

Abstract
The FMR1 gene contains a CGG-repeat present in the 5’UTR which can be unstable upon transmission to the next generation. The repeat is up to 55 CGGs long in the normal population. In patients with fragile X syndrome, a repeat length exceeding 200 CGGs (full mutation: FM) generally leads to methylation of the repeat and the promoter region, which is accompanied by silencing of the FMR1 gene. The absence of FMR1 protein, FMRP, seen in FM is the cause of the mental retardation in patients with fragile X syndrome. The premutation (PM) is defined as 55-200 CGGs. Female PM carriers are at risk of developing primary ovarian insufficiency. Elderly PM carriers might develop a progressive neurodegenerative disorder called fragile X-associated tremor/ataxia syndrome. Although arising from the mutations in the same gene, distinct mechanisms lead to fragile X syndrome (absence of FMRP), FXTAS (toxic RNA gain of function) and FXPOI. The pathogenic mechanisms thought to underlie these disorders are discussed. This review gives insight on the implications of all possible repeat length categories seen in fragile X families.
Keywords: CGG repeat, FMR1, FXPOI, FXTAS, mental retardation, treatment
Fragile X syndrome (FXS) (MIM #30955) is among the most common human single gene disorders, and is the leading cause of inherited cognitive disability. It is inherited as an X-linked dominant disorder with reduced penetrance. Both males and females can be affected, however the degree of cognitive disability is typically more severe in males. The disease has an estimated frequency of 1/4000 males and 1/7000 females worldwide. The gene involved in FXS was discovered in 1991 and it was called fragile X mental retardation 1 (FMR1) gene (1).
The primary features of an affected male are mild to severe cognitive disability, macroorchidism and a connective tissue dysplasia leading to a characteristic, yet mild, physical appearance of a long, narrow face and large ears. Patients with FXS often display autistic features and suffer from epilepsy. Other clinical signs, presumably due to the connective tissue disorder, include velvet-like skin, finger-joint hyperextensibility, recurrent otitis media, aortic root dilation, and mitral valve prolapse (2). Women may also be affected, however, affected females have a less severe phenotype than do males, and the severity of dysfunction is correlated to the degree of X-inactivation on the abnormal chromosome (3).
Fragile X Mental Retardation gene, FMR1
The FMR1 gene is mapped on the X chromosome at position q27.3 spaning approximately 40 kb of genomic sequence (1). The FMR1 gene contains 17 exons and its mRNA is ~ 4 kb long. Exons 12, 14, 15 and 17 can be alternatively spliced, resulting in different mRNAs and protein isoforms. Cloning of the FMR1 gene revealed that the fragile site of the X chromosome contains a CGG repeat in the 5’ untranslated region (UTR) of the gene. This CGG trinucleotide repeat is unstable, and therefore the repeat length is variable (polymorpyhic) in the normal population, ranging from 6-55 repeats. The repeat can become unstable upon maternal transmission, usually resulting in the expansion of the repeat in the next generation. When the repeat expands and ranges from 55-200, the individuals are considered premutation carriers (Figure 1). In patients with FXS, the CGG repeat has expanded above 200 units (FM) (4-5). Usually, a full mutation results in hypermethylation of the CpG site in the promoter region of the FMR1 gene (6). Methylation of DNA promoter sequences is associated with gene silencing and can be accompanied by a number of modifications in histone N-tails (7), while the promoters of actively transcribed genes typically have demethylated DNA and acetylated lysines in the N-tail of histones H3 and H4. Pietrobono et al. showed that the earliest events in the cascade leading to the inactivation of an expanded FMR1 gene seems to be histone deacetylation and H3-K9 methylation, which are followed by DNA methylation and H3-K4 demethylation (8). Additionally, using human embryonic stem cells, it has also been demonstrated that FMR1 inactivation is initiated by downregulation of transcription and chromatin modifications prior to DNA methylation (9). Hypermethylation of the promoter region of the FMR1 gene results in transcriptional silencing, leading to fragile X mental retardation protein (FMRP) deficiency and intellectual disability. Although different hypotheses have been postulated about CGG repeat instability, the exact mechanism is still not fully understood (10).
Figure 1. CGG repeat length, FMRP expression and clinical outcome.
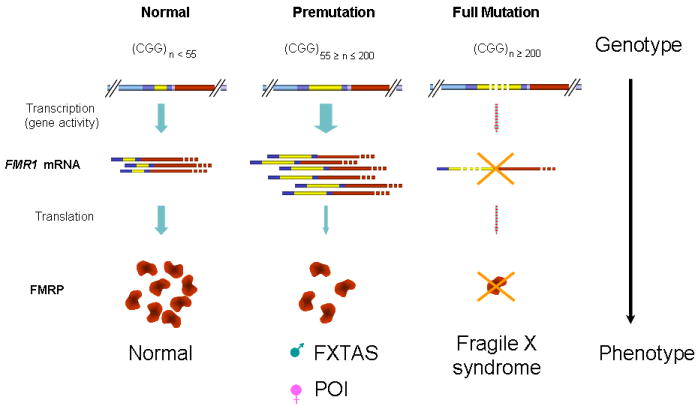
In unaffected individuals, the CGG repeat in the 5’ UTR ranges between 5-55, leading to normal FMR1 mRNA transcription and translation, and normal FMRP expression. The premutation repeat (55-200) results in elevated FMR1 mRNA transcription, but reduced FMRP expression. This increases the risk of developing FXTAS in males or FXPOI in females (111). A full mutation repeat (over 200) leads to silencing of the FMR1 gene due to hypermethylation. As a consequence FMRP is lacking resulting in fragile X syndrome (adapted from (112)).
FMRP
The protein contains different domains, including the RNA binding domains KH and RGG box and a nuclear localization and a nuclear export signal (NLS and NES), that facilitate its shuttling between nucleus and cytoplasm (11). Both RNA binding motifs contribute to the role of FMRP as suppressor of target mRNA translation via binding of noncoding RNA structures, including G-quartets and kissing complexes (12-13). In brain tissue, FMRP is mainly present in the cell soma of neurons and is packed together with other proteins and mRNAs in ribonucleoprotein (RNP) particles. Significant quantities of these FMRP-positive RNP particles are transported into the dendrite, and during development a minority of the protein is localized in the axons (14).
FMRP in brain
In neurons, many different types of cytoplasmic granules exist, i.e. stress granules, P-bodies and RNA-granules, and FMRP is mainly present in these cytoplasmic granules (reviewed in (15)). Briefly, stress granules are cytoplasmic foci where untranslated mRNAs accumulate when cells are subjected to a variety of environmental stressors. P-bodies on the other hand, are sites where decapping and mRNA decay occur. P-bodies are also important in microRNA processing and contain the ribonucleoprotein RNA-induced silencing complex (RISC). Finally, RNA-granules harbor translationally silenced mRNAs, and in neurons RNA-granules are transported to dendritic synapses. These RNA-granules can be divided in two types: RNA-granules containing ribosomal subunits (RNP particles) and RNA-granules not containing ribosomal subunits (transporting granules) (15).
Although the exact function of FMRP in these different types of granules is still unknown, FMRP seems to display many different functions in neurons, including repression of mRNA translation during dendritic transport and at the synapse (Figure 2).
Figure 2. Schematic model of the function of FMRP in neurons.

FMRP is synthesised in the cytoplasm. Dimers of FMRP (yellow hexagons) enter the nucleus via the NLS domain. In the nucleus, FMRP binds to target mRNAs and other proteins, forming an RNP particle. The FMRP-RNP particle is transported back to the cytoplasm, via the NES of FMRP. In the cytoplasm, the FMRP-RNP particle can interact with members of the RNA-induced silencing complex (RISC; green circle) and also associate with ribosomes (red ovals). FMRP-RNP particles regulate protein synthesis (string of blue circles) in the cell body of the neuron. A proportion of the FMRP-RNP particles is packed in mRNA granules and is transported into dendrites. During transport, FMRP fulfils its major role as a translational repressor of target mRNAs. Upon synaptic stimulation of the group I mGluRs, FMRP allows translation of its mRNA targets. The proteins that are synthesised seem to be involved in the cyclic internalisation of AMPA receptors and other neuronal processes.
Besides an important role in neurons, FMRP is also developmentally expressed in astrocytes (16). The presence of FMRP in astrocytes during development may be essential for the role of astrocytes in synaptogenesis. It has been demonstrated that FMRP expression in astrocytes is important for shaping the dendritic arbors of neurons in FXS (17) suggesting a functional role of astrocytic FMRP in the neurobiology of FXS.
Dendritic mRNA transport
The transport of FMRP-containing granules has been demonstrated to be microtubule-dependent, with the same speed as that of general dendritic RNA transport (18). When microtubules were disrupted by nocadazole treatment, FMRP-positive RNA-granules became immobile, whereas disruption of the actin network had no effect. Because FMRP binds mRNAs and is found in dendritic RNA-granules, it is hypothesized that FMRP is important for the transport of its target mRNAs into the dendrite (Figure 2). Electrical stimulation is known to induce transport of specific mRNAs such as CamkII and Arc, which are both target mRNAs of FMRP. Therefore, it was suggested that lack of FMRP resulted in differences in the amount of these target mRNAs in the dendrites at basal state or after stimulation. However, Steward et al. found no differences in the amount of these target mRNAs in the Fmr1 KO hippocampus at basal state or after stimulation (19). In contrast, Dictenberg et al. did find differences in the amount of dendritic target mRNAs in Fmr1 KO hippocampal neurons after chemical stimulation with DHPG, no differences were observed at basal state (20). After stimulation with DHPG, fluorescent in situ hybridization showed enhanced signals for CAMKII, MAP1B, SAPAP4 and GABAA-R-δ mRNAs in the dendrites of wild-type hippocampal neurons, while in Fmr1 KO neurons the signals did not differ compared to unstimulated conditions. It was also shown that the RNA-granules were less motile in Fmr1 KO neurons (20). These results suggest that FMRP is at least partially involved in activity-dependent dendritic transport of its target mRNAs, although more research is warranted to define the exact function of FMRP in mRNA transport (Figure 2).
Translational repressor
The majority of FMRP is present in the cytoplasm, associated with elongated polyribosomes in large messenger ribonucleoprotein particles, suggesting that FMRP is important for silencing mRNA translation (21). The importance of this association of FMRP with polyribosomes has been demonstrated in a severely mentally retarded patient with a missense mutation I304N in the second KH domain of FMRP (22). As a consequence of this mutation, FMRP (i) is unable to bind to the “kissing-complex” RNA sequence, (ii) can no longer associate with polyribosomes (13), and (iii) is predominantly found in small RNP particles (21, 23-24). It has been postulated that FMRP is associated with polyribosomes to mediate translation of target mRNAs at the synapse. In vitro experiments have demonstrated that FMRP acts as a translational repressor in rabbit reticulocyte lysates in a dose-dependent manner (25). Moreover, local translation in the spines after mGluR activation seems to be regulated by FMRP as well (26). The exact mechanism of translational repression is not fully understood, and different mechanisms involved in repression of translation have been proposed, including the phosphorylation status of FMRP and the microRNA pathway.
It has been demonstrated that FMRP can act as a translation repressor in a phosphorylation-dependent mechanism (27). Initially, it was shown that FMRP can be phosphorylated specifically at serine 499 (28). The phosphorylation status of FMRP influences the translation of target mRNAs since phosphorylated FMRP is associated with stalled polyribosomes and unphosphorylated FMRP with actively translating polyribosomes. Subsequently, it has been shown that FMRP is rapidly dephosphorylated by PP2A after specific stimulation at the synapse, probably resulting in local protein synthesis of target mRNAs (29). Simultaneously, after specific synaptic stimulation, a second cascade involving TSC1/TSC2 and the mTOR pathway is activated, which ultimately results in re-phosphorylation of FMRP by S6 kinase (27). Although this hypothesis is an attractive model, an alternative hypothesis has been proposed in which FMRP regulates translation at the initiation stage.
Another hypothesized mechanism of FMRP-dependent regulation of mRNA translation might be in cooperation with the microRNA pathway. The first indications came from Drosophila studies, where it was shown that dFmrp interacts directly or indirectly with two components of the RISC complex, Argonaute 2 (AGO2) and Dicer (30). In mammalian cells, FMRP seems to be incorporated in RNP particles that also contain Ago2, Dicer and miRNAs (31), and also in postsynaptic compartments, FMRP seems to interact with Dicer and Ago2 (32). More recently, FMRP was found to interact with specific miRNAs resulting in translational repression of corresponding target mRNAs (33). The results show that FMRP interacts with miRNA125b and thus regulates the translation of NMDA receptor subunit 2A (33). Evidence for the relationship between the miRNA pathway and FMRP is increasing. The exact role of FMRP in the miRNA pathway needs to be further investigated.
FMRP and spine abnormalities
Microscopic analysis of autopsy material from patients with FXS revealed significant morphological abnormalities in brain tissue (34). It was found that in some brain areas the dendrites of neurons show a higher protrusion density than neurons of control individuals and the dendritic spines exhibited a more immature phenotype (35). Due to the increased number and abnormal morphology of the protrusions, the balance in signal transmission is believed to be altered, resulting in the FXS phenotype. Generation of the first Fmr1 KO mouse model finally allowed examination of in vivo protrusion morphology in different brain areas at different ages. Comery et al. quantified dendritic protrusions of layer V pyramidal neurons in the visual cortex of adult Fmr1 KO mice (36). These neurons have more elongated protrusions and fewer shorter protrusions. Greenough and colleagues extended this examination by making eight classes of protrusions, ranging from immature to mature, and quantified the protrusions in mice in a C57BL/6 background (37).
Nimchinsky et al. showed that elongated protrusions in the Fmr1 KO mice were only present in the somatosensory cortex at young ages, i.e. 1 week and 2 weeks postnatal, whereas the abnormal morphology disappeared at 4 weeks of age (38). Fmr1 KO mice also showed significantly more protrusions in young animals. These data strongly suggest that differences in spine morphology and number are related to development. Therefore, the spine morphology was examined again in younger and older Fmr1 KO and wild-type mice in a C57BL/6 background (25 days and 75 days, respectively) (39). This study demonstrated that at P25 minimal protrusion abnormalities (expressed in length, morphology and number) were present in the somatosensory cortex of Fmr1 KO mice, whereas the abnormalities “reappeared” at 75 days of age, in accordance with Nimchinsky et al. Thus, at 75 days, dendritic protrusions of Fmr1 KO neurons in the somatosensory cortex are significantly longer, have a more immature morphology, and show increased density per apical dendrite. Grossman also investigated the CA1 region of the hippocampus (40). Pyramidal neurons in the hippocampus of Fmr1 KO mice showed more immature protrusions and fewer shorter protrusions, but the number of protrusions was not significantly different. Furthermore, cultured hippocampal primary neurons of Fmr1 KO showed protrusion abnormalities, illustrated by an increased number of immature protrusions and a decreased number of synapses (41), more filopodia (immature protrusions) (42), and increased length of protrusions.
Animal models for FXS
Different FXS animal models have been generated to study the effects of a lack of FMRP on behavior, mRNA translation, and pharmacological interventions. All animal models are not identical to the human genotype, because the silencing of the gene is not due to methylation of an expanded CGG repeat, but all animal models are generated using genetic modification resulting in a (almost) complete loss of Fmr1 transcription and therefore total lack of Fmrp expression.
Fmr1 KO mice have been studied extensively and are found to be a useful animal model to study FXS (43-44). Numerous behavioral tests have been performed to investigate behavioral deficits in the Fmr1 KO mice. Although not all studies show consistent results, it has been reported that Fmr1 KO mice show deficits in spatial learning, defect of prepulse inhibition of acoustic startle response (PPI), increased locomotor activity and that they are more susceptible for epileptic seizures (42, 45-47). Approximately 13-18% of patients with FXS suffer from epilepsy (45). This symptom is preserved in the Fmr1 KO mice, since they are highly susceptible for audiogenic seizures. As mentioned above, Fmr1 KO mice exhibit abnormal spine morphology in different brain areas, similar to what is found in patients with FXS. Moreover, this animal model has offered the opportunity to study synaptic plasticity in the brain and is altered in many brain areas, such as hippocampus, cortex, and cerebellum. After identification of the Drosophila orthologue, dFmr null mutant fly models were generated and shown to be viable (48). The dFmr null mutant flies are anatomically normal, but show altered behavior. It has been demonstrated that dFmrp deficiency results in abnormal circadian rhythm (49), locomotor defects (48), and altered courtship behavior (50). A closer look at the central nervous system also revealed morphological abnormalities of neurons. Loss of dFmrp resulted in mushroom body defects and altered axonal branching (48). To study the function of FMRP during development, one of the approaches which can be used is the morpholino antisense oligonucleotide knockdown technique in zebrafish. Injecting an antisense oligonucleotide morpholino in fertilized eggs will result in a transient knockdown of the target gene, i.e. Fmr1. Using this approach, an FXS phenotype has been observed including abnormal axonal branching, neuronal guidance, and defasciculation defects (51). However, these results are under debate since a genetic Fmr1 KO zebrafish model, developed with ENU-mutation screening, shows no obvious phenotype at all (52). This suggests that the phenotype of morpholino injected Fmr1 knockdown fish appears to result from potential experimental artifacts.
In conclusion, all animal models have shown to be valuable in elucidating the molecular mechanisms underlying the clinical symptoms in FXS.
Treatment of Fragile X syndrome
In the brain, many different types of receptors are present at the synaptic membranes which can be divided in two major classes of neurotransmitter receptors: ionotropic and metabotropic receptors. In FXS mainly two neurotransmitters seem to be relevant, i.e. glutamate and gamma-amino butyric acid (GABA). Glutamate receptors are expressed throughout the brain and are essential for excitatory neurotransmission and synaptic plasticity. Glutamate receptors are found both at the postsynaptic and presynaptic membranes. AMPA, NMDA and Kainate receptors form the major class of ionotropic glutamate receptors in the brain, mediating fast excitatory neurotransmission.
In 2004, Bear et al. defined the mGluR theory to explain many aspects of the symptoms found in patients with FXS and in the Fmr1 KO mouse, including: (1) higher density of spines and more immature spines, (2) electrophysiological deficits in Fmr1 KO mice, (3) exaggerated dendritic protein synthesis in Fmr1 KO mice after mGluR receptor activation, and (4) behavioral phenotypes in patients with FXS and Fmr1 KO mice (53). This mGluR theory states that AMPA receptor internalization, triggered by mGluR5 stimulation is exaggerated in FXS causing a weakening of the synapse (Figure 3). Many experiments showed defects in synaptic signaling transmission in the Fmr1 KO mice.
Figure 3. Therapeutic strategies for FXS.
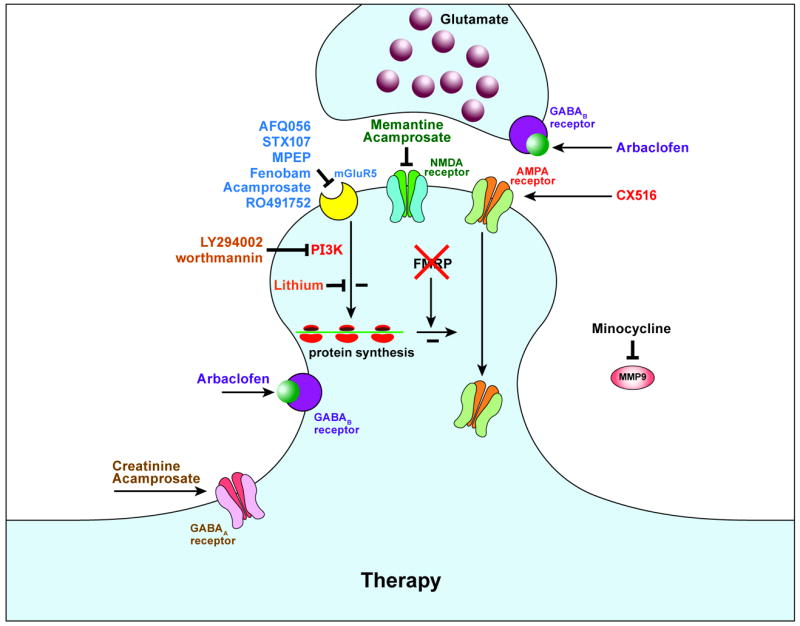
Schematic representation of a glutamatergic excitatory and GABAergic inhibitory synapse lacking FMRP. Several types of drugs can interact with different type of neuronal receptors which may result in rescue of the disturbed synaptic transmission found in FXS (adapted from (59)).
Besides the mGluR receptor theory, it is also hypothesized that GABA signaling is altered in patients with FXS. GABA receptors are known to be involved in phenotypes such as epilepsy, anxiety, hyperactivity, insomnia, learning and memory deficit, autism, all present in patients with FXS (54). Experimental evidence of the correlation between GABA-A receptors and FXS has been shown in Drosophila (55) and in mice (56). mRNA expression levels of 8 out of 18 known subunits of the GABA-A receptor are decreased in the cortex of the FXS mouse model, but not in the cerebellum.
New strategies for therapeutic intervention have been developed based on the mGluR and GABA theories (Figure 3). Most treatments are first tested in the FXS animal models, like the dFmr1 mutant (KO) fly or Fmr1 KO mice. Several clinical trials were started using a variety of existing and new drugs that interfere in either the mGluR or GABA pathways. A number of clinical trials are presently ongoing and the first results can be expected in 2011. (Detailed information is available at clinicaltrials.gov.) A most promising trial concerns AFQ056 (mGluR antagonist), that ameliorates the behavior of adults affected with FXS in which only fully methylated full mutation carriers are responders (57); This study is in line with a positive outcome of treatment by AFQ056 in Fmr1 KO mice (58). A second is treatment with arbaclofen (GABA receptor agonist) for which promising preliminary data were presented at the International Meeting for Autism Research (http://imfar.confex.com/imfar/2010/webprogram/Paper6707.html). The third promising compound is minocycline (MMP-9 inhibitor). Potential therapeutic interventions were recently reviewed extensively by Levenga et al (59), Dolen et al (60), Heulens et al (61).
It is likely that in the near future a treatment for FXS based on intervention in mGluR or GABA receptor pathways will become available.
Premutation
Carriers of the fragile X premutation (55-200 CGG repeats) are relatively common in the general population. Approximately 1:130-250 females and 1:800 males carry the premutation (62). Female carriers of the premutation can transmit either their normal or premutation allele in the next generation. However, upon maternal transmission the risk of expansion of the premutation to a full mutation allele increases with increasing CGG repeat number. In male carriers of the premutation relatively stable transmissions occur, and thus the risk of expansion to a full mutation is negligible. All individuals with a premutation allele have increased levels of FMR1 mRNA (2-8 fold in lymphocytes) and paradoxically slightly decreased FMRP levels (Figure 1) (63). The mechanism underlying the increased transcriptional activity of premutation range alleles is unknown, however, a recent study reported differential usage of transcriptional start sites and polyadenylation sites in FMR1 premutation alleles leading to different expression in expanded premutation alleles compared to normal alleles (64). The fragile X premutation is associated with two clinical disorders: fragile X-associated tremor/ataxia syndrome (FXTAS) and fragile X-associated primary ovarian insufficiency (FXPOI) disorder. Both disorders show an incomplete penetrance.
FXTAS
Clinical and neuropathological phenotype
In 2001, a late-onset neurodegenerative disorder that affects a subset of older adult carriers of premutation alleles was described (65). In FXTAS, a progressive intention tremor and cerebellar gait ataxia are the major clinical features. More advanced cases may be accompanied by progressive decline in cognition, peripheral neuropathy, Parkinsonism and autonomic dysfunctions (66). The clinical symptoms vary among subjects with FXTAS and also the progression is highly variable with life expectancy between 5 to 25 years after presentation (67-68). Generally, affected female carriers of the premutation present milder clinical involvement compared to affected male carriers (69). The cause of the incomplete penetrance of FXTAS within carriers of the premutation remains unclear, however, the penetrance increases with both age and CGG repeat size (70). Approximately 40% of male carriers of the premutation and 8-16% of the female carriers of the premutation aged 50 years and older, who are identified by family-based studies, develop FXTAS (71). Apparently the partial protective influence of the second non-expanded X-chromosome in female carriers results in a lower penetrance. Psychiatric dysfunction is also associated with FXTAS. In male carriers with and without FXTAS, mood and anxiety disorders are frequently reported (72) whereas in female carriers higher rates of depression, anxiety and ADHD have been found (72). Comprehensive examination of cognitive impairment in male premutation carriers suggests that FXTAS involves substantial executive impairment and diffuse deficits in other cognitive functions (72)Only recently, a broader phenotype has been documented in both male and female carriers of the premutation with FXTAS who display symptomatology beyond the central nervous system. Females with FXTAS present significantly more frequently thyroid problems, hypertension, fibromyalgia, seizures, autoimmune disease and migraine headache (72) Medical comorbidities that were commonly noticed in male carriers of the premutation with FXTAS include impotence and cardiac arrhythmias, however, the evidence is still anecdotal (73). Since these medical conditions have implications for treatment strategies more-detailed studies are warranted to provide further evidence for this expanded clinical phenotype in female and male carriers of the premutation with and without FXTAS.
In addition to the core clinical symptoms, magnetic resonance imaging (MRI) and neuropathological studies are part of the diagnostic criteria for FXTAS. MRI studies demonstrated a generalized brain atrophy, T2 hyperintensities of the middle cerebellar peduncle and adjacent white matter, illustrating white matter disease (74). Neuropathological analyses of postmortem brain tissue of symptomatic carriers of the premutation revealed the presence of eosinophilic, ubiquitin-positive intranuclear inclusions in both neurons and astrocytes throughout the brain, Purkinje cell loss, spongiosis of the deep cerebellar white matter and Bergman gliosis (Figure 4A) (68, 75).
Figure 4. Inclusions in human and mouse brain.


Paraffin sections of hippocampus from FXTAS patient (A) and KI mouse (B). The micrograph shows the presence of ubiquitin-positive intranuclear inclusions in neurons located in the CA3 region of the hippocampus using an indirect immunoperoxidase protocol.
Animal models for FXTAS
Several animal models have been generated as a putative model for FXTAS, including mouse and Drosophila. Both models have proven invaluable for the study of the pathophysiology of FXTAS and have provided critical information about the development of brain pathology.
Two different knock-in (KI) mouse models with expanded CGG RNA expression under the control of the endogenous Fmr1 promoter and one transgenic mouse model that express expanded CGG RNA in Purkinje cells outside the context of the Fmr1 gene have been generated (76-78). Both KI models show repeat instability (expansions and contractions) upon maternal and paternal transmission, intriguingly, increased frequency of repeat expansion on maternal transmission is ATR-sensitive, whereas an ATM-sensitive mechanism shows a male expansion bias (79-80). ATR is a protein important in the cellular response to stalled replication forks and bulky DNA lesions, while ATM has been shown to be involved in the rescue of replication forks stalled by depletion of intracellular dCTP pools. In addition, oxidizing agents may play a role in the frequency of CGG repeat expansion as well (81). To date, CGG repeat tracts have been reported over 200-300 CGGs long but surprisingly without methylation of the promoter region of the Fmr1 gene (77, 82). Both KI mouse strains phenocopy human FXTAS in the production of elevated levels of CGG RNA and the presence of specific neuropathological features associated with FXTAS, including intranuclear inclusions in both neurons and astrocytes and reduced numbers of Purkinje cells (Figure 4B) (76-77, 83-85). Also the Purkinje neuron-specific transgenic mice show intranuclear inclusions in Purkinje neurons and Purkinje neuron cell death (78). All three FXTAS mouse models have been extensively evaluated for cognitive and behavioral phenotypes. In summary, cognitive deficits include memory impairments on the Morris water maze, spatial processing for both metric and topological tasks and temporal ordering for visual objects (84, 86-87). Deficits in motor performance are observed on the accelerating rotarod (78, 86). In addition, KI mice are hyperactive, exhibited less anxiety and are impaired on the passive avoidance test (88). No effective therapy is currently available for FXTAS.
The presence of similarities in the behavioral phenotype between FXTAS mouse models and patients with FXTAS, although subtle, may provide behavioral translational endpoints to assess efficacy for new therapeutic intervention in FXTAS using these mouse models. Next to the FXTAS mouse models, transgenic flies are generated that overexpress untranslated riboCGG90 repeats upstream of the coding sequence for the eGFP reporter gene. Overexpression of the mutant mRNA in fly eyes results in the formation of inclusions in both nuclei and cytoplasm that are positive for ubiquitin and causes neurodegeneration (89). The fly model has proven extremely useful to perform biased genetic screens to identify genetic modifiers of the neurodegenerative phenotype (90). Recently, Todd et al. demonstrated that overexpressing histone deacetylases suppresses CGG repeat-induced neurodegeneration in a Drosophila model (91).
Although these mouse and fly models are not perfect, they have facilitated cellular studies on the underlying molecular basis of FXTAS and provided critical information about the molecular events that occur with the onset and progression of FXTAS.
Molecular basis of FXTAS
Compelling evidence indicates an RNA toxic gain-of-function mechanism for FXTAS (92). The increased levels of FMR1 mRNA, containing premutation-length CGG repeats and the presence of mutant mRNA within the intranuclear inclusions are the first indications for such a RNA-mediated neurodegeneration mechanism for FXTAS (63, 93). The absence of cases of FXTAS in the full mutation range is another indication. Later studies focused on the sequestration model, as demonstrated for CUG repeats in DMPK mRNA in myotonic dystrophy (DM). Indeed, experimental evidence showed the accumulation of a variety of proteins within the inclusions using in vitro and in vivo models, including Purα, hnRNP A2/B1, CUGBP1 and Sam68 (94-97). The sequestration of Sam68, an RNA binding protein involved in alternative splicing regulation, results in partial loss of Sam68-responsive splicing and consequently misregulation of splicing of target pre-mRNAs in FXTAS patients. Although, the sequestration hypothesis is currently the most favorable model, alternative models have been proposed, including antisense transcript model, mitochondrial dysfunction model and stress response model (98-100).
FXTAS is a late-onset disorder, however, the presence of functional and morphological abnormalities in cultured primary hippocampal neurons from KI pups (P0-2) and defects in neocortical development in KI embryos suggest a developmental component in the pathogenesis of FXTAS as well (101-102). Indeed, children carrying a premutation have been reported to develop psychopathology more frequently and this may very well be associated with abnormalities in the developing brain owing to the FMR1 premutation allele (103-105).
FXPOI
Only 1% of females experience menopause before the age of 40 years, termed primary ovarian insufficiency (POI) (106). POI is the “end-stage” of a continuum of ovarian dysfunction and despite the clinical importance the etiology of POI is often not identified. Females who carry the premutation are at risk for premature ovarian failure (fragile X associated primary ovarian insufficiency; FXPOI). It has been shown that females carrying a premutation ascertained through families with FXS have up to a 23% rate of POF, and as a group, experience earlier menopause by approximately 5 years (107-109). In female premutation carriers the ovarian dysfunction depends on repeat size, however the relationship is not linear, that is, females with mid-range repeats (80-100 repeats) experience POF earlier and at higher frequencies (32%) than other carrier groups (110). The underlying molecular mechanisms of FXPOI are not known, however, findings from FXTAS studies may be relevant to FXPOI as well. The key question in studies to obtain knowledge about the aetiology of ovarian insufficiency in females with the premutation is: how premutation mRNA may lead to early menopause. The hypothesis that the FMR1 premutation may have a toxic RNA gain-of-function effect on ovarian follicle dynamics is currently under study by close examination of the ovarian follicle population in female KI mice (R. Willemsen, unpublished data). Preliminary data show high expression of FMRP in both oocytes and granulosa cells in human ovaries during folliculogenesis (Figure 5). Perhaps that the mid-range repeat lengths result in the greatest amount of mRNA in ovaries and therefore the greatest toxicity. It is also possible that repeat size confers a specific mRNA conformation, like hairpins, and therefore interaction with different proteins or other mRNAs depending on repeat size.
Figure 5. FMRP expression in human ovary.

Parafin section of human ovary from a woman at age 20 years old. The micrograph shows FMRP expression in oocytes and granulosa cells during folliculogenesis using an indirect immunoperoxidase protocol. arrow = oocyte ; arrowhead = granulosa cells
In conclusion, it is extraordinary that expansions of the CGG repeat in the FMR1 gene are involved in three distinct disorders in which the size of the CGG repeat will ultimately define the clinical phenotype.
Acknowledgments
This work was supported by NIH-NINDS grant RL1NS062411 (RW), NIH Roadmap Initiative UL1DE019583 (RW), (NICHD R01 HD38038) (BAO), ZonMw 912-07-022 (RW), E-rare 113301069 (RW) and CBG (BAO).
Footnotes
Conflict of interest: no
References
- 1.Verkerk AJ, Pieretti M, Sutcliffe JS, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 2.Hagerman RJ. Physical and behavioral phenotype. In: Hagerman RJ, Silverman AC, editors. Fragile X syndrome: diagnosis, treatment and research. Baltimore and London: The John Hopkins University Press; 1996. pp. 3–87. [Google Scholar]
- 3.Merenstein SA, Sobesky WE, Taylor AK, et al. Molecular-clinical correlations in males with an expanded FMR1 mutation. Am J Med Genet. 1996;64:388–394. doi: 10.1002/(SICI)1096-8628(19960809)64:2<388::AID-AJMG31>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 4.Yu S, Pritchard M, Kremer E, et al. Fragile X genotype characterized by an unstable region of DNA. Science. 1991;252:1179–1181. doi: 10.1126/science.252.5009.1179. [DOI] [PubMed] [Google Scholar]
- 5.Oberlé I, Rousseau F, Heitz D, et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 6.Bell MV, Hirst MC, Nakahori Y, et al. Physical mapping across the fragile X: hypermethylation and clinical expression of the fragile X syndrome. Cell. 1991;64:861–866. doi: 10.1016/0092-8674(91)90514-y. [DOI] [PubMed] [Google Scholar]
- 7.Chiurazzi P, Neri G. Reactivation of silenced genes and transcriptional therapy. Cytogenet Genome Res. 2003;100:56–64. doi: 10.1159/000072838. [DOI] [PubMed] [Google Scholar]
- 8.Pietrobono R, Tabolacci E, Zalfa F, et al. Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum Mol Genet. 2005;14:267–277. doi: 10.1093/hmg/ddi024. [DOI] [PubMed] [Google Scholar]
- 9.Eiges R, Urbach A, Malcov M, et al. Developmental Study of Fragile X Syndrome Using Human Embryonic Stem Cells Derived from Preimplantation Genetically Diagnosed Embryos. Cell Stem Cell. 2007;1:568–577. doi: 10.1016/j.stem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Brouwer JR, Willemsen R, Oostra BA. Microsatellite repeat instability and neurological disease. Bioessays. 2009;31:71–83. doi: 10.1002/bies.080122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willemsen R, Oostra BA, Bassell GJ, et al. The fragile X syndrome: From molecular genetics to neurobiology. Ment Retard Dev Disabil Res Rev. 2004;10:60–67. doi: 10.1002/mrdd.20010. [DOI] [PubMed] [Google Scholar]
- 12.Darnell JC, Jensen KB, Jin P, et al. Fragile X Mental Retardation Protein Targets G Quartet mRNAs Important for Neuronal Function. Cell. 2001;107:489–499. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- 13.Darnell JC, Fraser CE, Mostovetsky O, et al. Kissing complex RNAs mediate interaction between the Fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev. 2005;19:903–918. doi: 10.1101/gad.1276805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christie SB, Akins MR, Schwob JE, et al. The FXG: a presynaptic Fragile X granule expressed in a subset of developing brain circuits. J Neurosci. 2009;29:1514–1524. doi: 10.1523/JNEUROSCI.3937-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiebler MA, Bassell GJ. Neuronal RNA granules: movers and makers. Neuron. 2006;51:685–690. doi: 10.1016/j.neuron.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 16.Pacey LK, Doering LC. Developmental expression of FMRP in the astrocyte lineage: Implications for fragile X syndrome. Glia. 2007;55:1601–1609. doi: 10.1002/glia.20573. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs S, Doering LC. Astrocytes prevent abnormal neuronal development in the fragile X mouse. J Neurosci. 2010;30:4508–4514. doi: 10.1523/JNEUROSCI.5027-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Diego Otero Y, Severijnen LA, Van Cappellen G, et al. Transport of Fragile X Mental Retardation Protein via Granules in Neurites of PC12 Cells. Mol Cell Biol. 2002;22:8332–8341. doi: 10.1128/MCB.22.23.8332-8341.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steward O, Bakker CE, Willems PJ, et al. No evidence for disruption of normal patterns of mRNA localization in dendrites or dendritic transport of recently synthesized mRNA in FMR1 knockout mice, a model for human fragile-X mental retardation syndrome. Neuroreport. 1998;9:477–481. doi: 10.1097/00001756-199802160-00022. [DOI] [PubMed] [Google Scholar]
- 20.Dictenberg JB, Swanger SA, Antar LN, et al. A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev Cell. 2008;14:926–939. doi: 10.1016/j.devcel.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng Y, Absher D, Eberhart DE, et al. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell. 1997;1:109–118. doi: 10.1016/s1097-2765(00)80012-x. [DOI] [PubMed] [Google Scholar]
- 22.De Boulle K, Verkerk AJ, Reyniers E, et al. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet. 1993;3:31–35. doi: 10.1038/ng0193-31. [DOI] [PubMed] [Google Scholar]
- 23.Levenga J, Buijsen RA, Rife M, et al. Ultrastructural analysis of the functional domains in FMRP using primary hippocampal mouse neurons. Neurobiol Dis. 2009;35:241–250. doi: 10.1016/j.nbd.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schrier M, Severijnen LA, Reis S, et al. Transport kinetics of FMRP containing the I304N mutation of severe fragile X syndrome in neurites of living rat PC12 cells. Exp Neurol. 2004;189:343–353. doi: 10.1016/j.expneurol.2004.05.039. [DOI] [PubMed] [Google Scholar]
- 25.Laggerbauer B, Ostareck D, Keidel EM, et al. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10:329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- 26.Weiler IJ, Spangler CC, Klintsova AY, et al. Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci U S A. 2004;101:17504–17509. doi: 10.1073/pnas.0407533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narayanan U, Nalavadi V, Nakamoto M, et al. S6K1 phosphorylates and regulates FMRP with the neuronal protein synthesis-dependent mTOR signaling cascade. J Biol Chem. 2008;283:18478–18482. doi: 10.1074/jbc.C800055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ceman S, O’Donnell WT, Reed M, et al. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet. 2003;12:3295–3305. doi: 10.1093/hmg/ddg350. [DOI] [PubMed] [Google Scholar]
- 29.Narayanan U, Nalavadi V, Nakamoto M, et al. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J Neurosci. 2007;27:14349–14357. doi: 10.1523/JNEUROSCI.2969-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishizuka A, Siomi MC, Siomi H. A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev. 2002;16:2497–2508. doi: 10.1101/gad.1022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin P, Zarnescu DC, Ceman S, et al. Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat Neurosci. 2004;7:113–117. doi: 10.1038/nn1174. [DOI] [PubMed] [Google Scholar]
- 32.Lugli G, Torvik VI, Larson J, et al. Expression of microRNAs and their Precursors in Synaptic Fractions of Adult Mouse Forebrain. J Neurochem. 2008;106:650–661. doi: 10.1111/j.1471-4159.2008.05413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edbauer D, Neilson JR, Foster KA, et al. Regulation of Synaptic Structure and Function by FMRP-Associated MicroRNAs miR-125b and miR-132. Neuron. 2010;65:373–384. doi: 10.1016/j.neuron.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greco CM, Navarro CS, Hunsaker MR, et al. Neuropathologic features in the hippocampus and cerebellum of three older men with fragile X syndrome. Mol Autism. 2011;2:2. doi: 10.1186/2040-2392-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Irwin SA, Patel B, Idupulapati M, et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: A quantitative examination. Am J Med Genet. 2001;98:161–167. doi: 10.1002/1096-8628(20010115)98:2<161::aid-ajmg1025>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 36.Comery TA, Harris JB, Willems PJ, et al. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc Natl Acad Sci USA. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irwin SA, Idupulapati M, Gilbert ME, et al. Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am J Med Genet. 2002;111:140–146. doi: 10.1002/ajmg.10500. [DOI] [PubMed] [Google Scholar]
- 38.Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in fmr1 knock-out mice. J Neurosci. 2001;21:5139–5146. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galvez R, Greenough WT. Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am J Med Genet A. 2005;135:155–160. doi: 10.1002/ajmg.a.30709. [DOI] [PubMed] [Google Scholar]
- 40.Grossman AW, Elisseou NM, McKinney BC, et al. Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 2006;1084:158–164. doi: 10.1016/j.brainres.2006.02.044. [DOI] [PubMed] [Google Scholar]
- 41.Antar LN, Li C, Zhang H, et al. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32:37–48. doi: 10.1016/j.mcn.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 42.De Vrij FMS, Levenga J, Van der Linde HC, et al. Rescue of behavioral phenotype and neuronal protrusion morphology in FMR1 KO mice. Neurobiol Dis. 2008;31:127–132. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bakker CE, Verheij C, Willemsen R, et al. Fmr1 knockout mice: A model to study fragile X mental retardation. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- 44.Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, et al. The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol Dis. 2006;21:549–555. doi: 10.1016/j.nbd.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 45.Musumeci SA, Bosco P, Calabrese G, et al. Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia. 2000;41:19–23. doi: 10.1111/j.1528-1157.2000.tb01499.x. [DOI] [PubMed] [Google Scholar]
- 46.Kooy RF. Of mice and the fragile X syndrome. Trends Genet. 2003;19:148–154. doi: 10.1016/s0168-9525(03)00017-9. [DOI] [PubMed] [Google Scholar]
- 47.Bakker CE, Oostra BA. Understanding fragile X syndrome: insights from animal models. Cytogenet Genome Res. 2003;100:111–123. doi: 10.1159/000072845. [DOI] [PubMed] [Google Scholar]
- 48.Zhang YQ, Bailey AM, Matthies HJ, et al. Drosophila Fragile X-Related Gene Regulates the MAP1B Homolog Futsch to Control Synaptic Structure and Function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- 49.Bushey D, Tononi G, Cirelli C. The Drosophila fragile x mental retardation gene regulates sleep need. J Neurosci. 2009;29:1948–1961. doi: 10.1523/JNEUROSCI.4830-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McBride SM, Choi CH, Wang Y, et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile x syndrome. Neuron. 2005;45:753–764. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 51.Tucker B, Richards RI, Lardelli M. Contribution of mGluR and Fmr1 Functional Pathways to Neurite Morphogenesis, Craniofacial Development and Fragile X Syndrome. Hum Mol Genet. 2006;15:3446–3458. doi: 10.1093/hmg/ddl422. [DOI] [PubMed] [Google Scholar]
- 52.den Broeder MJ, van der Linde H, Brouwer JR, et al. Generation and characterization of FMR1 knockout zebrafish. PLoS One. 2009;4:e7910. doi: 10.1371/journal.pone.0007910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 54.D’Hulst C, Kooy RF. The GABA(A) receptor: a novel target for treatment of fragile X? Trends Neurosci. 2007;30:425–431. doi: 10.1016/j.tins.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 55.Chang S, Bray SM, Li Z, et al. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol. 2008;4:256–263. doi: 10.1038/nchembio.78. [DOI] [PubMed] [Google Scholar]
- 56.D’Hulst C, De Geest N, Reeve SP, et al. Decreased expression of the GABA(A) receptor in fragile X syndrome. Brain Res. 2006;1121:238–245. doi: 10.1016/j.brainres.2006.08.115. [DOI] [PubMed] [Google Scholar]
- 57.Jacquemont S, Curie A, des Portes V, et al. Epigenetic Modification of the FMR1 Gene in Fragile X Syndrome Is Associated with Differential Response to the mGluR5 Antagonist AFQ056. Sci Transl Med. 2011;3:64ra61. doi: 10.1126/scitranslmed.3001708. [DOI] [PubMed] [Google Scholar]
- 58.Levenga J, Hayashi S, de Vrij FM, et al. AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol Dis. 2011;42:311–317. doi: 10.1016/j.nbd.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 59.Levenga J, de Vrij FM, Oostra BA, et al. Potential therapeutic interventions for fragile X syndrome. Trends Mol Med. 2010;16:516–527. doi: 10.1016/j.molmed.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dolen G, Carpenter RL, Ocain TD, et al. Mechanism-based approaches to treating fragile X. Pharmacol Ther. 2010;127:78–93. doi: 10.1016/j.pharmthera.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 61.Heulens I, Kooy F. Fragile X syndrome: from gene discovery to therapy. Front Biosci. 2011;16:1211–1232. doi: 10.2741/3785. [DOI] [PubMed] [Google Scholar]
- 62.Hagerman PJ. The Fragile X Prevalence Paradox. J Med Genet. 2008;45:498–499. doi: 10.1136/jmg.2008.059055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tassone F, Beilina A, Carosi C, et al. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA. 2007;13:555–562. doi: 10.1261/rna.280807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tassone F, De Rubeis S, Carosi C, et al. Differential usage of transcriptional start sites and polyadenylation sites in FMR1 premutation alleles. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr100. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hagerman RJ, Leehey M, Heinrichs W, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- 66.Leehey MA. Fragile X-Associated Tremor/Ataxia Syndrome: Clinical Phenotype, Diagnosis, and Treatment. J Investig Med. 2009;57:830–836. doi: 10.231/JIM.0b013e3181af59c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamm C, Healy DG, Quinn NP, et al. The fragile X tremor ataxia syndrome in the differential diagnosis of multiple system atrophy: data from the EMSA Study Group. Brain. 2005;128:1855–1860. doi: 10.1093/brain/awh535. [DOI] [PubMed] [Google Scholar]
- 68.Greco CM, Berman RF, Martin RM, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS) Brain. 2006;129:243–255. doi: 10.1093/brain/awh683. [DOI] [PubMed] [Google Scholar]
- 69.Hagerman RJ, Leavitt BR, Farzin F, et al. Fragile-X-Associated Tremor/Ataxia Syndrome (FXTAS) in Females with the FMR1 Premutation. Am J Hum Genet. 2004;74:1051–1056. doi: 10.1086/420700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jacquemont S, Hagerman RJ, Leehey MA, et al. Penetrance of the fragile x-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291:460–469. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- 71.Garcia Arocena D, Louis ED, Tassone F, et al. Screen for expanded FMR1 alleles in patients with essential tremor. Mov Disord. 2004;19:930–933. doi: 10.1002/mds.20043. [DOI] [PubMed] [Google Scholar]
- 72.Grigsby J, Brega AG, Seritan AL, et al. FXTAS: neuropsychological/neuropsychiatric phenotypes. In: Tassone F, Berry-Kravis EM, editors. The fragile X-Associated Tremor Ataxia Syndrome (FXTAS) New York: Springer; 2010. pp. 31–54. [Google Scholar]
- 73.Greco CM, Soontrapornchai K, Wirojanan J, et al. Testicular and pituitary inclusion formation in fragile X associated tremor/ataxia syndrome. J Urol. 2007;177:1434–1437. doi: 10.1016/j.juro.2006.11.097. [DOI] [PubMed] [Google Scholar]
- 74.Jacquemont S, Hagerman RJ, Leehey M, et al. Fragile X Premutation Tremor/Ataxia Syndrome: Molecular, Clinical, and Neuroimaging Correlates. Am J Hum Genet. 2003;72:869–878. doi: 10.1086/374321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Greco CM, Hagerman RJ, Tassone F, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125:1760–1771. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- 76.Willemsen R, Hoogeveen-Westerveld M, Reis S, et al. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet. 2003;12:949–959. doi: 10.1093/hmg/ddg114. [DOI] [PubMed] [Google Scholar]
- 77.Entezam A, Biacsi R, Orrison B, et al. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. 2007;395:125–134. doi: 10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hashem V, Galloway JN, Mori M, et al. Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum Mol Genet. 2009;18:2443–2451. doi: 10.1093/hmg/ddp182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Entezam A, Usdin K. ATR protects the genome against CGG*CGG-repeat expansion in Fragile X premutation mice. Nucleic Acids Res. 2007;36:1050–1056. doi: 10.1093/nar/gkm1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Entezam A, Usdin K. ATM and ATR protect the genome against two different types of tandem repeat instability in Fragile X premutation mice. Nucleic Acids Res. 2009;37:6371–6377. doi: 10.1093/nar/gkp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Entezam A, Lokanga AR, Le W, et al. Potassium bromate, a potent DNA oxidizing agent, exacerbates germline repeat expansion in a Fragile X premutation mouse model. Hum Mutat. 2010;31:611–616. doi: 10.1002/humu.21237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brouwer JR, Mientjes EJ, Bakker CE, et al. Elevated Fmr1 mRNA levels and reduced protein expression in a mouse model with an unmethylated Fragile X full mutation. Exp Cell Res. 2007;313:244–253. doi: 10.1016/j.yexcr.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brouwer JR, Huizer K, Severijnen LA, et al. CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J Neurochem. 2008;107:1671–1682. doi: 10.1111/j.1471-4159.2008.05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hunsaker MR, Wenzel HJ, Willemsen R, et al. Progressive spatial processing deficits in a mouse model of the fragile X premutation. Behav Neurosci. 2009;123:1315–1324. doi: 10.1037/a0017616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wenzel HJ, Hunsaker MR, Greco CM, et al. Ubiquitin-positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation. Brain Res. 2010;1318:155–166. doi: 10.1016/j.brainres.2009.12.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Van Dam D, Errijgers V, Kooy RF, et al. Cognitive decline, neuromotor and behavioural disturbances in a mouse model for Fragile-X-associated tremor/ataxia syndrome (FXTAS) Behavioural Brain Research. 2005;162:233–239. doi: 10.1016/j.bbr.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 87.Hunsaker MR, Goodrich-Hunsaker NJ, Willemsen R, et al. Temporal Ordering Deficits in Female CGG KI Mice Heterozygous for the Fragile X Premutation. Behav Brain Res. 2010;213:263–268. doi: 10.1016/j.bbr.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Qin M, Entezam A, Usdin K, et al. A mouse model of the fragile X premutation: Effects on behavior, dendrite morphology, and regional rates of cerebral protein synthesis. Neurobiol Dis. 2011;42:85–98. doi: 10.1016/j.nbd.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jin P, Zarnescu DC, Zhang F, et al. RNA-Mediated Neurodegeneration Caused by the Fragile X Premutation rCGG Repeats in Drosophila. Neuron. 2003;39:739–747. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- 90.Galloway JN, Nelson DL. Evidence for RNA-mediated toxicity in the fragile X-associated tremor/ataxia syndrome. Future Neurol. 2009;4:785. doi: 10.2217/fnl.09.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Todd PK, Oh SY, Krans A, et al. Histone Deacetylases Suppress CGG Repeat-Induced Neurodegeneration Via Transcriptional Silencing in Models of Fragile X Tremor Ataxia Syndrome. PLoS Genet. 2010;6:e1001240. doi: 10.1371/journal.pgen.1001240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Garcia-Arocena D, Hagerman PJ. Advances in understanding the molecular basis of FXTAS. Hum Mol Genet. 2010;19:R83–89. doi: 10.1093/hmg/ddq166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tassone F, Iwahashi C, Hagerman PJ. FMR1 RNA withinthe intranuclear inclusions of fragile X-associated Tremor/Ataxia syndrome (FXTAS) RNA biology. 2004;1:103–105. doi: 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- 94.Jin P, Duan R, Qurashi A, et al. Pur alpha Binds to rCGG Repeats and Modulates Repeat-Mediated Neurodegeneration in a Drosophila Model of Fragile X Tremor/Ataxia Syndrome. Neuron. 2007;55:556–564. doi: 10.1016/j.neuron.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sofola OA, Jin P, Qin Y, et al. RNA-Binding Proteins hnRNP A2/B1 and CUGBP1 Suppress Fragile X CGG Premutation Repeat-Induced Neurodegeneration in a Drosophila Model of FXTAS. Neuron. 2007;55:565–571. doi: 10.1016/j.neuron.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arocena DG, Iwahashi CK, Won N, et al. Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum Mol Genet. 2005;14:3661–3671. doi: 10.1093/hmg/ddi394. [DOI] [PubMed] [Google Scholar]
- 97.Sellier C, Rau F, Liu Y, et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. Embo J. 2010;29:1248–1261. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ladd PD, Smith LE, Rabaia NA, et al. An Antisense Transcript Spanning the CGG Repeat Region of FMR1 is Upregulated in Premutation Carriers but Silenced in Full Mutation Individuals. Hum Mol Genet. 2007;16:3174–3187. doi: 10.1093/hmg/ddm293. [DOI] [PubMed] [Google Scholar]
- 99.Garcia-Arocena D, Yang JE, Brouwer JR, et al. Fibroblast phenotype in male carriers of FMR1 premutation alleles. Hum Mol Genet. 2009;19:299–312. doi: 10.1093/hmg/ddp497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ross-Inta C, Omanska-Klusek A, Wong S, et al. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem J. 2010;429:545–552. doi: 10.1042/BJ20091960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen Y, Tassone F, Berman RF, et al. Murine Hippocampal Neurons Expressing Fmr1 gene Premutations Show Early Developmental Deficits and Late Degeneration. Hum Mol Genet. 2010;19:196–208. doi: 10.1093/hmg/ddp479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cunningham CL, Martinez Cerdeno V, Navarro E, et al. Premutation CGG-repeat expansion of the Fmr1 gene impairs mouse neocortical development. Hum Mol Genet. 2011;20:64–79. doi: 10.1093/hmg/ddq432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Aziz M, Stathopulu E, Callias M, et al. Clinical features of boys with fragile X premutations and intermediate alleles. Am J Med Genet. 2003;121:119–127. doi: 10.1002/ajmg.b.20030. [DOI] [PubMed] [Google Scholar]
- 104.Farzin F, Perry H, Hessl D, et al. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27:S137–144. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- 105.Bailey DB, Jr, Raspa M, Olmsted M, et al. Co-occurring conditions associated with FMR1 gene variations: Findings from a national parent survey. Am J Med Genet A. 2008;16:2060–2069. doi: 10.1002/ajmg.a.32439. [DOI] [PubMed] [Google Scholar]
- 106.Coulam CB, Adamson SC, Annegers JF. Incidence of premature ovarian failure. Obstet Gynecol. 1986;67:604–606. [PubMed] [Google Scholar]
- 107.Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, et al. Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study- -preliminary data. Am J Med Genet. 1999;83:322–325. [PMC free article] [PubMed] [Google Scholar]
- 108.Murray A. Premature ovarian failure and the FMR1 gene. Semin Reprod Med. 2000;18:59–66. doi: 10.1055/s-2000-13476. [DOI] [PubMed] [Google Scholar]
- 109.Sullivan AK, Marcus M, Epstein MP, et al. Association of FMR1 repeat size with ovarian dysfunction. Hum Reprod. 2005;20:402–412. doi: 10.1093/humrep/deh635. [DOI] [PubMed] [Google Scholar]
- 110.Allen EG, Sullivan AK, Marcus M, et al. Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum Reprod. 2007;22:2142–2152. doi: 10.1093/humrep/dem148. [DOI] [PubMed] [Google Scholar]
- 111.Brouwer JR, Willemsen R, Oostra BA. The FMR1 gene and fragile X-associated tremor/ataxia syndrome. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:782–798. doi: 10.1002/ajmg.b.30910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hagerman RJ, Hagerman PJ. The fragile X premutation: into the phenotypic fold. Curr Opin Genet Dev. 2002;12:278–283. doi: 10.1016/s0959-437x(02)00299-x. [DOI] [PubMed] [Google Scholar]