

Abstract
Akt activation is common in progressive thyroid cancer. In breast cancer, Akt1 induced primary cancer growth, but is reported to inhibit metastasis in vivo in several model systems. In contrast, clinical and in vitro studies suggest a metastasis-promoting role for Akt1 in thyroid cancer. The goal of this study was to determine the functional role of Akt1 in thyroid cancer growth and metastatic progression in vivo using thyroid hormone receptor βPV/PV knock-in (PV) mice which develop metastatic thyroid cancer. We crossed Akt1-/- and PV mice and compared tumor development, local progression, metastasis, and histology in TRβPV/PV/Akt1+/+ (PVPV-Akt1WT) and TRβPV/PV/Akt1-/- (PVPV-Akt1KO) mice. Mice were sacrificed at 3, 6, 9, 12, and 15 months; necropsy was performed and serum TSH was measured. Thyroid hyperplasia occurred in both groups beginning at three months; the thyroid size was greater in the PVPV-Akt1WT mice (p<0.001). In comparison with PVPV-Akt1WT mice, thyroid cancer development was delayed in the PVPV-Akt1KO mice (P=0.003) and the degree of tumor invasion was reduced. The PVPV-Akt1WT mice displayed pulmonary metastases at 12 and 15 months of age, by contrast PVPV-Akt1KO mice did not develop distant metastases at 15 months of age. Despite continued expression of Akt2 or Akt3, pAkt levels were decreased, and there was evidence of reduced Akt effect on p27 in the PVPV-Akt1KO thyroids. TSH levels were similarly elevated in PV mice regardless of Akt1 expression. In conclusion, thyroid cancer development and progression in TRβPV/PV mice are Akt1-dependent, consistent with a tumor progression-promoting role in this murine thyroid cancer model.
Key Terms: PI3 Kinase, Thyroid Hormone Receptor Beta, Thyrotropin, p27, gelsolin
Introduction
Akt (protein kinase b) represents a family of three proteins, Akt1, 2, and 3, encoded by unique genes with sequence and functional similarities. Akt family members regulate diverse cell functions including proliferation, apoptosis, motility, and glucose uptake and utilization. It has been recognized increasingly that the Akt isoforms have similar, but not entirely overlapping functions [reviewed in (Stambolic and Woodgett, 2006)]. Determinants of Akt isoform-specific activity may relate to affinity for specific intracellular chaperones, subcellular localization, availability for phosphatase interactions, or tissue or cell-type specific functions. For example altered subcellular localization was recently demonstrated to be critical for the specific role of Akt2 in insulin sensitivity in adipocytes (Gonzalez and McGraw, 2009), and Akt2 loss in mice causes a phenotype with type 2 diabetes (Cho et al., 2001a) while Akt1 loss causes small size but no evidence of diabetes (Chen et al., 2001). When considering cell motility and invasion, in some cell systems Akt1 enhances cell invasion and migration while in other systems, it inhibits migration and invasion and Akt2 has a pro-motility function (Dillon and Muller, 2010). These differences in vitro may be a function of the cell type being studied or other specific details of the model systems. The data have been extended to in vivo models in breast cancer, in which Akt1 loss inhibits primary tumor growth but results in increased metastatic progression in several models consistent with a potential metastasis inhibiting role (Cheng et al., 2007; Dillon et al., 2009; Hutchinson et al., 2004; Irie et al., 2005; Liu et al., 2006; Yoeli-Lerner et al., 2005). Interestingly, in this system, loss of Akt2 inhibits metastatic progression. These data suggest that isoform-specific inhibition of Akt may be needed for therapeutic targeting in breast cancer. Whether these results are applicable to other solid tumors is uncertain.
Akt1 is reported to have a pro-motility role in thyroid cancer, colon cancer, and melanoma cells in vitro (Saji et al., 2005; Vasko et al., 2004). Moreover, Akt1 null fibroblasts are relatively less motile and invasive than control fibroblasts (Saji et al., 2005; Zhou et al., 2006) raising the possibility that Akt1 effects on motility are cell type specific. In human thyroid carcinoma tissue samples, an increase in phosphorylated Akt is common in the invasive fronts in a manner that co-localizes with Akt1, but not Akt2 or 3 (Vasko et al., 2004). Moreover, mutations in the pleckstrin homology (PH) domain of Akt1, but not Akt2 or 3 have been identified in metastatic thyroid cancer tissue from patients with recurrence that does not respond to standard therapies (Ricarte-Filho et al., 2009). These data suggest that Akt1 may play an important role in promoting thyroid cancer progression.
In animal models, Akt activation induced by loss of expression of phosphate and tensin homolog located on chromosome ten (PTEN), either in all tissues or just in the thyroid, leads to goiter formation and in some cases, minimally invasive thyroid cancer (Di Cristofano et al., 2001; Podsypanina et al., 1999). Tumor formation is reduced by crossing the mice into an Akt1 deficient background (Chen et al., 2006). However, the impact of Akt isoforms on metastases could not be assessed using this system and most endogenous in vivo thyroid cancer models develop metastases only after the tumors are anaplastic (Kim and Zhu, 2009). One model of thyroid cancer in which mice develop metastatic thyroid cancer with differentiated features is the thyroid hormone receptor (TR) ß PV knock-in model (Suzuki et al., 2002). In this model, mice homozygous for a knock-in of the PV mutation of TRβ develop thyroid hormone resistance, high thyroid stimulating hormone (TSH) levels and metastatic thyroid cancer that maintains differentiated features. Although the TSH levels are supraphysiological, this model allows for identification of pathways involved in differentiated thyroid cancer metastatic progression. We previously reported that similar to human thyroid cancer, increased activation of Akt occurred in the primary tumor and distant metastatic tissues from these mice and that Akt1 co-localized with pAkt in the primary tumors (Kim et al., 2005). In vitro studies using primary cultured thyrocytes from the tumors demonstrated Akt-dependent invasiveness (Kim et al., 2005). Mechanistic studies demonstrate that the TRßPV has a high affinity for the regulatory domain of PI3K even when not bound to T3, thereby causing constitutive PI3K signaling (Furuya et al., 2006). In vivo, the tumor progression is dependent on PI3K activity based on pharmacological studies (Furuya et al., 2007) and to a lesser degree on mTOR using a TORC1 complex inhibitor (Guigon et al., 2010); however, Akt isoform specificity has not been assessed.
The goal of the present study was to determine if TRßPV thyroid cancer development and progression were dependent on Akt1. Our results demonstrate that loss of Akt1 reduced both tumor formation and metastatic progression. This effect occurred without Akt1-related changes in TSH levels. These data demonstrate Akt1 plays both tumor proliferative and pro-metastatic functions role in the TRßPV model system, similar to both correlative studies in human cancers and functional studies in vitro.
Results
Deletion of Akt1 reduces thyroid enlargement in the TRßPVPV mice
PVPV-Akt1WT mice developed goiters beginning at three months of age. Thyroid enlargement was also noted in the PVPV-Akt1KO mice, but it was less dramatic than in the presence of Akt1 (examples shown Figure 1A). This was also identified using ultrasound (supplement Figure 1). Statistically, when normalized for body weight, the increase in thyroid size in PVPV mice was delayed by Akt1 deletion (Figure 1B, p<0.001). Over time, similar body weight-normalized thyroid sizes were achieved. This was felt, in part, to be due to an increase in the death of the PVPV-Akt1WT mice with the largest goiters due to compression of local structures since the metastatic burden was very small. Consistent with this speculation, the rate of spontaneous death was greater in the PVPV-Akt1WT (32%) compared with the PVPV-Akt1KO (19%) mice although the study was not designed to evaluate spontaneous death rate as an end point.
Figure 1.

Thyroid growth in PVPV mice is Akt1-dependent. Panel A: Gross images of the thyroid glands removed at 12 months of age for wild type (WT), PVPV-Akt1WT and PVPV-Akt1KO mice. The marked enlargement of the thyroid in the PVPV-Akt1WT and the tumor vascularity are less evident in the PVPV-Akt1KO mice. Panel B: When normalized for body weight, the thyroid glands of the PVPV-Akt1KO mice are significantly (p<0.001) smaller than age-matched PVPV-Akt1WT mice.
Thyroid stimulating hormone levels are not changed by loss of Akt1
PVPV mice have high TSH levels due to their thyroid hormone resistance (Kaneshige et al., 2000). To assess whether differences in the thyroid histology or tumor behavior correlated with a change in circulating TSH levels, serum TSH was measured at 9, 12, and 15 months of age. PVPV-Akt1WT and −Akt1KO mice had similar TSH levels, and both were statistically greater than levels in the wild type (WT) and homozygous Akt1 null (Akt1KO) littermate control mice (Figure 2, p<0.001). The lack of effect of Akt1 loss on TSH levels is further supported by comparable levels in the WT and Akt1KO control mice. Consistent with the TSH levels, there was no difference in pituitary size or in the incidence of basophil hyperplasia by histology in the presence or absence of Akt1 in the PVPV mice. In both cases, the pituitaries were markedly larger than littermate controls (Supplement Figure 2A).
Figure 2.
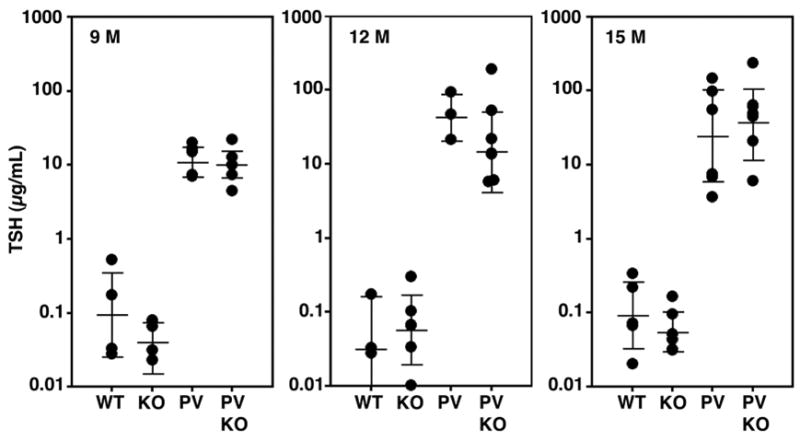
TSH levels are not affected by loss of Akt1. TSH levels were assessed at 9, 12 and 15 months (M) of age in wild-type (WT), Akt1 KO (KO), PVPV-Akt1WT (PV) and PVPV-Akt1KO (PVKO) mice. TSH levels in the WT and KO were similar. Levels in the PV and PVKO mice were both markedly elevated vs. the WT and KO mice (p<0.001) and were statistically similar to each other in all age groups.
Deletion of Akt1 delays tumor progression, vascular invasion, and distant metastases in PVPV mice
Histological examination revealed that ∼75% of the PVPV-Akt1WT mice developed thyroid carcinomas at 6 months of age (Figure 3A and Figure 4A) while PVPV-Akt1KO mice showed only adenomas. At 9 months of age, all PVPV-AktWT mice, but only 2/7 PVPV-Akt1KO, had thyroid carcinomas (Figure 3). The deletion of Akt1 significantly delayed the formation of thyroid carcinoma (p=0.003). Although both PVPV-Akt1WT and –Akt1KO mice developed thyroid carcinomas after 12 months, the histological findings differed between the two groups. Carcinomas in PVPV-Akt1WT mice were characterized by solid areas comprised of neoplastic cells with large nuclei (Figure 4D) in addition to intravascular thrombi (Figures 4B and 4C). In contrast, the majority of the PVPV-Akt1KO mice had adenomas at 9 months (Figure 4F). When carcinomas developed in these mice, only minimal capsular invasion was identified and the tumors maintained a follicular morphology (Figures 4G and 4H). Vascular invasion in thyroid carcinomas from the PVPV-Akt1KO mice was rare (only one mouse) even at 15 months (Figures 4g and h). Tumor associated macrophages were not identified by immunostaining in thyroid tissues from either genotype. Thyroglobulin mRNA levels were slightly lower and thyroid transcription factor 1 mRNA levels were similar in the PVPV-Akt1WT and PVPV-Akt1KO mice by quantitative RT-PCR (PVPV-Akt1KO/PVPV-Akt1WT ratio 0.655 ±0.144 and 1.309 ±0.438, respectively).
Figure 3.

Thyroid cancer development and metastases are Akt1-dependent. Panel A: There is a delay in the development of thyroid cancer in the PVPV-Akt1KO mice vs. PVPV-Akt1WT mice (p=0.003). Panel B: The incidence of distant metastases was reduced in PVPV-Akt1KO mice (p=0.04) and no distant metastases were identified at 15 months of age.
Figure 4.

Histological evaluation of thyroid cancers in PVPV-Akt1WT and −Akt1KO mice. Panels A-E are PVPV-Akt1WT mice; Panel A: Carcinoma with minimal invasion and retention of follicular morphology at 6 months; Panel B: Evidence of intravascular neoplastic thrombus (arrow) at 9 months; Panel C: Large intravascular tumor thrombus with retention of follicular structure and colloid (arrows) at 12 months; Panel D: Solid area with prominent mitotic figures within carcinoma at 12 months; Panel E: Example of a pulmonary micrometastatic lesion (arrows) demonstrating maintained follicular structure with colloid at 12 months of age. Panels F-H are from PVPV-Akt1 KO mice; Panel F: Adenoma at 9 months of age; Panel G: Minimally invasive carcinoma at 12 months (arrow demonstrates invasion) with no evidence of vascular invasion. H: Carcinoma at 15 months with no evidence of vascular invasion.
Distant metastases to the lungs were only observed in PVPV-Akt1WT (Figures 3B and 4E) at 12 and 15 months of age, thus metastases began to occur between 9 and 12 months. In contrast, no PVPV-Akt1KO mice had detectable distant metastases, up to 15 months of age, even after serial sectioning. Based on significant incidence difference above, we used a directional (single sided test) for metastasis, and this difference was significant (p=0.04). Together these data demonstrate that Akt1 promotes both tumor growth and progression in the PVPV mice.
Akt2 and 3 do not compensate for the loss of Akt Activity in Akt1 in PVPV thyroid carcinomas
Because of the potential for compensatory overexpression and activation of Akt2 and 3 in the thyroid cancers in the PVPV-Akt1KO mice, we examined gene and protein expression of Akt1, Akt2 and Akt3 by real-time RT-PCR, IHC, and Western blot. Akt1 mRNA and protein were not detectable in the Akt1KO and PVPV-Akt1KO mice. Akt2 and 3 mRNA levels were not significantly different between WT and PVPV-Akt1WT in thyroid tissues at the mRNA level; although Akt2 level tended to be higher in the Akt1KO mice. Similar results were obtained in the pituitary gland at the mRNA level (Table 1) and protein levels on IHC (Supplement 2B).
Table 1. Gene expression of Akt isoforms in thyroid and pituitary tissues.
mRNA levels of each Akt isoform were examined by quantitative RT-PCR. For each genotype group for thyroid and pituitary, 4 mice were included. Results were normalized to expression levels in either wild-type (thyroid) or PVPV-Akt1WT (pituitary).
| A : Thyroid | |||
|---|---|---|---|
| Akt1 | Akt2 | Akt3 | |
| WT | 1 | 1 | 1 |
| Akt1 KO | 0 | 1.79 ± 0.84 | 1.12 ± 0.53 |
| PVPV-Akt1 WT | 0.86 ± 0.39 | 0.96 ± 0.55 | 0.81 ± 0.34 |
| PVPV–Akt1 KO | 0 | 1.24 ± 0.57 | 1.11 ± 0.50 |
| B : Pituitary | |||
|---|---|---|---|
| Akt1 | Akt2 | Akt3 | |
| PVPV-Akt1 WT | 1 | 1 | 1 |
| PVPV–Akt1 KO | 0 | 1.46 ± 0.43 | 1.07 ± 0.80 |
IHC of the thyroid revealed Akt1, 2, and 3 immunoactivity in the PVPV-WT thyroid carcinomas and WT controls. IHC also demonstrated loss of Akt1 in the thyroid tissue with no convincing evidence of compensatory enhanced expression of Akt2 or Akt 3 in the absence of Akt1, although there was some evidence of an increase in Akt2 localization to the nucleus in the absence of Akt1 (Figure 5A). Interestingly, despite retained expression of Akt2 and Akt3, immunoactive pAkt levels were markedly reduced in the absence of Akt1 in the thyroid carcinomas (Figure 5B), suggesting that Akt2 and 3 were not able to compensate for loss of Akt1 function in the thyroid carcinomas. Western blot confirmed the loss of Akt1, the absence of compensatory overexpression of Akt2 and 3, and the lower levels of phosphorylated Akt in the thyroid tumors (supplement Figure 3).
Figure 5.

Akt isoform expression and Akt activation in PVPV-Akt1KO mice. Panel A: Akt isoform expression in thyroid tissues from PVPV-Akt1WT, PVPV-Akt1KO, and wild type (WT) mice at 12 months of age. There is loss of Akt1 expression in the PVPV-Akt1KO mice with retention of Akt2 and 3 immunoreactivity. Panel B: PVPV-Akt1KO mouse thyroid carcinomas at 12 months demonstrate near complete loss of pAkt, redistribution of p27 from the cytosol to the nucleus, and re-expression of gelsolin.
p27Kip1 and gelsolin, are regulated by Akt1 in PVPV thyroid carcinomas
Akt phosphorylates p27 leading to its cytosolic sequestration (Fujita et al., 2002; Liang et al., 2002; Shin et al., 2002; Viglietto et al., 2002). This effect has been demonstrated to be important for Akt pro-motility effects in vitro (Saji et al., 2005; Wu et al., 2006). We therefore assessed expression levels and localization of p27 in the thyroid cancers. In comparison to PVPV-Akt1WT mice that had cytosolic-predominant p27 immunoactivity, the PVPV-Akt1KO mice demonstrated nuclear-predominant p27, consistent with loss of Akt function despite continued expression of Akt 2 and 3 (Figure 5B). Moreover, expression of gelsolin, a stabilizer of actin that is activated by phosphoinositides (Chellaiah et al., 2001), is markedly reduced in the PVPV-Akt1WT mice at the mRNA and protein levels and its re-expression reduces PVPV thyroid cancer cell invasiveness in vitro (Kim et al., 2007). To determine if Akt1 regulated gelsolin levels in vivo, we assessed gelsolin IHC and demonstrated that protein levels were restored in PVPV-Akt1KO (Figure 5B). Taken together with the loss of pAkt immunoactivity, these results are consistent with the loss of Akt function in the thyroid tumors in the PVPV-Akt1KO mice.
Discussion
The three Akt proteins exhibit structural and functional similarities and together function to increase glucose utilization, promote cell growth, regulate cell motility, and inhibit apoptosis. Opposing Akt1 isoform-specific functions on cell motility, invasion, and metastatic tumor progression have been reported in breast cancer models (Dillon and Muller, 2010; Héron-Milhavet et al., 2006; Irie et al., 2005; Liu et al., 2006). In addition, Akt2 has been shown to be a key stimulator of neutrophil migration (Chen et al., 2010). These observations are particularly important given the development of both generalized and isoform specific Akt inhibitors for clinical trials. Prior work in thyroid cancer cells, cancer cells from other organs, vascular endothelial cells, and fibroblasts, have identified a pro-motility function for Akt1 in vitro (Fernandez-Hernando et al., 2009; Meng et al., 2006; Saji et al., 2005; Zhou et al., 2006). These differences could be related to the specific cell lines and assays or they might represent tissue-specific responses to Akt isoforms.
In the present study, we report that Akt1 deletion delays thyroid cancer formation in the PVPV mouse model and reduces vascular invasion and distant metastases over 15 months in vivo. The loss of Akt1 was associated with a marked reduction in immunoreactive phosphorylated Akt in the tumors despite continued expression of Akt2 and 3. The re-localization of p27 to the nucleus provides further evidence for the loss of total Akt activation in thyroid cancer tissues. It is not clear why loss of Akt1 is not rescued by Akt2 or 3 in the thyroid; however, the data are consistent with those from PTEN null mice in which 80% of mice with a Pten+/- genotype develop thyroid tumors while only 43% develop tumors when they lack Akt1 (Chen et al., 2006). In the same study, prostate cancer incidences in both groups were similar supporting the notion of tissue specific effects of the Akt isoforms.
TSH levels are elevated in the TRβ PV mouse model [Figure 2 and (Suzuki et al., 2002)]. TSH is a major growth factor for thyroid cells (reviewed in (Kimura et al., 2001)) that cooperates with Akt signaling in inducing cell proliferation (Coulonval et al., 2000; Saito et al., 2001). The effect of Akt1 deletion in the thyroid carcinomas occurred despite similar TSH levels in the PVPV-Akt1WT and PVPV-Akt1KO. Thus, it is not likely that changes in TSH alone are responsible for the results. However, the Akt1 effects in the thyroid may still require increased TSH receptor signaling. Indeed, TSH receptor signaling has been shown to play an important growth promoting role in thyroid cancer in the PV mouse model, but it is not sufficient to induce thyroid cancer in the mouse (Lu et al., 2010). This may account for the difference between the thyroid and pituitary changes, since pituitary tissue does not express the TSH receptor. Thus, in this model system, co-activation of TSH receptors and other signaling pathways are required for the aggressive phenotype to develop. The principal advantage of this model for the present study is the reliable development of pulmonary micrometastases that retain differentiated features; however, it is also recognized that the very high TSH levels and the PV mutation in the TRβ receptor are not typical in human thyroid cancer.
The mechanisms by which Akt1 loss reduces thyroid cancer metastasis in this model are not certain and were not directly tested in this study. It is possible that novel interacting proteins may exist for Akt and TRβ in the mice. This possibility was not tested in the present manuscript. However, two potential mechanisms were explored based on prior functional studies. We previously reported that Akt-induced cell motility is dependent on p27 expression in vitro (Saji et al., 2005). Akt is known to phosphorylate p27 at several locations leading to its cytosolic sequestration and ability to bind to actin filaments (Fujita et al., 2002; Larrea et al., 2009; Liang et al., 2002; McAllister et al., 2003; Shin et al., 2002; Viglietto et al., 2002; Wu et al., 2006). In the PV mouse thyroid cancers, we demonstrate that increased levels of nuclear pAkt and cytoplasmic-predominant p27, consistent with Akt activation. In the absence of Akt1, pAkt levels were reduced and p27 relocalized to the nucleus, consistent with loss of Akt function. Gelsolin is a phosphoinositide-regulated protein involved in capping of actin filaments which reduces cell motility (Chellaiah et al., 2001; Tanaka et al., 2006). Loss of gelsolin expression is associated with aggressive tumor behavior or tumor development and its reexpression reduces cancer cell invasiveness and mesenchymal features (Mielnicki et al., 1999; Ni et al., 2008; Tanaka et al., 2006). Previously published microarray studies demonstrated a reduction of gelsolin mRNA levels in PVPV tumors (Ying et al., 2003). Furthermore, primary cultures of PVPV thyroid cancer cell invasion requires loss of gelsolin (Kim et al., 2007). Therefore, we were interested in determining whether loss of Akt1 altered gelsolin levels in the tumors. Indeed the PVPV-Akt1KO thyroid tumors displayed markedly increased levels of gelsolin (Figure 5). The role of Akt1 in regulating gelsolin transcription, translation, or protein stability requires further study.
It is possible that distant metastases will be identified as the mice continue to age and that the reduction in distant metastases is related directly to the delay in primary tumor progression. The absence of vascular invasion and the absence of distant metastases even 3-6 months after the cancers developed suggest that Akt1 has an effect on the metastatic process; however, this has not been directly proven. Nonetheless, it is important to recognize that in contrast to breast cancer, the results do not support a metastasis suppressing role for Akt1 in the PVPV model of thyroid cancer.
It is also important to recognize that we have utilized a generalized Akt1 null system and the genetic background is not pure c57/BL6J. Thus, while we have identified effects of Akt1 loss in the thyroid cancer cells, Akt1 is also deleted in other tissues that can impact on cancer progression. As with all mouse models, there also may be strain-specific modifier effects. Indeed, recent data demonstrate that activation of Akt signaling in the stroma can independently influence the behavior of breast cancer in mice (Trimboli et al., 2009). These non-cancer cell or strain-specific effects may also influence the apparent tissue specificity of the Akt1 effects.
In summary, we have demonstrated for the first time that thyroid cancer development, vascular invasion, and distant metastases in the TRβ PV mouse model are dependent on Akt1. These Akt1 isoform-specific effects occur with similarly high TSH levels, in the presence of Akt2 and 3, and in association with changes in p27 localization and gelsolin expression. These results demonstrate that Akt1 functions to promote tumor development and progression in the PVPV model of thyroid cancer.
Materials and Methods
Mice
Akt1+/- mice on a C57BL/6J background were obtained from Jackson Laboratory (#004912, Bar Harbor, MI) (Cho et al., 2001b) and homozygous Akt1 knockout (Akt1-/-) mice were generated. The homozygous Akt1-/- mice were mated with heterozygous TRßPV (TRßPV/-) on a C57BL/6Jx129SV (Kaneshige et al., 2000) to create homozygous TRßPV (TRßPV/PV) and TRßPV/PVAkt1-/- mice. In this manuscript, we use the term PVPV-Akt1WT for TRßPV/PV, PVPV-Akt1KO for TRßPV/PV-Akt1-/-. For genotyping, crude DNA was isolated from tail biopsies and PCR was performed using previously described methods and primers (Cho et al., 2001b; Kaneshige et al., 2000). Mice were sacrificed at 3, 6, 9, 12, and 15 months of age and the thyroid, lung, pituitary, and liver tissues were harvested, and fixed with 10% formalin and stored in -80 freezer until protein and RNA were isolated. Body weight (BW) and thyroid size were measured at necropsy. Thyroid volume was calculated by the following formula: [Volume] = [Longest length] × [Shortest length] ˆ2 × 0.52. Mouse studies were approved by the Ohio State University Institutional Animal Care and Use Committee.
Antibodies
Antibodies against total Akt (#9272), H40, and GAPDH (#2118) were purchased from Cell Signaling Technology (Danvers, MA). Antibodies against phosphorylated (Ser 473)-Akt (sc-7985-R), Akt3 (sc-11520), gelsolin (sc-48769) and α-tubulin (sc-5546) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibody against GAPDH (NB300-327) was from NOVUS Biologicals, Inc. (Littleton, CO). Antibody against Akt1 (#610860) and p27(Kip1) (#554069) were from BD Biosciences (Billerica, MA). Antibody against Akt2 (#06-606) was from Upstate Biotechnology (Lake Placid, NY). Anti-CD34 was from Abcam (Cambridge, MA) and anti F4/80 was from Invitrogen (Carlsbad, CA). Specificity of isoform-specific anti-Akt antibodies was previously confirmed (Saji et al., 2005; Vasko et al., 2004). Secondary antibodies against rabbit and mouse were from Cell Signaling, and against sheep, from Upstate Biotechnology.
Histology and Immunohistochemistry
Thyroid, lung, and pituitary tissues were fixed in 10% zinc formalin (Fisher Scientific, Pittsburgh, PA) for 3 days, processed by routine methods, embedded in paraffin, and 4-μm-sections were made. Hematoxylin and eosin (H&E) stained slides were evaluated by light microscopy by one veterinary pathologist (KLP). When metastases were not identified in lung tissues, an additional 10 sections from different depths were cut and subjected to H&E staining. For immunohistochemistry (IHC), sections were dewaxed and soaked in alcohol. After microwave treatment in antigen unmasking solution (Vector Laboratory, Inc., Burlingame, CA) for 10 min, endogenous peroxidase activity was inactivated by incubating in 3% hydrogen peroxide for 15 min and sections were incubated with primary antibody in PBS at 4°C overnight. After washing with PBS, immunostaining was performed using the Vectastain Universal Quick Kit and Peroxidase Staining Kit (DAB) according to the manufacturer's instructions. Antiserum was omitted in the negative control. H&E staining and IHC were reviewed by at least three investigators including KLP, MS, MDR, and VVV; IHC results were interpreted independently and consensus was obtained if qualitative interpretations differed.
TSH measurement
Blood was harvested at necropsy, and serum was collected after centrifugation of blood at 3,000 × g for 15 min and stored at -80 C until the assay was performed. Since higher levels of TSH were expected in PVPV mice (Kaneshige et al., 2000), serum was diluted with TSH-free mouse serum (Lu et al., 2010). Serum TSH levels were determined as described (Pohlenz J Fau - Maqueem et al., 1999).
RNA isolation and analysis
Half of the thyroid tissue from each mouse and the entire pituitary gland from randomly selected mice were stored at -80°C. Tissues were homogenized in 1 ml of Trizol (Invitrogen), and after addition of 200 μl of chloroform, tubes were shaken, incubated for 2 min at room temperature and centrifuged at 16,000 g for 15 min at 4°C. The supernatant was mixed with 500 μl of isopropanol, and centrifuged at 16,000 g for 15 min at 4°C. The supernatant was aspirated and the pellets containing RNA were washed using 70% ethanol and air-dried. The RNA was then reconstituted in DEPC-treated water and concentration was measured by spectrophotometry (230/260/280).
For RT-PCR, 880 ng of RNA were treated with DNase I (Invitrogen) for 15 min and 400 ng of DNase-treated RNA were reversed transcribed using the TaqMan® Reverse Transcription Reagents Kit (Applied Biosystems). Quantitative PCR was performed in 96 sample plates using cDNA equivalent to 24 ng of total RNA (3 μl of RT reaction mixture) per 20 μl per well. As an internal control to normalize gene expression, 18S ribosomal RNA was amplified using Taqman Ribosomal RNA Control Reagents Kit as described (Ringel et al., 2001). Specific primers for Akt1 were the following: forward 5′-AGC TCT TCT TCC ACC TGT CTC (Exon 7-8) and reverse CGG CGT TCC GCA GAA TGT (Exon 9). Specific primers and probes for Akt2 (Mm00545827_m1), Akt3 (Mm00442194_m1), thyroglobulin (Mm00447525_m1), TTF-1 (Mm00657018_m1), and gelsolin (Mm00456679_m1 were purchased from Applied Biosystems and PCR was performed with SYBR Green Mix (Applied Biosystems) for Akt1 and TaqMan® Universal PCR Master Mix (Applied Biosystems) for other genes using the following parameters: after an initial 2 min incubation at 50°C for inactivation of AmpEASE UNG activity, the cDNA was denatured at 94°C for 10 min and samples were subjected to 40 cycles of a two-step amplification protocol that included 15 seconds at 94°C and a one-minute annealing-elongation step at 60°C. Target gene and 18S were amplified in all samples in duplicate in two separate reactions. Negative controls were included for the RT (RT negative) and PCR (Non-Template Control) reactions. To compare expression levels, difference of threshold (ΔCt) were calculated by subtracting threshold (Ct-18S) of 18S from threshold (Ct-gene) of the target gene, and expression levels were estimated by 2ˆ(-ΔCt). All samples were electrophoresed to confirm the sizes of the amplicons and reaction specificity.
Statistical Analysis
Thyroid volume, adjusted for body weight, and TSH levels were assessed by linear models to estimate and test differences due to the presence or absence of Akt1. The primary hypothesis was that Akt1 loss would inhibit tumor formation. The effect of Akt1 on tumor formation at fixed ages was assessed using logistic regression modeling. To assess subsequent effects of Akt1 on the development of distant metastases, logistic regression was performed beginning at 12 months of age, as that was the age at which distant metastases were expected in the PVPV mice. Significance tests were performed at the α=0.05 level (double-sided) for the primary endpoints of thyroid size and incidence of tumor formation. The number of mice used for each of the age groups provided 83% power to detect an effect size of Akt1 on tumor incidence consistent with the prior studies which demonstrated complete penetrance of thyroid cancer formation in the PVPV mice by 9 months of age (Suzuki et al., 2002) and the anticipated reduction in tumor formation with Akt1 loss (Chen et al., 2006). Power for thyroid tumor volume as a continuous indicator of growth was much higher. Finally, significance testing for distant metastases was one-sided, because its direction depended on the results from the test of incidence. For the logistic regression modeling, exact tests were used to accommodate the small sample sizes and the potential for zero events.
Supplementary Material
Acknowledgments
Funding for the work is from NIH grants (P01CA124570 and R01CA 102572) to MDR. We appreciate the technical assistance of Michael Ostrowski, John Thompson and Jun Liu.
Footnotes
Conflict of Interest: Dr. Ringel has received honoraria for advisory boards from Veracyte, Inc and Astra Zeneca. This was not related to the work presented in the manuscript. He has received funding from NIH for this work. M. Saji, K. Narahara, S. McCarty, V. Vasko, K. La Perle, K. Porter, D. Jarjoura, C. Lu, and S-Y Cheng all declare no conflicts of interest.
References
- Chellaiah MA, Biswas RS, Yuen D, Alvarez UM, Hruska KA. Phosphatidylinositol 3,4,5-trisphosphate directs association of Src homology 2-containing signaling proteins with gelsolin. J Biol Chem. 2001;276:47434–44. doi: 10.1074/jbc.M107494200. [DOI] [PubMed] [Google Scholar]
- Chen J, Tang H, Hay N, Xu J, Ye RD. Akt isoforms differentially regulate neutrophil functions. Blood. 2010;115:4237–4246. doi: 10.1182/blood-2009-11-255323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ML, Xu PZ, Peng XD, Chen WS, Guzman G, Yang X, et al. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/- mice. Genes Dev. 2006;20:1569–1574. doi: 10.1101/gad.1395006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007;67:1979–1987. doi: 10.1158/0008-5472.CAN-06-1479. [DOI] [PubMed] [Google Scholar]
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001a;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001b;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- Coulonval K, Vandeput F, Stein RC, Kozma SC, Lamy F, Dumont JE. Phosphatidylinositol 3-kinase, protein kinase B and ribosomal S6 kinases in the stimulation of thyroid epithelial cell proliferation by cAMP and growth factors in the presence of insulin. Biochem J. 2000;348(Pt 2):351–358. [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222–224. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- Dillon RL, Marcotte R, Hennessy BT, Woodgett JR, Mills GB, Muller WJ. Akt1 and akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer Res. 2009;69:5057–5064. doi: 10.1158/0008-5472.CAN-08-4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon RL, Muller WJ. Distinct Biological Roles for the Akt Family in Mammary Tumor Progression. Cancer Res. 2010;70:4260–4264. doi: 10.1158/0008-5472.CAN-10-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Hernando C, Jozsef L, Jenkins D, Di Lorenzo A, Sessa WC. Absence of Akt1 reduces vascular smooth muscle cell migration and survival and induces features of plaque vulnerability and cardiac dysfunction during atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:2033–40. doi: 10.1161/ATVBAHA.109.196394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2002;277:28706–28713. doi: 10.1074/jbc.M203668200. [DOI] [PubMed] [Google Scholar]
- Furuya F, Hanover JA, Cheng Sy. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone ß receptor. Proc Natl Acad Sci USA. 2006;103:1780–1785. doi: 10.1073/pnas.0510849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya F, Lu C, Willingham MC, Cheng SY. Inhibition of phosphatidylinositol 3′ kinase delays tumor progression and blocks metastatic spread in a mouse model of thyroid cancer. Carcinogenesis. 2007;28:2451–2458. doi: 10.1093/carcin/bgm174. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, McGraw TE. Insulin-modulated Akt subcellular localization determines Akt isoform-specific signaling. Proc Natl Acad Sci U S A. 2009;106:7004–9. doi: 10.1073/pnas.0901933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigon CJ, Fozzatti L, Lu C, Willingham MC, Cheng SY. Inhibition of mTORC1 signaling reduces tumor growth but does not prevent cancer progression in a mouse model of thyroid cancer. Carcinogenesis. 2010;31:1284–1291. doi: 10.1093/carcin/bgq059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Héron-Milhavet L, Franckhauser C, Rana V, Berthenet C, Fisher D, Hemmings BA, et al. Only Akt1 Is Required for Proliferation, while Akt2 Promotes Cell Cycle Exit through p21 Binding. Mol Cell Biol. 2006;26:8267–8280. doi: 10.1128/MCB.00201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson JN, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt-1 (PKB-α) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer Res. 2004;64:3171–3178. doi: 10.1158/0008-5472.can-03-3465. [DOI] [PubMed] [Google Scholar]
- Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter TA, et al. Mice with a targeted mutation in the thyroid hormone beta receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci U S A. 2000;97:13209–13214. doi: 10.1073/pnas.230285997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CF, Vasko VV, Kato Y, Kruhlak M, Saji M, Cheng SY, et al. AKT Activation Promotes Metastasis in a Mouse Model of Follicular Thyroid Carcinoma. Endocrinology. 2005;146:4456–4463. doi: 10.1210/en.2005-0172. [DOI] [PubMed] [Google Scholar]
- Kim CS, Furuya F, Ying H, Kato Y, Hanover JA, Cheng Sy. Gelsolin: A Novel Thyroid Hormone Receptor-ß Interacting Protein that Modulates Tumor Progression in a Mouse Model of Follicular Thyroid Cancer. Endocrinology. 2007;148:1306–1312. doi: 10.1210/en.2006-0923. [DOI] [PubMed] [Google Scholar]
- Kim CS, Zhu X. Lessons from Mouse Models of Thyroid Cancer. Thyroid. 2009;19:1317–1331. doi: 10.1089/thy.2009.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of Thyroid Cell Proliferation by TSH and Other Factors: A Critical Evaluation of in Vitro Models. Endocr Rev. 2001;22:631–630. doi: 10.1210/edrv.22.5.0444. [DOI] [PubMed] [Google Scholar]
- Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D, et al. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci U S A. 2009;106:9268–9273. doi: 10.1073/pnas.0805057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- Liu H, Radisky DC, Nelson CM, Zhang H, Fata JE, Roth RA, et al. Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proc Natl Acad Sci U S A. 2006;103:4134–4139. doi: 10.1073/pnas.0511342103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Zhao L, Ying H, Willingham MC, Cheng Sy. Growth Activation Alone Is Not Sufficient to Cause Metastatic Thyroid Cancer in a Mouse Model of Follicular Thyroid Carcinoma. Endocrinology. 2010;151:1929–1939. doi: 10.1210/en.2009-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister SS, Becker-Hapak M, Pintucci G, Pagano M, Dowdy SF. Novel p27(kip1) C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol. 2003;23:216–228. doi: 10.1128/MCB.23.1.216-228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q, Xia C, Fang J, Rojanasakul Y, Jiang BH. Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell Signal. 2006;18:2262–2271. doi: 10.1016/j.cellsig.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Mielnicki LM, Ying AM, Head KL, Asch HL, Asch BB. Epigenetic regulation of gelsolin expression in human breast cancer cells. Exp Cell Res. 1999;249:161–76. doi: 10.1006/excr.1999.4461. [DOI] [PubMed] [Google Scholar]
- Ni XG, Zhou L, Wang GQ, Liu SM, Bai XF, Liu F, et al. The ubiquitin-proteasome pathway mediates gelsolin protein downregulation in pancreatic cancer. Mol Med. 2008;14:582–9. doi: 10.2119/2008-00020.Ni. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96:1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlenz J, Maqueem A, Cua K, Weiss RE, Van Sande J, Refetoff S. Improved radioimmunoassay for measurement of mouse thyrotropin in serum: strain differences in thyrotropin concentration and thyrotroph sensitivity to thyroid hormone. Thyroid. 1999;9:1265–1271. doi: 10.1089/thy.1999.9.1265. [DOI] [PubMed] [Google Scholar]
- Ricarte-Filho JC, Ryder M, Chitale DA, Rivera M, Heguy A, Ladanyi M, et al. Mutational Profile of Advanced Primary and Metastatic Radioactive Iodine-Refractory Thyroid Cancers Reveals Distinct Pathogenetic Roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69:4885–4893. doi: 10.1158/0008-5472.CAN-09-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringel MD, Hayre N, Saito J, Saunier B, Schuppert F, Burch H, et al. Overexpression and Overactivation of Akt in Thyroid Carcinoma. Cancer Res. 2001;61:6105–6111. [PubMed] [Google Scholar]
- Saito J, Kohn AD, Roth RA, Noguchi Y, Tatsumo I, Hirai A, et al. Regulation of FRTL-5 thyroid cell growth by phosphatidylinositol (OH) 3 kinase-dependent Akt-mediated signaling. Thyroid. 2001;11:339–351. doi: 10.1089/10507250152039073. [DOI] [PubMed] [Google Scholar]
- Saji M, Vasko V, Kada F, Allbritton EH, Burman KD, Ringel MD. Akt1 contains a functional leucine-rich nuclear export sequence. Biochem Biophys Res Commun. 2005;332:167–173. doi: 10.1016/j.bbrc.2005.04.109. [DOI] [PubMed] [Google Scholar]
- Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27Kip1 at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Woodgett JR. Functional distinctions of protein kinase B/Akt isoforms defined by their influence on cell migration. Trends Cell Biol. 2006;16:461–466. doi: 10.1016/j.tcb.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Willingham MC, Cheng SY. Mice with a mutation in the thyroid hormone receptor beta gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid. 2002;12:963–969. doi: 10.1089/105072502320908295. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Shirkoohi R, Nakagawa K, Qiao H, Fujita H, Okada F, et al. siRNA gelsolin knockdown induces epithelial-mesenchymal transition with a cadherin switch in human mammary epithelial cells. Int J Cancer. 2006;118:1680–91. doi: 10.1002/ijc.21559. [DOI] [PubMed] [Google Scholar]
- Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461:1084–1091. doi: 10.1038/nature08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasko V, Saji M, Hardy E, Kruhlak M, Larin A, Savchenko V, et al. Akt activation and localization correlate with tumor invasion and oncogene expression in thyroid cancer. J Mol Genet. 2004;41:161–170. doi: 10.1136/jmg.2003.015339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viglietto G, Motti ML, Bruni P, Melillo RM, D'Alessio A, Califano D, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27Kip1 by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8:1136–1144. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- Wu FY, Wang SE, Sanders ME, Shin I, Rojo F, Baselga J, et al. Reduction of cytosolic p27(Kip1) inhibits cancer cell motility, survival, and tumorigenicity. Cancer Res. 2006;66:2162–2172. doi: 10.1158/0008-5472.CAN-05-3304. [DOI] [PubMed] [Google Scholar]
- Ying H, Suzuki H, Furumoto H, Walker R, Meltzer P, Willingham MC, et al. Alterations in genomic profiles during tumor progression in a mouse model of follicular thyroid carcinoma. Carcinogenesis. 2003;24:1467–1479. doi: 10.1093/carcin/bgg111. [DOI] [PubMed] [Google Scholar]
- Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A. Akt Blocks Breast Cancer Cell Motility and Invasion through the Transcription Factor NFAT. Mol Cell. 2005;20:539–550. doi: 10.1016/j.molcel.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Zhou GL, Tucker DF, Bae SS, Bhatheja K, Birnbaum MJ, Field J. Opposing roles for Akt1 and Akt2 in Rac/Pak signaling and cell migration. J Biol Chem. 2006;281:36443–36453. doi: 10.1074/jbc.M600788200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.