

Abstract
This review discusses the most important current methods employing mass spectrometry (MS) analysis for the study of protein affinity interactions. The methods are discussed in depth with particular reference to MS-based approaches for analyzing protein–protein and protein–immobilized ligand interactions, analyzed either directly or indirectly. First, we introduce MS methods for the study of intact protein complexes in the gas phase. Next, pull-down methods for affinity-based analysis of protein–protein and protein–immobilized ligand interactions are discussed. Presently, this field of research is often called interactomics or interaction proteomics. A slightly different approach that will be discussed, chemical proteomics, allows one to analyze selectivity profiles of ligands for multiple drug targets and off-targets. Additionally, of particular interest is the use of surface plasmon resonance technologies coupled with MS for the study of protein interactions. The review addresses the principle of each of the methods with a focus on recent developments and the applicability to lead compound generation in drug discovery as well as the elucidation of protein interactions involved in cellular processes. The review focuses on the analysis of bioaffinity interactions of proteins with other proteins and with ligands, where the proteins are considered as the bioactives analyzed by MS.
Figure.
Approach for analysis of protein complexes with MS
Keywords: Mass spectrometry, Protein–protein interactions, Chemical proteomics, affinity, Native mass spectrometry and interaction proteomics
Introduction
This review addresses mass spectrometry (MS) methods for the study of (intact) protein complexes and so-called protein–protein-interaction-based pull-down strategies to elucidate these bioactive interactions. In addition, it discusses automated digestion steps after affinity purifications and surface plasmon resonance technologies coupled with MS. The methods described are used mainly in two research areas. Firstly, they are used in fundamental studies of protein interactions with other proteins and with small molecules [1]. This is an important research area aimed at gaining a better understanding of biological processes in general and more specifically their cellular processes. Looking more elaborately at cellular processes, signaling cascades involve many coordinated multiprotein binding events, production and metabolism of signaling molecules, modifications of proteins, and binding of small signaling molecules to proteins [2]. These processes facilitate the cellular machinery needed for homeostasis and, for example, allow coordinated tissue growth. Furthermore, other processes such as cellular localization of proteins and cellular morphology processes are mediated by protein binding events and are pivotal for cells and the functioning of the body. Secondly, the study or screening of small molecules that bind to proteins can also be used to screen for hits or lead compounds in drug discovery. With the emergence of protein–protein interactions and biopharmaceuticals in drug discovery and development, screening and studying these interactions are becoming increasingly important in this research area as well [3].
The different distinct MS approaches which are discussed in this review are as follows. First, the study of noncovalent complexes directly from solution by MS (native MS). The second topic is the study of cellular protein complexes involved in signaling events by pull-down (“fishing”)-based interaction proteomics. Third, chemical proteomics is looked at; this allows one to screen selectivity profiles of ligands for multiple drug targets and off-targets. Finally, surface plasmon resonance (SPR) coupled with MS for protein affinity analysis, quantification, and identification is discussed. Many reviews have been published about the four distinct approaches that are discussed here. This review, however, provides an overview of the different MS-based approaches with recent examples and focuses on the protein binding aspects and methods behind them in relation to biological binding events and less on the MS technologies. For every method, recent applications and specific examples of studies performed are briefly discussed.
Electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI) are the most suitable methods for generating gas-phase ions of large biomolecules. In the case of ESI, many different intact noncovalent protein complexes can be studied in the gas phase under certain conditions. These studies are typically called native MS studies. They also allow one to study protein–ligand complexes (receptors as well as enzymes). Ganem et al. [4] and Katta and Chait [5] were among the first to use ESI-MS to study noncovalent receptor–ligand complexes and biological myoglobin interactions. For the analysis of noncovalent complexes by MS, a great variety of biological interactions have been studied: receptor–ligand, enzyme–substrate, DNA duplex and quadruplex species, intact multimeric proteins, host–guest, oligonucleotide–ligand and protein-ligand complexes, and protein–protein complexes [5–14]. Even intact virus assemblies are currently analyzed with native MS approaches [15].
The advantage of native MS for structural-biology-oriented studies compared with other approaches, such as crystallography, protein nuclear magnetic resonance (NMR) and isothermal titration calorimetry, is the possibility to look directly at protein–protein and protein–ligand interactions in solution. This permits scientists to rapidly effectuate changes (e.g., add ligand or protein) to the in vitro system and thereby study directly the effects on the protein complexes dynamically under real-time conditions. Also, MS allows the study of extremely large protein complexes and even virus assemblies, which is out of scope for, e.g., crystallography and protein NMR. Furthermore, for MS approaches in the native MS area, only low amounts of proteins are needed as they are commonly introduced via a nano-ESI source. With sufficient analytical resolution, the successful analysis of picomolar amounts of large heterogenous protein complexes becomes reality. In addition, it allows real-time addition of cofactors, substrates, and ligands while monitoring the resulting changes to the complexes. This contrasts with the limited possibilities of other methods used for analyzing the stoichiometry of protein complexes, such as crystallography and protein NMR. To obtain sufficient MS signals, however, high protein concentrations (e.g., generally low micromolar concentration range, while only consuming low volumes introduced via nano-ESI capillaries) are required because the overall MS signal is divided over isotopic patterns and differently charged protein complexes.
The study of noncovalent complexes directly by MS relies on extensive optimization to obtain sufficiently stabile protein–protein or protein–ligand complexes in solution and in the gas phase. Furthermore, for the study of protein–ligand interactions it is mandatory to distinguish the protein–ligand complex from the unbound protein without a separation step. Presently, progress in the development of MS and its application to protein analysis in the gas-phase have led to major improvements in this field. Also, these methods provide the most direct evidence of protein–ligand and protein–protein complexation and can be used as a model for in vivo complexation, with the caveat that gas-phase complexation in the mass spectrometer is a good representation of in vivo binding, and is not an analytical artifact caused by the analysis [16]. Contrary to ESI, MALDI techniques are less suited for the study of noncovalent protein–protein and protein–ligand interactions as these interactions are often disrupted under the conditions needed and the procedures followed to produce the MALDI matrix.
A distinctly different way of analyzing protein–protein interactions is performed indirectly after so-called pull-down assays. For this, the protein (or ligand) of interest is immobilized to create an affinity column in a manner similar to that for affinity chromatography approaches. After all bound proteins have been trapped and subsequently released, MS analysis is often performed after a 1D gel electrophoresis separation and a (tryptic) digestion of the separated proteins [17]. Instead of a normal affinity-chromatography-based protein complex purification, immunoprecipitation [18] or tandem affinity purification [19] can be used. The protein-“fishing”-based MS approaches discussed in this review are not suited for the dynamic study of protein binding events, but rather allow the identification of large multiprotein complexes involving many different proteins.
Finally, SPR has been coupled with MS to study protein binding events on an SPR chip directly followed by MS identification of the bound proteins. This approach allows protein quantification combined with structural characterization/identification of the proteins. Consequently, MS complements the SPR detection and may reveal structural modifications not detected by SPR [20].
The study of noncovalent complexes by native MS
Analysis of noncovalent complexes by MS, also known as native MS, requires ESI-compatible buffer solutions. This implies that in a number of cases maximum sensitivity is not achieved and/or nonphysiological conditions have to be used. Although noncovalent complexes observed by MS are not analyzed directly from real cellular systems, most often the stoichiometry of complexes determined by native MS matches that determined in other ways, such as electron microscopy, X-ray crystallography, and NMR. There are, however, a few recognized exceptions [21].
In general, native MS is a very powerful technique for the study of protein complexes, complementary to more traditional approaches. Other approaches such as crystallography and NMR have different analytical capabilities. Whereas crystallography allows a detailed 3D image of a protein–ligand complex to be obtained, the analysis of multiprotein complexes, large protein complexes, and several types of protein classes in general is difficult or the protein complexes may be impossible to crystallize. Furthermore, crystallography only permits the analysis of a static crystal and real-life dynamic analysis in vitro is therefore not possible. Protein NMR, on the other hand, is an emerging technology but the study of large and complex protein structures, such as whole virus assemblies, is still not feasible. For protein NMR, protein sizes in the 0.1–1-MDa range have been studied [22, 23]. With NMR studies, homomeric complexes are analyzed more conveniently than heteromeric protein complexes as the NMR spectra then become much more difficult to interpret, which is not an issue with native MS. Currently, among the largest protein structures resolved by protein NMR are the 300-kDa aspartate transcarbamoylase [24] and the 670-kDa proteasome [23] obtained by the group of Kay. With native MS, among the currently largest protein complexes (over 10 MDa) studied is the Norwalk virus assembly [15].
All technologies used to study protein complexes have their own intrinsic advantages and disadvantages. In this regard, MS allows the dynamic real-life study of protein complexes, the study of very large complexes, and the stoichiometric determination of the complexes analyzed, and importantly only requires small amounts of protein. In contrast, high-resolution 3D protein structures cannot be determined by MS techniques.
Advances in MS, however, do provide new opportunities regarding the analysis of these complexes. One can think of advanced nano-ESI sources, new ionization techniques such as ambient temperature ionization, and implementation of ion mobility spectrometry (IMS) technologies, but also new and adapted MS configurations and hardware [25]. Still, the buffers used for biochemical studies that mimic physiological conditions often contain phosphates and other nonvolatile salts [e.g., phosphate-buffered saline or tris(hydroxymethyl)aminomethane buffer with NaCl] and cannot be used in combination with ESI-MS because they are nonvolatile. Also, the use of a low pH for efficient ESI-MS in positive ionization mode is not an option when studying biological noncovalent complexes, nor is the commonly used high percentage of organic modifier. Instead, physiological buffer conditions have to be substituted with MS-compatible buffers, such as ammonium formate, acetate and bicarbonate. Furthermore, the percentage of methanol, acetonitrile, or 2-propanol has to be low (usually lower than 5%) to prevent dissociation or denaturation of the noncovalent complexes to be studied. Finally, nonvolatile additives such as detergents and blocking reagents may cause ionization suppression, and their use should be avoided or they should only be used in very low concentrations. Another factor to take into account is that the complexes that are formed and studied in native MS depend on both protein–protein and protein–ligand affinities and their concentrations. For most proteins (and ligands), much higher concentrations have to be used than those present under physiological conditions. Therefore, one must be aware of the physiological relevance of the complexes studied as in the body the concentrations of proteins studied are much lower. This implies that low-affinity protein complexes may be seen under the artificial conditions with high protein concentrations in the mass spectrometer, but might have less relevance in the body (or only under specific conditions) when they are not formed or are only formed at very low percentages.
A way to study these possible effects might be by analyzing the protein complexes in different ratios and concentrations and by omitting specific binding partners. Although analysis of the protein complexes in lower concentrations will give a worse signal-to-noise ratio, observed changes in the ratios of the complexes seen might give indications of the affinities of the different binding partners in these complexes. Also, chemical cross-linking at lower protein concentrations followed by analysis of the complexes under denaturing conditions can by utilized to verify if the complexes are relevant at low concentration. In-solution dissociation experiments can also give valuable additional information about the binding interactions of the interaction partners. Nonspecific oligomerization, for example, can be distinguished from specific interactions by looking at the distribution of the molecules, which is related to the initial concentration and droplet sizes in the ESI source [26]. All these considerations dictate the balance required between efficient and representative analyses.
ESI-MS can be seen as a complementary tool to established biochemical methods for investigating protein structure and conformation under nondenaturating conditions. Types of information that can be obtained by ESI-MS include protein conformation properties and molecular interactions, protein–protein interactions, protein–ligand interactions, and protein–cofactor interactions. Some typical examples are now briefly discussed.
Noncovalent interactions between low molecular weight antiamyloid agents and amyloid β peptides were studied by Martineau et al. [27] to rank binders that may be able to modulate/inhibit the amyloid β aggregation process. Jecklin et al. [28] compared different approaches, ESI-MS, SPR, and isothermal titration calorimetry, for label-free quantitative assessment of binding strengths of the protein human carbonic anhydrase I with small ligands. Real-time monitoring of enzymatic conversions and inhibition and formation of complexes is also possible with MS [29]. Hydrogen–deuterium exchange experiments can be used to study proteins by MS and allow one to monitor protein dynamics and binding interactions over time [30]. Bich et al. [31] applied MS to study the retinoic acid induced heterodimerization of the nuclear retinoid X receptor (RXR), resulting in formation of an activated dimer that binds to DNA hormone response elements, mimicked by DNA-based direct repeat configurations. This is exemplified in Fig. 1. The dots in the MS spectrum represent the m/z values of the differentially charged ions corresponding to the complex of the RXR–retinoic acid receptor dimer bound to the double-helix DNA fragment DR5. The dots on top of the peaks represent the same ions with different charge states. After cross-linking, the authors also successfully studied these complexes with high-mass MALDI-MS. Van Duijn et al. [12] have used native MS for the analysis of complexes involved in the chaperonin-assisted refolding of the major capsid protein (gp23) of bacteriophage T4. Intermediate complexes that are involved in chaperonin (GroEL-GroES) folding were studied as such. It was found that chaperonin complexes can bind up to two unfolded gp23 proteins. When in complex with the cochaperonin gp31, only one gp23 can bind. Figure 2 shows typical results obtained for this study. Ions with different charge states corresponding to the 801-kDa complex (GroEL; blue dots), the 857-kDa complex (one gp23 molecule bound to GroEL; yellow dots), and the 913-kDa complex (two gp23 molecules bound to GroEL; red dots) are seen in Fig. 2a. Figure 2b shows the deconvoluted spectrum of the three different complexes. These results nicely illustrate the capabilities of native MS for the study of protein–protein interactions.
Fig. 1.

Interaction of the nuclear hormone receptor dimer retinoic acid receptor (RAR) − retinoid X receptor (RXR) binding to a short strand of DNA (induced by retinoic acid binding). All RXR heterodimers preferentially bind DNA at two sites of a direct repeat (DR) configuration, separated by one to five nucleic acids, called DR1,DR2, DR3, DR4, and DR5. The binding to a DR5 configuration is shown, resembling binding to hormone response elements (HREs). Upon binding to actual HREs, gene transcription can occur. (Reprinted from Bich et al. [31]. With permission)
Fig. 2.

Typical native mass spectrometry (MS) results from van Duijn et al. [12] a Nano-electrospray ionization mass spectrum of a mixture of GroEL and unfolded polypeptide gp23 (1:4). Charge-state series of GroEL (800 kDa; blue circles), one gp23 molecule bound to GroEL (856 kDa; yellow circles) and two gp23 molecules bound to GroEL (912 kDa; magenta circles) are seen. b The corresponding deconvoluted spectrum, which reveals the three chaperonin complexes with their binding stoichiometries. (Reprinted from van Duijn et al. [12]. With permission)
Following the advent of native MS, very large protein complexes such as ribosomes and even whole viruses can now be studied in the gas phase [32–34]. With the recent addition of ion mobility to MS analysis, new doors have been opened for the study of such large complexes [35]. In IMS, biomolecules and noncovalent complexes are separated in the gas phase according to their differences in size, shape, and charge prior to actual MS analysis. For IMS, new possibilities lie, for example, in the analysis of heterogeneous protein complexes, providing information on the topology, stoichiometry, and cross section. This new addition to the available MS tools does, however, require additional and extensive data handling to have feasible data interpretation [36]. Research has already shown that results obtained by IMS-MS for noncovalent complexes show good correlation with results obtained by traditionally applied methods, such as cryoelectron microscopy and X-ray crystallography [37]. However, there is also evidence that proteins and protein complexes may become more compact or collapse in the gas phase in the absence of water [38]. Besides the technologies mentioned, electron microscopy is an alternative method to MS to look at protein complexes, and also protein complexes consisting of many different proteins, provided that the complexes are very large [39]. With crystallography approaches, often ligand binding to a receptor or enzyme is studied.
A selected set of very recent typical examples of studies involving native MS are now discussed. By combining IMS-MS with tandem MS, one can characterize non-covalently bound macromolecular complexes (mass, cross-sectional area, and stability) with only one experiment, which was demonstrated by Knapman et al. [40] by determining the topology of virus assembly intermediates. Boeri-Erba et al. [41] used IMS-MS to study the influence of subunit packing and the charge on the dissociation of multiprotein complexes of heat shock protein 16.9 and stable protein 1. Also, native MS can be used to identify protein aggregates after (size-exclusion) chromatography, which was demonstrated for human monoclonal antibody aggregates [42]. Recent advances in native MS, including specifically applied surface-induced dissociation approaches, allow one to get a closer look at quaternary structures of protein complexes [43]. The central glycolytic genes repressor (CggR) plays a role in glycolysis in Bacillus subtilis. The effector sugar fructose 1,6-bisphosphate (FBP) abolishes binding cooperativity of CggR and DNA. Atmanene et al. [44] used native MS to investigate FBP-dependent CggR–DNA interactions using automated chip-based nano-ESI MS and traveling wave IMS-MS. Among others findings, it was revealed that tetrameric CggR dissociates into dimers upon FBP binding. In a more recent study, the assembly states of the nucleosome assembly protein 1 were studied by sedimentation velocity and native MS [45]. From this it was concluded that the basic assembly was a dimer from which even-numbered higher-assembly states formed. Phosphatidylethanolamine-binding protein (PEBP) can be associated with morphine and morphine glucoronides. In a native MS study, Atmanene et al. [46] characterized these interactions and finally suggested that PEBP might protect morphine 6-glucuronide following its secretion into blood, which leads to a longer half-life. The ribosomal stalk complex has a role in the delivery of translation factors to the ribosome. The stoichiometry of these complexes is important to further understand their functioning, which was investigated by Gordiyenko et al. [47].
The study of cellular protein complexes involved in signaling events with pull-down-based interaction proteomics
For many drug target systems, genomics approaches, e.g., by RNA array analysis of gene expression, have revealed that receptor stimulation results in numerous pathway regulations. More recent advances in proteomics also allow not only the unraveling of the complex protein regulations mediated by ligand signaling, but moreover give insights into protein phosphorylation processes that precede this. These processes are efficiently studied with, e.g., phosphoproteomics approaches based on phosphopeptide purification with affinity chromatography [with, e.g., immobilized metal affinity chromatography (IMAC) or TiO2] performed after proteolytic digestion of all proteins. The affinity-purified phosphopeptides are then separated by liquid chromatography (LC) and analyzed by MS [48].
We are starting to understand that ligand-mediated signaling is not a one-directional linear process, but rather a parallel process with different dependent, independent, cross-linking, correlating, and influencing pathways and eventual effects [49–51]. Protein kinases play pivotal roles in transmitting ligand-mediated signals through many different pathways. Before eventual gene expression in cells takes place, phosphorylation and dephosphorylation steps of subsequent protein kinases and other proteins occur, thereby activating and/or deactivating them, in order to pass on signaling events. Also, protein localization processes, e.g., based on phosphorylation state, are important mediators in (localized) cellular processes. Finally, key proteins in different pathways are upregulated or downregulated and dictate the final cellular (desired or undesired) effects. Through all these signaling cascades, protein complexes play crucial roles. In other words, the way that proteins interact with each other, form noncovalent complexes, and localize, internalize, and recruit other proteins is fundamental to cellular signaling [49, 52, 53]. Some typical examples include G-protein-(in)dependent and/or β-arrestin signaling for G-protein-coupled receptors (GPCRs), coactivator and/or repressor protein recruitment for the nuclear receptors, and localization of certain proteins (e.g., protein kinases and GPCRs) by binding/complexation to/with, e.g., anchoring proteins [50, 51, 54–57]. These important processes in signaling are difficult to study with traditional biological/biochemical approaches as they comprise a complex interplay of many different events. One relatively new way of studying these complexes in a more comprehensive manner is by interactome proteomics.
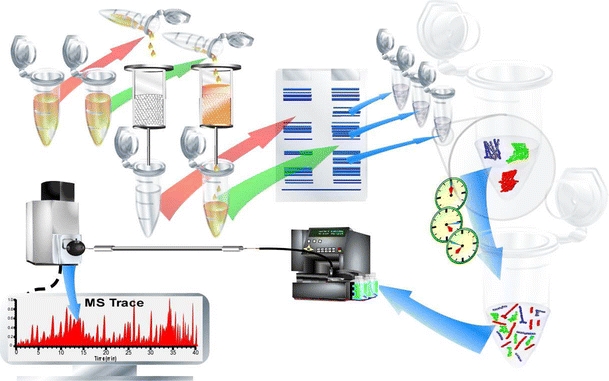
In interactome proteomics, an affinity purification of the protein complexes to be studied precedes the actual analysis. The affinity purification uses the key interactor to be studied for its interactome in a relevant biological surrounding. Here, the key interactor can be a ligand, an inhibitor, a protein, DNA, RNA, or another biomolecule, whereas the biological surroundings are often lysates of (cultured) cells, but also tissues, subcellular compartments, organs, and insects have been studied this way. The key interactor is immobilized onto a solid support, such as a (spin) affinity column or (magnetic) beads. When the key interactor is incubated with the lysate, complexes are formed with the key interactor under the conditions used for the study, thereby selectively extracting or “fishing” the interacting proteins from the complex mixture. Figure 3 gives an overview of a typical interaction proteomics workflow. The exemplary procedure depicted starts with an affinity purification step (2a and 2b) of the sample (1a) and the control (1b). After the affinity purification, the proteins bound are separated on a gel (3a and 3b). The two experiments can be performed in one experiment if, e.g., a stable isotope labeling with amino acids in cell culture (SILAC) is used. Subsequently, protein bands are excised and digested in-gel. Then, MS-based analysis occurs. In the example shown, nano-LC-MS is used for peptide separation and analysis. This approach is essentially based on the reversed principle of affinity-selection MS approaches discussed by Jonker et al. [58]: Rather than immobilizing the target proteins to retain ligands, one immobilizes the ligands to retain the target proteins. Subsequent washing away of all nonbinding entities (e.g., proteins, membranes, and small molecules) followed by release of all binding proteins (by, e.g., heat shock, pH shock, chaotropic agents, organic modifiers, ligand-based displacement, or tryptic digestion when a bottom-up approach is used) allows the proteomic analysis of the interactome. Here it is mandatory that the cell lysate used is carefully prepared in order to maintain the cellular conditions in terms of the interactome/noncovalent complexes under study. Cellular conditions or cell-mimicking conditions are easily disturbed chemically by buffer conditions/lysis conditions, as well as biologically and are cell-mediated during the initiation of, e.g., cell lysis. Furthermore, often cellular conditions are unknown (and cannot be specified for different cell compartments) and thus it is difficult to mimic them. The use of homogenized tissues or even whole organs can be problematic. First, to investigate cellular pathways it is important to know the corresponding type of cell to which the proteins belong. An association of different cell types should only be used to give the first hints. Furthermore, if different cell types are used in one batch, uncontrolled reactions may be initiated. It is difficult to control changes occurring when only one type of cell is homogenized because cell compartments are destroyed. For example, disrupting the vacuole might cause significant artefacts in the study of plant cells.
Fig. 3.

Pull-down proteomics, interactomics, or interaction proteomics. The typical workflow is illustrated. The sample to be analyzed (1a) and often a control sample (1b) are subjected to affinity chromatography (2a and 2b). Alternatively, immunoprecipitation can also be used (not shown). After trapping, all bound proteins are eluted (by, e.g., disruption or sequential elution with affinity displacers) and (often) subjected to gel electrophoresis (3a and 3b). Sample and control proteins are compared and all (or only interesting) proteins are excised from the gel slab, and are in-gel digested (in gel bands) (4). The resulting peptides can then be separated by (nano)-liquid chromatography (LC) (6) and detected by MS (7). Alternatively, matrix-assisted laser desorption ionization MS may also be used. The resulting total ion current of a chromatogram (8), the MS spectrum of a peptide (9), the MS/MS spectrum of the fragmented peptide (10), and a database search (11) are shown
Alternatively, immunoprecipitation can be performed instead of interactome fishing with immobilized key interactors. An advantage of immunoprecipitation is the possibility of in-solution incubation, which can avoid steric hindrance of (co-)binders to the complexes formed with the key interactor when it is not immobilized (the indirect approach). A limitation is that the method depends on the specificity of the antibodies used, the choice of the antigen, and the accessibility of the antibodies for binding the antigens when the complexes are formed in solution.
With interactome proteomics, a control experiment is often compared with the actual experiment in which the biological matrix under study is stimulated by a certain chemical, biological, or physical stimulus. For this approach, both biological matrixes can be isotopically labeled. Although this is an elegant way of incorporating the control experiment and stimulated experiment in one analysis with a labeling approach, controls can be performed without isotopic labeling. The advantage of labeling is that both the control and the stimulated experiments can be pooled and analyzed in one simultaneous proteomics experiment where the peptide ratios used to calculate protein ratios can be distinguished in MS owing to the differences in molecular masses between the differently labeled peptides. Labeling can be achieved in situ during cell growth via SILAC for proteomic comparison of the stimulus’ effect later on [59, 60]. Other applicable labeling approaches can be performed, such as isobaric tags for relative and absolute quantitation (iTRAQ; based on a combination of 18O, 15N, and 13C to achieve isobars) [61], isotope-coded affinity tags (ICAT) [62], labeling with 18O [63], and [2H6]dimethyl labeling [64]. These labeling approaches have to be performed after the interactome fishing process, that is, after or during a digestion step of the isolated proteins, in contrast to SILAC. The actual proteomics experiment can be performed in a bottom-up approach in which first all proteins are digested, followed by LC-MS analysis and database searching [48]. The common approach involves protein separation by 1D sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Here, gel bands are subsequently excised, in-gel digested, and the peptides formed are then analyzed by LC-MS (or potentially MALDI-MS). An advantage of this approach is the possibility for additional Western blotting for confirmation of the identity of specific proteins. Also the molecular mass of the excised proteins in denaturating gels can be estimated, thus yielding additional confirmation. A disadvantage is that this method is quite labor-intensive.
An example of a typical pull-down proteomics study involves the use of an immobilized inhibitor for phosphodiesterase 5 to study its interactome [65]. To gain specificity, selective precleaning and elution protocols were developed for efficient discrimination between specific and nonspecific or less-specific binding proteins. A similar approach was used with cyclic AMP affinity column materials to study a specific protein kinase anchoring protein for type I cyclic AMP dependency [66]. This protein plays important roles in localization processes for specific kinases during complex interplays of signaling events. For studying protein–protein complexes, a method was developed in which the “bait protein” was constructed as a glutathione S-transferase fusion protein for interactome pull-down chromatography with glutathione beads. These pull downs can be envisioned as the protein-based version of yeast two-hybrid screens [67]. Prior to eventual MS analysis and data handling for protein identification, different sample preparations of interacting proteins (e.g., specialized gel staining techniques and in-gel tryptic digestions) were evaluated and used. An example of immunoprecipitation pull downs is given for GTP cyclohydrolase I, which is an important enzyme in the biosynthesis of tetrahydrobiopterin, an essential cofactor for aromatic amino acid hydroxylase and nitric oxide synthase [68]. It was found that 29 proteins from different subcellular components interacted with GTP cyclohydrolase I. In an example where affinity protein columns were manufactured for the pull down, proteins targeted by the thioredoxin superfamily in Plasmodium falciparum were identified, yielding 21 potential target proteins [69]. Another example targeted the phosphatidylinositol 3,4,5-trisphosphate interactome [70], important in regulations of cell physiological processes, e.g., via GPCR-mediated signaling. In this study, 282 proteins were found to directly or indirectly interact with phosphatidylinositol 3,4,5-trisphosphate.
The family of 14-3-3 proteins are regulatory proteins conserved across species with the ability to bind many different proteins involved in signaling, such as kinases and membrane receptors. The importance of these proteins in signaling processes renders them excellent candidates for interactomics studies helping to unravel their exact binding partners. This is true not only for mammalian 14-3-3 proteins, but also for plant proteins. Paul et al. [71] studied Arabidopsis 14-3-3 complexes, which revealed highly conserved interactions between humans and plants. Among other important proteins, also in plants, are the protein kinases. One study focused on transgenic rice plants to identify binders to rice-leaf-expressed protein kinases fused to tandem affinity purification (TAP) tags [19]. TAPs are two-step affinity purification protocols which allow isolation of protein complexes under close-to-physiological conditions with the help of fusion proteins. These fusion proteins can have a “bait” part, an enzymatic cleavage part, and a trapping part, for example, protein A which binds to immobilized IgG. After initial purification, the TAP tag can be broken enzymatically for further processing and eventual MS analysis. One manner of efficient quantification of proteins after affinity trapping procedures is the recently developed quantitative bacterial artificial chromosomes interactomics [72]. In this approach, tagged full-length baits are employed which are expressed under endogenous control. Different cell lines with tagged proteins are available for this approach. Actual quantification occurs by SILAC, but it can also be performed by a label-free approach.
The α7 nicotinic receptor, which is an important potential drug target against several brain-residing diseases, has also been studied indirectly with an interactomics approach. Bungarotoxin, which has a high affinity for this nicotinic receptor, was used as a key binding partner. For this, isolated carbachol-sensitive α-bungarotoxin-binding complexes from total mouse brain tissue were selectively eluted and analyzed [73]. By comparison of results obtained from wild-type mice and from α7 nicotinic receptor knockout mice, binding proteins were identified from the brain tissues used.
The importance of interactomics studies in life science today is reflected by the many different studies performed to investigate the binding partners of specifically selected interaction proteins. Other recent examples include an interactomics study towards the most widely expressed isoforms of p63, a transcription factor for the p53 tumor suppression protein [74]. Relevant binding partners of helicases, which are important in the unwinding of the strands of DNA double helixes, have been studied by Jessulat et al. [75] by a TAP approach in an in vivo study. Another TAP approach, by Guo et al. [76], centered on the identification of human tuberous sclerosis protein 1 complexes. In the case of binding partners of the estrogen receptor as a key mediator in certain breast cancer cells, knowledge of binding partners of the ligand-activated receptor is important for a better understanding of transduction of the hormonal signal that allows the cancer cells to grow. These binding partners were revealed in an interaction proteomics study using TAP by Tarallo et al. [77]. Integrins are transmembrane proteins that are involved in regulation of cellular mobility, shape, and cell cycle processes. To look closely at associated proteins that might be involved in these processes, Raab et al. [78] looked at the interactome of the platelet integrin αIIb regulatory motif. The use of tethered RNAs to detect RNA–protein interactions was described by Lioka et al. [79], revealing specific protein binders. The nuclear lamina is among other factors an important regulator of the structural integrity of the nucleus. It is involved in nuclear processes, including DNA replication. Unraveling of binding partners of the lamina can allow scientists to further understand the processes behind the regulations involved. For this, protein interactors with lamin A and progerin were studied by Kubben et al. [80]. The immune adapter protein adhesion and degranulation promoting adapter protein (ADAP) is involved in integrin-dependent migration and adhesion processes after T-cell stimulation. To investigate and differentiate between phosphorylation-specific and nonspecific protein interactions, Lange et al. [81] used SILAC and enzymatic 18O-labeling to identify ADAP interaction partners. Jäger et al. [82] described an affinity purification method to characterize HIV protein complexes. The interaction partners of dysferlin, an important protein involved in muscle membrane repair, were recently also studied [83]. It was shown that dysferlin is not only involved in membrane repair, but that it is also important for maintenance and integrity of muscle membranes. For proteins that interact with muscarinic receptor, Borroto-Escuela et al. [84] revealed many protein interactions in various signaling pathways that will allow a better understanding of the muscarinic interactome.
An alternative, attractive approach uses protein trapping with reactive chemical affinity tags which efficiently traps proteins for MS-based analysis. Fischer et al. [85] and Luo et al. [86] used this so-called capture compound MS approach which involves binding of a small reactive molecule (e.g., a druglike compound) to interacting proteins followed by covalent reaction (e.g., after photoactivation) with the binding proteins. An incorporated biotin function then allows selective purification for MS analysis. In a typical example, the broad-range and high-affinity protein kinase binder staurosporine was used to trap and study protein kinases in the hepatocyte cell line HepG2 [87]. To study chemical cross-linking of covalently connected binding partners in order to allow identification of interacting proteins directly in cells, Sinz [88] discussed different in vivo cross-linking strategies that allow protein–protein interactions to be looked at under physiological conditions. Other capture compound MS approaches include the profiling of methyltransferases and S-adenosyl-l-homocysteine-binding proteins [89] and the use of a genetically incorporated photo-cross-linkable amino acid to study protein complexes of protein 2 bound to mammalian growth factor receptor [90].
Chemical proteomics to screen selectivity profiles of ligands for multiple drug targets and off-targets
Affinity beads or columns can be used in a more pharmaceutically oriented fashion than interactome proteomics. Here, an additional step is included involving the addition of different concentrations of a ligand (e.g., lead compound) to cell lysates prior to processing. This technology is the so-called chemical proteomics approach [91]. Figure 4 gives an overview of a typical chemical proteomics approach. In the exemplary figure, Petri dishes with cells are shown (1a, 1b, 1c, and 1d), each incubated with a different stimulus (different ligand concentrations in this case). After cell lysis and sample preparation, the lysed cells are incubated with affinity beads. The proteins bound for each incubation (2a-d) are subsequently isolated by washing the beads (3 to 4) followed by elution with help of, e.g., a disruption step (5a to 5b). Different approaches for protein separation can be used prior to analysis (10a to 10b, or potentially 11a to 11b). iTRAQ labeling is performed, allowing the different experiments (1a, 1b, 1c, and 1d) to be combined after digestion and labeling. With this approach, the samples can then be pooled prior to LC-MS analysis (9). The technology uses affinity beads with immobilized ligand to fish out target proteins for MS-based analysis. By addition of solution-phase test ligands of pharmaceutical interest (which bind with different affinities to the target proteins), the target proteins, when bound to the test ligands, do not bind anymore or bind in a lower percentage to the beads, depending on their intrinsic test ligand affinities. For specific interactions, this results in a reduced amount of target protein extracted and consequently lower amounts quantified per target protein by MS. Using this approach, proteins complexing with the target proteins bound are not looked at, but can theoretically be looked at if desired.
Fig. 4.

Chemical proteomics. The typical workflow is shown. The sample with different concentrations of the test ligand (four concentrations; 1a, 1b, 1c, and 1d) are subjected to lysates of cultured cells of interest and incubated with immobilized ligand affinity beads (2a-d). After incubation (3), the affinity beads with bound target proteins are removed from the incubation (4), washed, resuspended (5a), and finally a disruption step (5b) releases the bound target proteins from the beads. The crude protein mixtures are then prepared for 1D (10a) or potentially 2D (11a) gel electrophoresis followed by excision of the proteins (10b, 11b). The proteins can then be in-gel digested prior to analysis. Alternatively, proteins can be directly prepared for digestion (6), digested (7 and 8) and subsequently labeled with, e.g., isobaric tags for relative and absolute quantitation (iTRAQ). After iTRAQ labeling, samples are combined for straightforward eventual relative protein quantification from MS/MS spectra (see Fig. 5) obtained by LC-MS/MS analysis (9)
A well-known example uses affinity beads (Kinobeads) that are able to trap most, if not all, protein kinases via their binding pocket(s) [92]. The approach uses immobilized broad-selectivity kinase inhibitors that bind protein kinases (and related proteins) mainly at their ATP binding sites and related sites. In the presence of increasing concentrations of a ligand, the ligand and the affinity material compete for a binding site on the protein kinases present in cellular lysates. This means that at low ligand concentrations, only the high-affinity-binding kinases are not trapped on the affinity material anymore as they are bound to the ligand, whereas at higher ligand concentrations, also the lower-affinity kinases are unable to bind to the affinity material anymore. Experiments with different ligand concentrations are done and after pull downs followed by proteomics analysis, decreasing amounts of affinity-material-trapped kinases are detected with increasing ligand concentrations. This is used to construct typical IC50 dose–response curves for all kinases studied (up to hundreds at once). There are 518 human protein kinases, and all protein kinases that bind can theoretically be detected as can other proteins that interact with the affinity material. Of course, their individual cellular concentrations and activation states in specific cell types might prevent binding and/or detection. For this specific process, medically interesting target cells are used where the endogenous protein kinases are the target proteins. After lysis, the ligand is added, followed by affinity trapping of the protein kinases, washing steps, release and digestion of the protein kinases, labeling, and finally LC-MS. An iTRAQ labeling reagent is commonly used for this [92, 93]. The method also allows for measurement of ligand-induced changes in phosphorylation states of the isolated protein kinases. The main advantage of this method is that it is capable of analyzing inhibitory panel profiles of protein targets instead of aiming at a single drug target. This possibility allows drug discovery projects to start aiming at drugs capable of selectively inhibiting several drug targets in a panel fashion. One challenge in protein kinase affinity screening is that the most interesting selective inhibitors for protein kinases are expected to bind allosterically (at non-ATP binding sites) and consequently might not be detected. These methods, however, are also starting to aim more specifically at multiple binding sites.
To give one example, a quantitative chemical proteomics approach was used to study the effects of small molecule ABL kinase inhibitor drugs on hundreds of endogenously expressed protein kinases and purine-binding proteins [92]. Furthermore, drug-induced changes in the captured proteome’s phosphorylation state were also looked at. Typical results obtained are shown in Fig. 5. Figure 5a shows cultured cells with different chemical stimuli (ligands; in this case drugs) at the top, and below this the schematic process of binding of protein kinases to the affinity beads in the presence of different concentrations of ligand is depicted. The MS/MS spectrum at the bottom left of Fig.5a shows the four characteristic iTRAQ reporter signals indicative of the relative amount of protein kinase trapped and thus of the percentage of ligand binding to the respective protein kinase. Shown at the bottom right of Fig. 5a are schematic binding curves of four different protein kinases (for which the ligand has no affinity for one protein kinase). Figure 5b shows 16 graphs, showing the results for one typical protein kinase per graph, with each graph having three actual binding curves for the same three different drugs: bosutinib, dasatinib, and imatinib. The bars in Fig. 5c represent relative affinities of each protein kinase for the three drugs. Similar work by Sharma et al. [94]described the analysis of small-molecule kinase inhibitors, an antibody, and a tyrosine-phosphorylated peptide as inhibitors of panels of protein kinases. As inhibitors of protein kinases are used as research tools in elucidating signal transduction cascades, characterizing these inhibitory profiles with chemical proteomics approaches can also aid progress in fundamental research.
Fig. 5.

a Overview of the Kinobeads assay. Either lysates or cells are treated with the compound (ligand or lead compound) over a range of concentrations (top). Subsequently, proteins are captured on Kinobeads. The in-solution ligand competes with the immobilized ligands for ATP binding or related ligand binding sites of its targets (middle). Bound proteins are digested with trypsin and each peptide pool is labeled with iTRAQ reagent (not shown). All four samples are combined and analyzed by MS. Each peptide gives rise to four characteristic iTRAQ reporter signals (scaled to 100%) indicative of the inhibitor concentration used (bottom left). For each peptide detected, the decrease of signal intensity compared with the control reflects competition by the in-solution ligand for its target (bottom right). b Examples of competition binding curves calculated from iTRAQ reporter signals. Binding of several known and novel targets to Kinobeads depends on the addition of the ligands imatinib (blue curves), dasatinib (green curves), and bosutinib (red curves) to K562 cell lysate. For each ligand, three independent quadruplexed experiments (control plus three ligand concentrations each) were performed in duplicate, and iTRAQ reporter signal data were combined to display the dose response over nine concentrations. c Kinase-binding profiles of the ABL ligands (kinase-inhibiting drugs) imatinib, dasatinib, and bosutinib across a set of protein kinases simultaneously identified from K562 cells. The bars indicate the IC50 values, defined as the concentration of drug at which half-maximal competition of binding of Kinobeads is observed. DMSO = dimethyl sulfoxide. (Reprinted from Bantscheff et al. [92]. With permission)
Finally, three very recent examples of typical chemical proteomics studies are discussed. Rix et al. [95] used chemical proteomics to study the binding kinases of BCR-ABL kinase inhibitor INNO-406 in myeloid leukemia cells. Better knowledge of its full target spectrum can help predict side effects and novel treatment applications, and can possibly provide information for next-generation therapies involving kinase inhibitors.
CB30865 is a selective and potent cytotoxic agent of previously unknown action mechanism. Chemical proteomics revealed that its cytotoxicity is due to high-affinity inhibition of nicotinamide phosphoribosyltransferase (Nampt) [96]. As cancer cells develop dependence on Nampt, because of an elevated energy requirement (involving NAD-consuming enzymes), it was implied that Nampt-inhibiting molecules provide a starting point for drug discovery with Nampt as a potential target in cancer therapy.
Lastly, in a chemical proteomics approach an affinity matrix was made from antiresorptive 5-chloro-1-(2,6-dimethylpiperidin-1-yl)-N-tosylpentan-1-imine, where prohibitin was identified as strong binding protein [97]. This might provide new handles for drug discovery aimed at antiresorptive drugs.
Where most proteomics approaches use off-line digestion steps, efforts to automate these have been made. In these experiments, (online) affinity protein purification followed by elution to online digestion reactors/chambers and finally elution of the peptides formed to MS or LC-MS can form fully automated analytical systems. This technique, however, has not gained widespread use, probably owing to compatibility problems between conditions needed for affinity purification, digestion, and (LC-)MS analysis, and the short digestion times required. This review briefly discusses a few selected examples.
A schematic view of an exemplifying setup used for used for online affinity trapping of proteins followed by in-solution online digestion and again trapping on a solid-phase extraction column prior to LC-MS is depicted in Fig. 6. In the complete setup, proteins are applied to immunoaffinity chromatography. Bound proteins are subsequently disrupted and eluted to an online bioreactor based on continuous-flow protein digestion with a protease. Peptides formed are subsequently trapped on a solid-phase extraction column. After desalination, the trapped peptides are subjected to LC-MS analysis, and the rest of the system is reequilibrated for the next run. Hoos et al. [98] accomplished successful online immunoaffinity chromatography with human serum albumin (HSA) antibodies and chemically adducted HSA followed by use of a solution-phase digestion chamber, after which the samples were analyzed online by LC-MS analysis. This technology was capable of detecting reactive chemical agents or drugs able to covalently bind to the cysteine-34 residue of HSA via the respective adducted peptide analyzed after digestion.
Fig. 6.

View of an exemplifying setup used for online affinity trapping of proteins followed by in-solution online digestion and again trapping on a solid-phase extraction (SPE) column prior to LC-MS. Proteins are injected (1) onto an immunoaffinity chromatography column (2) and nonbinders are eluted to the waste (W2; switching valve S2 is therefore temporarily switched). The disruption buffer (3) elutes the bound proteins by switching valve S1. A protease in solution present in a cooled superloop (4) is mixed in continuously with help of an LC pump (5). The digestion now takes place online in a thermostated reaction coil (6) and the peptides formed are trapped on an SPE column (7). After switching valve S2, the gradient LC pumps (8) desalinate the trapped peptides and elute the peptides over the LC column (9) to the mass spectrometer (10). Switching valve S3 and LC pump 11 are used for reequilibration of the LC column
Another example includes the selective analysis of online-immunoaffinity-purified cytochrome c [99]. A slightly different technique can allow detection of proteins with large similarities, such as polymorphic proteins or proteins with different posttranslational modifications, when an initial protein separation step is incorporated [100]. With use of this method, proteins are first separated by nano-LC prior to postcolumn online digestion and MS analysis. Similar results were obtained with a method based on column-switching recycling size-exclusion chromatography, microenzymatic online digestion, and LC-MS [101].
The complexity of automation and/or the lack of sensitivity are still serious drawbacks in these methods. They can function efficiently, however, when the number of proteins to be analyzed is limited. For the online digestion part only, however, many different setups have been developed [102–104]. Furthermore, also microfluidics on-chip [105, 106], on-capillary zone electrophoresis [107], and monolithic disk or bioreactor [108, 109] digestion procedures after affinity-capturing are entering the affinity screening arena. The development of a Sepharose material with a small immobilized peptide for metalloprotease selection based on affinity by Freije and Bischoff [110] allowed sample enrichment in online bioaffinity selection–trypsin digestion–MS proteomics in order to identify proteases and rank affinities [111].
IMAC comprises different methods that are similar to protein affinity chromatography. The main difference is that IMAC separates proteins, small organic ligands, or phosphopeptides on the basis of their affinity for the immobilized metal ion. This separation then depends on the number and location of histidine (His) residues in proteins, and cysteine and tryptophan residues to a lesser extent [112]. For the already briefly discussed phosphoproteomics approaches (see “The study of cellular protein complexes involved in signaling events with pull-down-based interaction proteomics”), the affinity interaction is between the phosphate group of phosphopeptides (which are formed after a bottom-up digestion step) and the affinity material. For His-tagged proteins, a very high affinity between the His-tag and the immobilized metal ion efficiently traps them from any matrix. For this reason, IMAC is the most widely used method for purification of proteins. Recently, Cheeks et al. [113] demonstrated a monolithic IMAC column for purification of His-tagged lentiviral vectors, and Zhang et al. [114] used a so-called affinity peptidomics method to affinity-capture bioactive proteins or peptides on hydrogels and microarrays followed by MALDI-MS for identification of the endogenous peptides trapped or the proteolytic peptides that result from trapped proteins. Because the IMAC affinity interaction is not based on biological affinity interactions, it will not be discussed further. By “biological affinity,” binding of ligands or proteins to specific binding sites or pockets present in target proteins (e.g., receptors or enzymes) is meant.
SPR coupled with MS for protein affinity analysis, quantification, and identification
SPR biosensors consist of a prism against a thin metal layer (e.g., gold) and a flow-through chamber containing analyte solution at the opposite side of the metal layer. Surface plasmons occurring at the metal–solution interface have a certain wave vector, which depends on the structure and composition of the metal surface and of the solution and analytes near the surface. A light beam directed at the prism undergoes total internal reflection at the prism–metal interface and photons excite the electrons in the metal film. The wave vector of these excited electrons at a specific angle of incidence is equal to that of the surface plasmons, which results in a total energy transfer. At this angle, the light beam is no longer reflected. The biosensor mechanism depends on the fact that the angle at which this happens depends on the composition of the solution near the surface. Ligand binding to an immobilized protein on the metal sensor changes the wave vector of the surface plasmons and the angle of incidence at which SPR occurs. As the technology only allows analysis of binding events without identification capabilities, SPR is sometimes combined with a mass spectrometer in order to identify the binding partners.
Since the launch of commercially available SPR biosensors, such as the Biacore [115], SPR has become an increasingly prominent technology in drug discovery. Presently, multiplexed SPR biosensors enable the recording of multiple binding experiments simultaneously [116]. In the Biacore and most competing SPR biosensors, an analyte is infused onto a chip with immobilized target protein and starts to bind until all possible binding sites are occupied, or until equilibrium is reached. Then, the chip is saturated or equilibrated. The speed at which this happens depends on association and dissociation rates of the complex, analogous to frontal affinity chromatography [58]. Likewise, the resulting data and data processing are analogous to those in frontal affinity chromatography. In SPR, the change in signal depends on analyte concentration and molecular weight. This is why SPR bioassays are especially suitable for the study of protein–protein interactions, or other large molecules that bind to an immobilized target protein or ligand.
Not surprisingly, the first major applications of SPR biosensors were antibody–antigen interactions [117], the study of DNA hybridization and dehybridization [118], and some other macromolecular interactions. Additionally, SPR has been coupled with MALDI time-of-flight MS in a number of cases, in which the SPR chip was directly used as the MALDI chip [119]. In this SPR-MALDI-based analytical setup, different target proteins have been looked at [20, 119, 120]. SPR-MS coupling has been used for a few years and interesting articles on recent SPR-MS method development include the work of Bouffartigues et al. [121] on a high-throughput SPR–surface-enhanced laser desorption ionization–MS method to identify protein bound to DNA, and that of Marchesini et al. [122] on an online coupling of an SPR method and a nano-LC system, resulting in an online ESI interface between the SPR-LC system and the mass spectrometer. An elegant solution for semiquantitatively analyzing and identifying protein binders to small molecules is to immobilize the small molecules on the chip, and subsequently measure the binding of the target proteins followed by elution to LC-MS [123].
For measurement of binding kinetics (k on and k off) or signaling events, besides SPR [124], also other technologies, such as total internal reflection fluorescence, can be applied, but these are commonly performed without compound identification by a parallel-placed mass spectrometer. In most cases, the use of MS is actually not needed as the compounds analyzed are known and are pure compounds. When unknown compounds are present in a mixture, the mass spectrometer can be placed efficiently after SPR technologies, then rendering the SPR more as an affinity-selection method since the characteristic binding kinetics information obtained by SPR is less useful for (protein) mixtures when multiple unknown binders are expected. However, the protein quantification capabilities of SPR are powerful in combination with MS. Then the MS data complement the SPR detection. The combination can reveal intrinsic protein structural modifications that cannot be analyzed by SPR detection alone.
Borch and Roepstorff described protocols for SPR-based protein capture, elution, and robust sample preparation for sensitive MS-based identification [125] and Nedelkov [126] described protocols and know-how for efficient coupling of SPR and MS. Similar to SPR-MALDI approaches, high-resolution localized SPR sensors have also been used in combination with MALDI and besides similar detection and identification possibilities have the additional advantage of showing fewer interferences from changes in the bulk refractive index. In a typical case, this combination was used by Anker at al. [127] to study amyloid β oligomers, important players in Alzheimer’s disease.
In conclusion, SPR-MS is a highly specific tool for the quantification and identification of proteins. It is efficient for high molecular weight compounds, and therefore particularly suitable for the study of protein–protein interactions and proteins interacting with immobilized ligands.
Conclusions and perspectives
For the analysis of noncovalent protein complexes or assemblies, advances in MS hardware and scientific expertise have opened up new ways of investigation. Key advantages of these so-called native MS approaches are the speed of analysis and the potential for directly analyzing complexes in solution. Although in vitro samples (e.g., cell lysates) can in theory be studied directly, artificially elevated concentrations of binding proteins have to be used for a study and other potential (unknown) proteins present under physiological conditions are omitted from the experiment. An important advantage of native MS compared with other structural biology methods is the significantly smaller amount of sample required. It is essential to keep in mind that all complexes are analyzed in the gas phase and not under physiological conditions. Furthermore, method development is crucial and can be elaborate. However, the results obtained by studying protein–protein and protein–ligand interactions most often do resemble the results obtained with traditional biological/biochemical approaches. In fact, these MS approaches are complementary to traditional methods in terms of data obtained. In many current cases, MS is the primary analytical technique of choice because of convenience, speed, and also its capability to study extremely large and diverse protein assemblies. Evidently, it is not possible to obtain high-resolution images including the shapes of protein complexes. But new ion mobility capabilities prior to the MS readout increasingly allow more detailed information on protein complexes to be obtained.
Considerable progress has also been made in protein–protein-interaction-based pull-down strategies followed by MS analysis, and the field is rapidly expanding. The data obtained require careful interpretation, because nonspecific binding processes during the pull-down experiments may introduce artifacts and thereby jeopardize the quality and validity of the end results regarding the eventual protein binding partners identified. Clearly, the pull-down part or “protein fishing” process has to be optimized and validated thoroughly prior to actual biological experiments. Furthermore, after protein complexes have been identified by MS, the use of traditional biochemical approaches is encouraged to confirm the MS-based results, followed by additional research with other approaches for further characterization. As such, MS provides an efficient means of screening for relevant protein–protein or protein–ligand interactions to be characterized further in depth by more traditional technologies.
When using pull-down proteomics studies from a slightly different angle, by pulling down drug target proteins via immobilized lead compounds or ligands (e.g., on an affinity column), the technology has opened up avenues to elaborate pharmaceutical selectivity analysis of lead compounds. In these methods, increasing concentrations of ligand added to the drug targets studied (in whole cell lysates) prevent the drug targets from binding to the immobilized ligand and thus preclude them from subsequent MS-based detection. In this way, many different drug targets and off-targets (antitargets) are screened at once (such as protein kinases), where the higher-affinity drug targets are displaced first and the lower-affinity drug targets are displaced later. Consequently, the technology allows inhibitory profiles to be analyzed for lead compounds in whole panels of drug targets. In the future, this might facilitate the vision of developing drugs that target panels of disease-involved drug targets instead of the accepted view of having one drug for one target. Perhaps, in the far future, this technology will even aid in drug discovery strategies leading to personalized medicines.
Finally, SPR is a very strong technology for the analysis of protein–protein and protein–immobilized ligand interactions, but is of less importance for the study of small ligands. The combination of SPR and MS is a strong asset because it allows the identification of the binding partners in complex mixtures. However, one must bear in mind that one of the main capabilities of SPR, the analysis of the binding kinetics of single known binders, is lost when analyzing mixtures of (unknown) proteins when it is coupled with MS. Then, SPR can only be used for semiquantitation of all combined binders to the SPR surface.
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- ADAP
adhesion and degranulation promoting adapter protein
- CggR
central glycolytic genes repressor
- ESI
electrospray ionization
- FBP
fructose 1,6-bisphosphate
- GPCR
G-protein-coupled receptor
- His
histidine
- ICAT
isotope-coded affinity tags
- IMAC
immobilized metal affinity chromatography
- IMS
ion mobility spectrometry
- iTRAQ
isobaric tags for relative and absolute quantitation
- LC
liquid chromatography
- HSA
human serum albumin
- MALDI
matrix-assisted laser desorption ionization
- MS
mass spectrometry
- Nampt
nicotinamide phosphoribosyltransferase
- NMR
nuclear magnetic resonance
- PEBP
phosphatidylethanolamine-binding protein
- RXR
retinoid X receptor
- SILAC
stable isotope labeling by amino acids in cell culture
- SPR
surface plasmon resonance
- TAP
tandem affinity purification
References
- 1.Zhou M, Robinson CV. Trends Biochem Sci. 2010;35(9):522–529. doi: 10.1016/j.tibs.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Oeljeklaus S, Meyer HE, Warscheid B. FEBS Lett. 2009;583(11):1674–1683. doi: 10.1016/j.febslet.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 3.Bantscheff M, Scholten A, Heck AJ. Drug Discov Today. 2009;14(21–22):1021–1029. doi: 10.1016/j.drudis.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Ganem B, Li Y, Henion JD. J Am Chem Soc. 1991;113:6294–6296. [Google Scholar]
- 5.Katta V, Chait BT. J Am Chem Soc. 1991;113(22):8534. [Google Scholar]
- 6.Chowdhury SK, Katta V, Chait BT. J Am Chem Soc. 1990;112:9012–9013. [Google Scholar]
- 7.Craig TA, Veenstra TD, Naylor S, Tomlinson AJ, Johnson KLMS, Juranic N, Kumar R. Biochemistry. 1997;36(34):10482–10491. doi: 10.1021/bi970561b. [DOI] [PubMed] [Google Scholar]
- 8.Kheterpal I, Cook KD, Wetzel R. Methods Enzymol. 2006;413:140–166. doi: 10.1016/S0076-6879(06)13008-6. [DOI] [PubMed] [Google Scholar]
- 9.Whitelegge J, Halgand F, Souda P, Zabrouskov V. Expert Rev Proteomics. 2006;3(6):585–596. doi: 10.1586/14789450.3.6.585. [DOI] [PubMed] [Google Scholar]
- 10.Evers TH, van Dongen JL, Meijer EW, Merkx M. J Biol Inorg Chem. 2007;12(6):919–928. doi: 10.1007/s00775-007-0246-6. [DOI] [PubMed] [Google Scholar]
- 11.Mazon H, Gabor K, Leys D, Heck AJ, van der Oost J, van den Heuvel RH. J Biol Chem. 2007;282(15):11281–11290. doi: 10.1074/jbc.M611177200. [DOI] [PubMed] [Google Scholar]
- 12.van Duijn E, Bakkes PJ, Heeren RM, van den Heuvel RH, van Heerikhuizen H, van der Vies SM, Heck AJ. Nat Methods. 2005;2(5):371–376. doi: 10.1038/nmeth753. [DOI] [PubMed] [Google Scholar]
- 13.Bovet C, Wortmann A, Eiler S, Granger F, Ruff M, Gerrits B, Moras D, Zenobi R. Protein Sci. 2007;16(5):938–946. doi: 10.1110/ps.062664107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffith WP, Kaltashov IA. Biochemistry. 2003;42(33):10024–10033. doi: 10.1021/bi034035y. [DOI] [PubMed] [Google Scholar]
- 15.Shoemaker GK, van Duijn E, Crawford SE, Uetrecht C, Baclayon M, Roos WH, Wuite GJ, Estes MK, Prasad BV, Heck AJ. Mol Cell Proteomics. 2010;9(8):1742–1751. doi: 10.1074/mcp.M900620-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nesatyy VJ. Int J Mass Spectrom. 2002;221(2):147–161. [Google Scholar]
- 17.Kim JY, Kim YG, Baik JY, Joo EJ, Kim YH, Lee GM. Biotechnol Prog. 2010;26(1):246–251. doi: 10.1002/btpr.323. [DOI] [PubMed] [Google Scholar]
- 18.Kang YJ, Jang M, Park YK, Kang S, Bae KH, Cho S, Lee CK, Park BC, Chi SW, Park SG. Biochem Biophys Res Commun. 2010;393(4):794–799. doi: 10.1016/j.bbrc.2010.02.084. [DOI] [PubMed] [Google Scholar]
- 19.Rohila JS, Chen M, Chen S, Chen J, Cerny RL, Dardick C, Canlas P, Fujii H, Gribskov M, Kanrar S, Knoflicek L, Stevenson B, Xie M, Xu X, Zheng X, Zhu JK, Ronald P, Fromm ME. PLoS One. 2009;4(8):e6685. doi: 10.1371/journal.pone.0006685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nedelkov D. Anal Chem. 2007;79(15):5987–5990. doi: 10.1021/ac070608r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Duijn E. J Am Soc Mass Spectrom. 2010;21(6):971–978. doi: 10.1016/j.jasms.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 22.Kay LE. J Magn Reson. 2011;210(2):159–170. doi: 10.1016/j.jmr.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Ruschak AM, Religa TL, Breuer S, Witt S, Kay LE. Nature. 2010;467(7317):868–871. doi: 10.1038/nature09444. [DOI] [PubMed] [Google Scholar]
- 24.Velyvis A, Yang YR, Schachman HK, Kay LE. Proc Natl Acad Sci USA. 2007;104(21):8815–8820. doi: 10.1073/pnas.0703347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Heuvel RH, van Duijn E, Mazon H, Synowsky SA, Lorenzen K, Versluis C, Brouns SJ, Langridge D, van der Oost J, Hoyes J, Heck AJ. Anal Chem. 2006;78(21):7473–7483. doi: 10.1021/ac061039a. [DOI] [PubMed] [Google Scholar]
- 26.Sun N, Sun J, Kitova EN, Klassen JS. J Am Soc Mass Spectrom. 2009;20(7):1242–1250. doi: 10.1016/j.jasms.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 27.Martineau E, de Guzman JM, Rodionova L, Kong X, Mayer PM, Aman AM (2010) J Am Soc Mass Spectrom 21(9):1506-1514 [DOI] [PubMed]
- 28.Jecklin MC, Schauer S, Dumelin CE, Zenobi R. J Mol Recognit. 2009;22(4):319–329. doi: 10.1002/jmr.951. [DOI] [PubMed] [Google Scholar]
- 29.Dennhart N, Letzel T. Anal Bioanal Chem. 2006;386(3):689–698. doi: 10.1007/s00216-006-0604-1. [DOI] [PubMed] [Google Scholar]
- 30.Yan X, Maier CS. Methods Mol Biol. 2009;492:255–271. doi: 10.1007/978-1-59745-493-3_15. [DOI] [PubMed] [Google Scholar]
- 31.Bich C, Bovet C, Rochel N, Peluso-Iltis C, Panagiotidis A, Nazabal A, Moras D, Zenobi R. J Am Soc Mass Spectrom. 2010;21(4):635–645. doi: 10.1016/j.jasms.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 32.McKay AR, Ruotolo BT, Ilag LL, Robinson CV. J Am Chem Soc. 2006;128(35):11433–11442. doi: 10.1021/ja061468q. [DOI] [PubMed] [Google Scholar]
- 33.Uetrecht C, Versluis C, Watts NR, Roos WH, Wuite GJ, Wingfield PT, Steven AC, Heck AJ. Proc Natl Acad Sci USA. 2008;105(27):9216–9220. doi: 10.1073/pnas.0800406105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bothner B, Siuzdak G. ChemBioChem. 2004;5(3):258–260. doi: 10.1002/cbic.200300754. [DOI] [PubMed] [Google Scholar]
- 35.Uetrecht C, Rose RJ, van Duijn E, Lorenzen K, Heck AJ. Chem Soc Rev. 2010;39(5):1633–1655. doi: 10.1039/b914002f. [DOI] [PubMed] [Google Scholar]
- 36.Ruotolo BT, Benesch JL, Sandercock AM, Hyung SJ, Robinson CV. Nat Protoc. 2008;3(7):1139–1152. doi: 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
- 37.Kaddis CS, Lomeli SH, Yin S, Berhane B, Apostol MI, Kickhoefer VA, Rome LH, Loo JA. J Am Soc Mass Spectrom. 2007;18(7):1206–1216. doi: 10.1016/j.jasms.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruotolo BT, Giles K, Campuzano I, Sandercock AM, Bateman RH, Robinson CV. Science. 2005;310(5754):1658–1661. doi: 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]
- 39.Wiedenheft B, van Duijn E, Bultema J, Waghmare S, Zhou K, Barendregt A, Westphal W, Heck A, Boekema E, Dickman M, Doudna JA (2011) Proc Natl Acad Sci USA (in press) [DOI] [PMC free article] [PubMed]
- 40.Knapman TW, Morton VL, Stonehouse NJ, Stockley PG, Ashcroft AE. Rapid Commun Mass Spectrom. 2010;24(20):3033–3042. doi: 10.1002/rcm.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boeri Erba E, Ruotolo BT, Barsky D, Robinson CV. Anal Chem. 2010;82(23):9702–9710. doi: 10.1021/ac101778e. [DOI] [PubMed] [Google Scholar]
- 42.Kukrer B, Filipe V, van Duijn E, Kasper PT, Vreeken RJ, Heck AJ, Jiskoot W. Pharm Res. 2010;27(10):2197–2204. doi: 10.1007/s11095-010-0224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blackwell AE, Dodds ED, Bandarian V, Wysocki VH. Anal Chem. 2011;83(8):2862–2865. doi: 10.1021/ac200452b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Atmanene C, Chaix D, Bessin Y, Declerck N, Van Dorsselaer A, Sanglier-Cianferani S. Anal Chem. 2010;82(9):3597–3605. doi: 10.1021/ac902784n. [DOI] [PubMed] [Google Scholar]
- 45.Noda M, Uchiyama S, McKay AR, Morimoto A, Misawa S, Yoshida A, Shimahara H, Takinowaki H, Nakamura S, Kobayashi Y, Matsunaga S, Ohkubo T, Robinson CV, Fukui K. Biochem J. 2011;436(1):101–112. doi: 10.1042/BJ20102063. [DOI] [PubMed] [Google Scholar]
- 46.Atmanene C, Laux A, Glattard E, Muller A, Schoentgen F, Metz-Boutigue MH, Aunis D, Van Dorsselaer A, Stefano GB, Sanglier-Cianferani S, Goumon Y. Med Sci Monit. 2009;15(7):BR178–BR187. [PubMed] [Google Scholar]
- 47.Gordiyenko Y, Videler H, Zhou M, McKay AR, Fucini P, Biegel E, Muller V, Robinson CV. Mol Cell Proteomics. 2010;9(8):1774–1783. doi: 10.1074/mcp.M000072-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gundry RL, White MY, Murray CI, Kane LA, Fu Q, Stanley BA, Van Eyk JE. Curr Protoc Mol Biol. 2009;88:10251–102523. doi: 10.1002/0471142727.mb1025s88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graves PR, Haystead TA. Recent Prog Horm Res. 2003;58:1–24. doi: 10.1210/rp.58.1.1. [DOI] [PubMed] [Google Scholar]
- 50.Michel MC, Alewijnse AE. Mol Pharmacol. 2007;72(5):1097–1099. doi: 10.1124/mol.107.040923. [DOI] [PubMed] [Google Scholar]
- 51.Pluder F, Morl K, Beck-Sickinger AG. Pharmacol Ther. 2006;112(1):1–11. doi: 10.1016/j.pharmthera.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 52.Niederberger E, Geisslinger G. Expert Rev Proteomics. 2010;7(2):189–203. doi: 10.1586/epr.10.1. [DOI] [PubMed] [Google Scholar]
- 53.Mastellos D, Andronis C, Persidis A, Lambris JD. Clin Immunol. 2005;115(3):225–235. doi: 10.1016/j.clim.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 54.Chen MH, Malbon CC. Cell Signal. 2009;21(1):136–142. doi: 10.1016/j.cellsig.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 55.Wang HY, Tao J, Shumay E, Malbon CC. Eur J Cell Biol. 2006;85(7):643–650. doi: 10.1016/j.ejcb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 56.Vicent GP, Zaurin R, Ballare C, Nacht AS, Beato M. Nucl Recept Signal. 2009;7:e008. doi: 10.1621/nrs.07008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lygren B, Tasken K. Biochem Soc Trans. 2006;34(4):489–491. doi: 10.1042/BST0340489. [DOI] [PubMed] [Google Scholar]
- 58.Jonker N, Kool J, Irth H, Niessen WM. Anal Bioanal Chem. 2011;399(8):2669–2681. doi: 10.1007/s00216-010-4350-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol Cell Proteomics. 2002;1(5):376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 60.Blagoev B, Kratchmarova I, Ong SE, Nielsen M, Foster LJ, Mann M. Nat Biotechnol. 2003;21(3):315–318. doi: 10.1038/nbt790. [DOI] [PubMed] [Google Scholar]
- 61.Ross PL, Huang YLN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Mol Cell Proteomics. 2004;3(12):1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 62.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Nat Biotechnol. 1999;17(10):994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 63.Fenselau C, Yao X. J Proteome Res. 2009;8(5):2140–2143. doi: 10.1021/pr8009879. [DOI] [PubMed] [Google Scholar]
- 64.Boersema PJ, Foong LY, Ding VM, Lemeer S, van Breukelen B, Philp R, Boekhorst J, Snel B, den Hertog J, Choo AB, Heck AJ. Mol Cell Proteomics. 2010;9(1):84–99. doi: 10.1074/mcp.M900291-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dadvar P, O'Flaherty M, Scholten A, Rumpel K, Heck AJ. Mol Biosyst. 2009;5(5):472–482. doi: 10.1039/b815709j. [DOI] [PubMed] [Google Scholar]
- 66.Kovanich D, van der Heyden MA, Aye TT, van Veen TA, Heck AJ, Scholten A. ChemBioChem. 2010;11(7):963–971. doi: 10.1002/cbic.201000058. [DOI] [PubMed] [Google Scholar]
- 67.Brymora A, Valova VA, Robinson PJ (2004). Curr Protoc Cell Biol 17.5.1–17.5.51 [DOI] [PubMed]
- 68.Du J, Xu H, Wei N, Wakim B, Halligan B, Pritchard KA, Jr, Shi Y. Biochem Biophys Res Commun. 2009;385(2):143–147. doi: 10.1016/j.bbrc.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sturm N, Jortzik E, Mailu BM, Koncarevic S, Deponte M, Forchhammer K, Rahlfs S, Becker K. PLoS Pathog. 2009;5(4):e1000383. doi: 10.1371/journal.ppat.1000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Catimel B, Yin MX, Schieber C, Condron M, Patsiouras H, Catimel J, Robinson DE, Wong LS, Nice EC, Holmes AB, Burgess AW. J Proteome Res. 2009;8(7):3712–3726. doi: 10.1021/pr900320a. [DOI] [PubMed] [Google Scholar]
- 71.Paul AL, Liu L, McClung S, Laughner B, Chen S, Ferl RJ. J Proteome Res. 2009;8(4):1913–1924. doi: 10.1021/pr8008644. [DOI] [PubMed] [Google Scholar]
- 72.Hubner NC, Mann M. Methods. 2011;53(4):453–459. doi: 10.1016/j.ymeth.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 73.Paulo JA, Brucker WJ, Hawrot E. J Proteome Res. 2009;8(4):1849–1858. doi: 10.1021/pr800731z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Amoresano A, Di Costanzo A, Leo G, Di Cunto F, La Mantia G, Guerrini L, Calabro V. J Proteome Res. 2010;9(4):2042–2048. doi: 10.1021/pr9011156. [DOI] [PubMed] [Google Scholar]
- 75.Jessulat M, Buist T, Alamgir M, Hooshyar M, Xu J, Aoki H, Ganoza MC, Butland G, Golshani A. Methods Mol Biol. 2010;587:99–111. doi: 10.1007/978-1-60327-355-8_7. [DOI] [PubMed] [Google Scholar]
- 76.Guo L, Ying W, Zhang J, Yuan Y, Qian X, Wang J, Yang X, He F. Acta Biochim Biophys Sin. 2010;42(4):266–273. doi: 10.1093/abbs/gmq014. [DOI] [PubMed] [Google Scholar]
- 77.Tarallo R, Bamundo A, Nassa G, Nola E, Paris O, Ambrosino C, Facchiano A, Baumann M, Nyman TA, Weisz A. Proteomics. 2011;11(1):172–179. doi: 10.1002/pmic.201000217. [DOI] [PubMed] [Google Scholar]
- 78.Raab M, Daxecker H, Edwards RJ, Treumann A, Murphy D, Moran N. Proteomics. 2010;10(15):2790–2800. doi: 10.1002/pmic.200900621. [DOI] [PubMed] [Google Scholar]
- 79.Iioka H, Loiselle D, Haystead TA, Macara IG. Nucleic Acids Res. 2011;39(8):e53. doi: 10.1093/nar/gkq1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kubben N, Voncken JW, Demmers J, Calis C, van Almen G, Pinto Y, Misteli T. Nucleus. 2010;1(6):513–525. doi: 10.4161/nucl.1.6.13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lange S, Sylvester M, Schumann M, Freund C, Krause E. J Proteome Res. 2010;9(8):4113–4122. doi: 10.1021/pr1003054. [DOI] [PubMed] [Google Scholar]
- 82.Jager S, Gulbahce N, Cimermancic P, Kane J, He N, Chou S, D'Orso I, Fernandes J, Jang G, Frankel AD, Alber T, Zhou Q, Krogan NJ. Methods. 2011;53(1):13–19. doi: 10.1016/j.ymeth.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Morree A, Hensbergen PJ, van Haagen HH, Dragan I, Deelder AM, t Hoen PA, Frants RR, van der Maarel SM (2010) PLoS One 5(11):e13854 [DOI] [PMC free article] [PubMed]
- 84.Borroto-Escuela DO, Correia PA, Romero-Fernandez W, Narvaez M, Fuxe K, Ciruela F, Garriga P. J Neurosci Methods. 2011;195(2):161–169. doi: 10.1016/j.jneumeth.2010.11.025. [DOI] [PubMed] [Google Scholar]
- 85.Fischer JJ, Michaelis S, Schrey AK, Graebner OG, Glinski M, Dreger M, Kroll F, Koester H. Toxicol Sci. 2010;113(1):243–253. doi: 10.1093/toxsci/kfp236. [DOI] [PubMed] [Google Scholar]
- 86.Luo Y, Fischer JJ, Baessler OY, Schrey AK, Ungewiss J, Glinski M, Sefkow M, Dreger M, Koester H. J Proteomics. 2010;73(4):815–819. doi: 10.1016/j.jprot.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 87.Fischer JJ, Graebner Baessler OY, Dalhoff C, Michaelis S, Schrey AK, Ungewiss J, Andrich K, Jeske D, Kroll F, Glinski M, Sefkow M, Dreger M, Koester H (2010) J Proteome Res 9(2):806-817 [DOI] [PubMed]
- 88.Sinz A. Anal Bioanal Chem. 2009;397(8):3433–3440. doi: 10.1007/s00216-009-3405-5. [DOI] [PubMed] [Google Scholar]
- 89.Lenz T, Poot P, Gräbner O, Glinski M, Weinhold E, Dreger M, Köster H (2010) J Vis Exp (46). http://www.jove.com/details.php?id=2264 [DOI] [PMC free article] [PubMed]
- 90.Hino N, Oyama M, Sato A, Mukai T, Iraha F, Hayashi A, Kozuka-Hata H, Yamamoto T, Yokoyama S, Sakamoto K. J Mol Biol. 2011;406(2):343–353. doi: 10.1016/j.jmb.2010.12.022. [DOI] [PubMed] [Google Scholar]
- 91.Rix U, Superti-Furga G. Nat Chem Biol. 2009;5(9):616–624. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- 92.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, Drewes G. Nat Biotechnol. 2007;25(9):1035–1044. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 93.Jin GZ, Li Y, Cong WM, Yu H, Dong H, Shu H, Liu X, Yan GQ, Zhang L, Zhang Y, Kang X, Guo K, Wang ZD, Yang PY, Liu YK (2011) J Proteome Res (in press) [DOI] [PubMed]
- 94.Sharma K, Weber C, Bairlein M, Greff Z, Keri G, Cox J, Olsen JV, Daub H. Nat Methods. 2009;6(10):741–744. doi: 10.1038/nmeth.1373. [DOI] [PubMed] [Google Scholar]
- 95.Rix U, Remsing Rix LL, Terker AS, Fernbach NV, Hantschel O, Planyavsky M, Breitwieser FP, Herrmann H, Colinge J, Bennett KL, Augustin M, Till JH, Heinrich MC, Valent P, Superti-Furga G. Leukemia. 2010;24(1):44–50. doi: 10.1038/leu.2009.228. [DOI] [PubMed] [Google Scholar]
- 96.Fleischer TC, Murphy BR, Flick JS, Terry-Lorenzo RT, Gao ZH, Davis T, McKinnon R, Ostanin K, Willardsen JA, Boniface JJ. Chem Biol. 2010;17(6):659–664. doi: 10.1016/j.chembiol.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 97.Chang SY, Bae SJ, Lee MY, Baek SH, Chang S, Kim SH. Bioorg Med Chem Lett. 2011;21(2):727–729. doi: 10.1016/j.bmcl.2010.11.123. [DOI] [PubMed] [Google Scholar]
- 98.Hoos JS, Damsten MC, de Vlieger JS, Commandeur JN, Vermeulen NP, Niessen WM, Lingeman H, Irth H. J Chromatogr B. 2007;859(2):147–156. doi: 10.1016/j.jchromb.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 99.Cingoz A, Hugon-Chapuis F, Pichon V. J Chromatogr B. 2010;878(2):213–221. doi: 10.1016/j.jchromb.2009.07.032. [DOI] [PubMed] [Google Scholar]
- 100.Bruyneel B, Hoos JS, Smoluch MT, Lingeman H, Niessen WM, Irth H. Anal Chem. 2007;79(4):1591–1598. doi: 10.1021/ac0616761. [DOI] [PubMed] [Google Scholar]
- 101.Yuan H, Zhou Y, Zhang L, Liang Z, Zhang Y. J Chromatogr A. 2009;1216(44):7478–7482. doi: 10.1016/j.chroma.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 102.Carol-Visser J, van der Schans M, Fidder A, Hulst AG, van Baar BL, Irth H, Noort D. J Chromatogr B. 2008;870(1):91–97. doi: 10.1016/j.jchromb.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 103.Lopez-Ferrer D, Petritis K, Lourette NM, Clowers B, Hixson KK, Heibeck T, Prior DC, Pasa-Tolic L, Camp DG, 2nd, Belov ME, Smith RD. Anal Chem. 2008;80(23):8930–8936. doi: 10.1021/ac800927v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Temporini C, Calleri E, Cabrera K, Felix G, Massolini G. J Sep Sci. 2009;32(8):1120–1128. doi: 10.1002/jssc.200800596. [DOI] [PubMed] [Google Scholar]
- 105.Liuni P, Rob T, Wilson DJ (2010) Rapid Commun Mass Spectrom 24(3):315-320 [DOI] [PubMed]
- 106.Seok HJ, Hong MY, Kim YJ, Han MK, Lee D, Lee JH, Yoo JS, Kim HS. Anal Biochem. 2005;337(2):294–307. doi: 10.1016/j.ab.2004.10.042. [DOI] [PubMed] [Google Scholar]
- 107.Zeisbergerova M, Adamkova A, Glatz Z. Electrophoresis. 2009;30(13):2378–2384. doi: 10.1002/elps.200800757. [DOI] [PubMed] [Google Scholar]
- 108.Nicoli R, Gaud N, Stella C, Rudaz S, Veuthey JL. J Pharm Biomed Anal. 2008;48(2):398–407. doi: 10.1016/j.jpba.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 109.Calleri E, Temporini C, Perani E, De Palma A, Lubda D, Mellerio G, Sala A, Galliano M, Caccialanza G, Massolini G. J Proteome Res. 2005;4(2):481–490. doi: 10.1021/pr049796h. [DOI] [PubMed] [Google Scholar]
- 110.Freije JR, Bischoff R. J Chromatogr A. 2003;1009(1–2):155–169. doi: 10.1016/s0021-9673(03)00920-8. [DOI] [PubMed] [Google Scholar]
- 111.Freije R, Klein T, Ooms B, Kauffman HF, Bischoff R. J Chromatogr A. 2008;1189(1–2):417–425. doi: 10.1016/j.chroma.2007.10.059. [DOI] [PubMed] [Google Scholar]
- 112.Gutierrez R, del Valle EMM, Galan MA. Sep Purif Rev. 2007;36(1):71–111. [Google Scholar]
- 113.Cheeks MC, Kamal N, Sorrell A, Darling D, Farzaneh F, Slater NKH. J Chromatogr A. 2009;1216(13):2705–2711. doi: 10.1016/j.chroma.2008.08.029. [DOI] [PubMed] [Google Scholar]
- 114.Zhang F, Dulneva A, Bailes J, Soloviev M. Methods Mol Biol. 2010;615:313–344. doi: 10.1007/978-1-60761-535-4_23. [DOI] [PubMed] [Google Scholar]
- 115.Oshannessy DJ, Brighamburke M, Peck K. Anal Biochem. 1992;205(1):132–136. doi: 10.1016/0003-2697(92)90589-y. [DOI] [PubMed] [Google Scholar]
- 116.Rucker J, Davidoff C, Doranz BJ. Methods Mol Biol. 2010;617:445–456. doi: 10.1007/978-1-60327-323-7_32. [DOI] [PubMed] [Google Scholar]
- 117.Malmqvist M, Karlsson R. Curr Opin Chem Biol. 1997;1(3):378–383. doi: 10.1016/s1367-5931(97)80077-4. [DOI] [PubMed] [Google Scholar]
- 118.Peterlinz KA, Georgiadis RM, Herne TM, Tarlov MJ. J Am Chem Soc. 1997;119(14):3401–3402. [Google Scholar]
- 119.Bellon S, Buchmann W, Gonnet F, Jarroux N, Anger-Leroy M, Guillonneau F, Daniel R. Anal Chem. 2009;81(18):7695–7702. doi: 10.1021/ac901140m. [DOI] [PubMed] [Google Scholar]
- 120.Grote J, Dankbar N, Gedig E, Koenig S. Anal Chem. 2005;77(4):1157–1162. doi: 10.1021/ac049033d. [DOI] [PubMed] [Google Scholar]
- 121.Bouffartigues E, Leh H, Anger-Leroy M, Rimsky S, Buckle M. Nucleic Acids Res. 2007;35(6):e39. doi: 10.1093/nar/gkm030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Marchesini GR, Buijs J, Haasnoot W, Hooijerink D, Jansson O, Nielen MW. Anal Chem. 2008;80(4):1159–1168. doi: 10.1021/ac071564p. [DOI] [PubMed] [Google Scholar]
- 123.Visser NFC, Scholten A, van den Heuvel RHH, Heck AJR. ChemBioChem. 2007;8(3):298–305. doi: 10.1002/cbic.200600449. [DOI] [PubMed] [Google Scholar]
- 124.Alves ID, Park CK, Hruby VJ. Curr Protein Pept Sci. 2005;6(4):293–312. doi: 10.2174/1389203054546352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Borch J, Roepstorff P. SPR/MS Methods Mol Biol. 2010;627:269–281. doi: 10.1007/978-1-60761-670-2_19. [DOI] [PubMed] [Google Scholar]
- 126.Nedelkov D. Methods Mol Biol. 2010;627:261–268. doi: 10.1007/978-1-60761-670-2_18. [DOI] [PubMed] [Google Scholar]
- 127.Anker JN, Hall WP, Lambert MP, Velasco PT, Mrksich M, Klein WL, Van Duyne RP. J Phys Chem C Nanomater Interfaces. 2009;113(15):5891–5894. doi: 10.1021/jp900266k. [DOI] [PMC free article] [PubMed] [Google Scholar]