FE, Rodriguez-Soriano J, et al. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet. 1996;14:152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 20.Lorenz JN, Baird NR, Judd LM, et al. Impaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter’s syndrome. J Biol Chem. 2002;277:37871–37880. doi: 10.1074/jbc.M205627200. [DOI] [PubMed] [Google Scholar]
- 21.Lu M, Wang T, Yan Q, et al. Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Bartter’s) knockout mice. J Biol Chem. 2002;277:37881–37887. doi: 10.1074/jbc.M206644200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22•.Cantone A, Yang X, Yan Q, et al. Mouse model of type II Bartter’s syndrome. Upregulation of thiazide-sensitive Na-Cl cotransport activity. Am J Physiol Renal Physiol. 2008;294:F1366–F1372. doi: 10.1152/ajprenal.00608.2007. This paper analyzes urinary excretion of Na+ and K+ in response to diuretics in control and ROMK knockout mice. It reports that NCC is upregulated in knockout mice and thiazide causes a greater natriuresis in the knockout than in the wild type. Yet, thiazide-induced increase in K+ secretion is less in the knockout compared to the control, supporting that ROMK as well as maxi-K are important for flow-stimulated K+ secretion. [DOI] [PubMed] [Google Scholar]
- 23.Bailey MA, Cantone A, Yan Q, et al. Maxi-K channels contribute to urinary potassium excretion in the ROMK-deficient mouse model of type II Bartter’s syndrome and in adaptation to a high-K diet. Kidney Int. 2006;70:51–59. doi: 10.1038/sj.ki.5000388. [DOI] [PubMed] [Google Scholar]
- 24.Finer G, Shalev H, Birk OS, et al. Transient neonatal hyperkalemia in the antenatal (ROMK defective) Bartter syndrome. J Pediatr. 2003;142:318–323. doi: 10.1067/mpd.2003.100. [DOI] [PubMed] [Google Scholar]
- 25.Satlin LM. Developmental regulation of expression of renal potassium secretory channels. Curr Opin Nephrol Hypertens. 2004;13:445–450. doi: 10.1097/01.mnh.0000133979.17311.21. [DOI] [PubMed] [Google Scholar]
- 26.Grimm PR, Sansom SC. BK channels in the kidney. Curr Opin Nephrol Hypertens. 2007;16:430–436. doi: 10.1097/MNH.0b013e32826fbc7d. [DOI] [PubMed] [Google Scholar]
- 27.Pluznick JL, Wei P, Grimm PR, Sansom SC. BK-β1 subunit: immunolocalization in the mammalian connecting tubule and its role in the kaliuretic response to volume expansion. Am J Physiol Renal Physiol. 2005;288:F846–F854. doi: 10.1152/ajprenal.00340.2004. [DOI] [PubMed] [Google Scholar]
- 28.Pluznick JL, Wei P, Carmines PK, Sansom SC. Renal fluid and electrolyte handling BKCa-β1-/- mice. Am J Physiol Renal Physiol. 2003;284:F1274–F1279. doi: 10.1152/ajprenal.00010.2003. [DOI] [PubMed] [Google Scholar]
- 29.Rieg T, Vallon V, Sausbier M, et al. The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int. 2007;72:566–573. doi: 10.1038/sj.ki.5002369. [DOI] [PubMed] [Google Scholar]
- 30.Hropot M, Fowler N, Karlmark B, Giebisch G. Tubular action of diuretics: distal effects on electrolyte transport and acidification. Kidney Int. 1985;28:477–489. doi: 10.1038/ki.1985.154. [DOI] [PubMed] [Google Scholar]
- 31.Peters M, Jeck N, Reinalter S, et al. Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am J Med. 2002;112:183–190. doi: 10.1016/s0002-9343(01)01086-5. [DOI] [PubMed] [Google Scholar]
- 32.Mayan H, Vered I, Mouallem M, et al. Pseudohypoaldosteronism type II: marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J Clin Endocrinol Metab. 2002;87:3248–3254. doi: 10.1210/jcem.87.7.8449. [DOI] [PubMed] [Google Scholar]
- 33.Achard J-M, Warnock DG, Disse-Nicodeme S, et al. Familial hyperkalemic hypertension: phenotypic analysis in a large family with the WNK1 deletion mutation. Am J Med. 2003;114:495–498. doi: 10.1016/s0002-9343(03)00054-8. [DOI] [PubMed] [Google Scholar]
- 34.Arnold JE, Healy JK. Hyperkalemia, hypertension and systemic acidosis without renal failure associated with a tubular defect in potassium excretion. Am J Med. 1969;47:461–472. doi: 10.1016/0002-9343(69)90230-7. [DOI] [PubMed] [Google Scholar]
- 35.Brautbar N, Levi J, Rosler A, et al. Familial hyperkalemia, hypertension and hyporeninemia with normal aldosterone levels: a tubular defect in potassium handling. Arch Intern Med. 1978;138:607–610. [PubMed] [Google Scholar]
- 36.Farfel Z, Iaina A, Rosenthal T, et al. Familial hyperpotassemia and hypertension accompanied by normal plasma aldosterone levels: possible hereditary cell membrane defect. Arch Intern Med. 1978;138:1828–1832. [PubMed] [Google Scholar]
- 37.Farfel Z, Iaina A, Levi J, Gafni J. Proximal renal tubular acidosis: association with familial normaldosteronemic hyperpotassemia and hypertension. Arch Intern Med. 1978;138:1837–1840. doi: 10.1001/archinte.138.12.1837. [DOI] [PubMed] [Google Scholar]
- 38.Schambelan M, Sebastian A, Rector FC. Mineralocorticoid-resistant renal hyperkalemia without salt wasting (type II pseudohypoaldosteronism): role of increased renal chloride reabsorption. Kidney Int. 1981;19:716–727. doi: 10.1038/ki.1981.72. [DOI] [PubMed] [Google Scholar]
- 39.Sanjad SA, Mansour FM, Hernandez RH, Hill LL. Severe hypertension, hyperkalemia, and renal tubular acidosis responding to dietary sodium restriction. Pediatrics. 1982;69:317–324. [PubMed] [Google Scholar]
- 40.Gordon RD. Syndrome of hypertension and hyperkalemia with normal glomerular filtration rate. Hypertension. 1986;8:93–102. doi: 10.1161/01.hyp.8.2.93. [DOI] [PubMed] [Google Scholar]
- 41.Wilson FH, Disse-Nicodeme S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 42.Lalioti MD, Zhang J, Volkman HM, et al. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet. 2006;38:1124–1131. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 43.Yang S-S, Morimoto T, Rai T, et al. Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4D561A/+ knockin mouse model. Cell Metab. 2007;5:331–344. doi: 10.1016/j.cmet.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 44.Kahle KT, Wilson FH, Leng Q, et al. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet. 2003;35:372–376. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- 45.Lazrak A, Liu Z, Huang C-L. Antagonistic regulation of ROMK by long and kidney-specific WNK1 isoforms. Proc Natl Acad Sci USA. 2006;103:1615–1620. doi: 10.1073/pnas.0510609103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He G, Wang H-R, Huang S-K, Huang C-L. Intersectin links WNK kinases to endocytosis of ROMK1. J Clin Invest. 2007;117:1078–1087. doi: 10.1172/JCI30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Licht JH, Hsueh WA, Lombardo JV. Familiar hyperkalemic acidosis. Q J Med. 1985;54:161–176. [PubMed] [Google Scholar]