

Abstract
As core skeletons of lamellarins: 5,6-Dihydropyrrolo[2,1-a]isoquinolines are one of the important alkaloids that exhibit significant biological activities, in this study, an efficient synthetic route was described for two novel compounds, 5,6-dihydropyrrolo[2,1-a]isoquinolines I and II. Compound I was synthesized from isovanillin with 28.3% overall yield by a six-step reaction while II from 2-(3, 4-dimethoxyphenyl) ethanamine was with 61.6% overall yield by a three-step reaction. And the structures of these two compounds were confirmed by means of IR spectrum, 1H NMR, 13C NMR, MS, HRMS, and melting point measurements.
1. Introduction
Lamellarins are a group of hexacyclic marine alkaloids that were initially isolated from a prosobranch mollusk by Faulkner and coworkers in 1985 [1]. Since then, over 70 compounds belonging to this group have been isolated and identified [2].
Some of these lamellarins and related compounds exhibit interesting biological activities in multidrug resistance (MDR) and their corresponding parental cell lines [3]. As well known [4], Lamellarin D (LMD) exhibits a significant cytotoxicity against a large panel of cancer cell lines and is a potential non-CPT (camptothecin) topoisomerase 1 poison [5, 6]. LMD affects cell cycle and acts on cancer cell mitochondria to induce apoptosia [7].
Due to the fascinating novel structures and biological activities, more and more researchers have devoted into the synthetic studies of lamellarins [8] and related 3,4-diarylpyrrolo derivatives. As one of the important alkaloids, 5,6-Dihydropyrrolo[2,1-a]isoquinolines exhibits pronounced biological activities. The biological activity of 5,6-dihydropyrrolo[2,1-a]isoquinolines I and II was evaluated by their effects on the proliferation of MDA-MB-231 (breast cancer cell line) by MTT assay. Our results showed that compound I could significantly inhibit the proliferation of MDA-MB-231 at the concentration of 40 μg/mL, in contrast, compound II could enhance the proliferation of the MDA-MB-231 at the same concentration. In addition, they are also scaffolds for synthesis of lamellarin analogues [9].
Increasingly elegant synthetic routes have been developed. An efficient synthetic route for two compounds, 8-benzyloxy-9-methoxy-2-(2,4,5-trimethoxyphenyl)-5,6-dihydropyrrolo[2,1-a]isoquinoline (I) and 8,9-dimethoxy-2-(2,4,5-trimethoxyphenyl)-5,6-dihydro[2,1-a]isoquinoline (II), is mainly introduced in this study. The synthetic procedure is very valuable because it employs 5,6-dihydropyrrolo[2,1-a]isoquinolines as starting materials and represents an easy and direct approach to a wide variety of 3,4-dihydroisoquinolines. The synthetic strategy is outlined in Scheme 1.
Scheme 1.
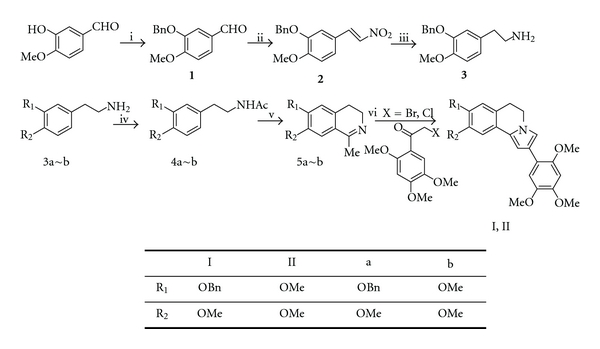
Reagents and conditions: (i) BnCl, K2CO3, EtOH, reflux, 5 h, 94%; (ii) CH3NO2, NH4OAc, AcOH, reflux, 4 h, 80.5%; (iii) LiAlH4, THF, reflux, 6 h, 84.9%; (iv) CH3COCl, Et3N, CH2Cl2, 0°C, 2 h, 4a: 81.9%, 4b: 90.7%; (v) POCl3, CH2Cl2, reflux, 3 h, 5a: 80.0%, 5b: 83.6%; (vi) 2-halogen-1-(2,4,5-trimethoxyphenyl)ethanone, CH3CN, K2CO3, reflux, 20 h, I: 74.7%, II: 72.6%.
Isovanillin was protected with benzyl chloride to get 3-benzyloxy-4-methoxy-benzaldehyde (1) with 84.4% yield [10], which was condensed with nitromethane giving 2-benzyloxy-1-methoxy-4-(2-nitrovinyl)-benzene (2) with 80.5% yield [11]. Compound 2 was then reduced with LiAlH4 to get 2-(3-benzyloxy-4-methoxyphenyl)-vinylamine (3) with 84.9% yield [12]. Treatment of 3a~b with acetylchloride (n(3a~b) : n(CH3COCl) : n(Et3N) = 1 : 1.8 : 4.0) afforded acetamide (4a~b) with 81.9% and 90.7% yield, respectively, followed by cyclization with phosphorous oxychloride to get 3,4-dihydroisoquinoline (5a~b) with 80.0% and 83.6% yield (n(4a) : n(POCl3) = 1 : 8). A solution of 5a~b, 2-bromo-1-(2,4,5-trimethoxy-phenyl)-ethanone and anhydrous K2CO3 in anhydrous acetonitrile was refluxed for 15 h. After a series of treatment, 5,6-dihydropyrrolo[2,1-a]isoquinoline I and II were obtained with 28.3% and 61.6% total yield, respectively.
2. Material and Methods
2.1. Analysis Means of Compounds
Melting points (uncorrected) were determined by a Gongyi X-4 apparatus. Infrared spectra(IR) were determined by Nicolet 550 spectrometer. NMR spectra were recorded by Bruker DRX500 or Bruker DRX400 spectrometer. All data were calibrated at δ 0.00 ppm for 1H spectra and 13C spectra from the original spectra (TMS). Low resolution mass spectra (LRMS) were recorded with an HP 6890/5973 GC-MS mass spectrometer. High resolution mass (HRMS) for unreported compounds were recorded with a Micromass GTC Gas Chromatography/TOF Mass spectrometer. All solvent were redistilled prior to use, unless otherwise stated, all other commercially available chemicals were used without further purification.
2.2. Chemical Synthesis
2.2.1. 3-(benzyloxy)-4-methoxybenzaldehyde (1)
A mixture of isovanillin (10.0 g, 66 mmol), benzyl chloride (16 mL, 139 mmol), and anhydrous K2CO3 (6.5 g, 47 mmol) in EtOH (150 mL) was refluxed for 5 h. After being stirred, the reaction mixture was concentrated to dry and redissolved in 70 mL CH2Cl2, and then 5% aqueous NaOH (3 × 100 mL) was added. The organic layer was washed with brine (2 × 50 mL) and H2O (2 × 50 mL), dried with anhydrous Na2SO4, and evaporated to dryness. Needles were obtained after crystallization from MeOH/CH2Cl2 corresponding to 3-(benzyloxy)-4-methoxybenzaldehyde (15.0 g, 94%): m.p. 61~62°C (lit.13 m.p. 61~62°C); 1H NMR (400 MHz, CDCl3) δ 9.82 (s, 1H), 7.45~7.47 (m, 4H), 7.38 (t, 2H, J = 7.34 Hz), 7.32 (t, 1H, J = 7.34 Hz), 6.99 (d, 1H, J = 8.24), 5.19 (s, 2H), 3.96 (s, 3H); MS (EI, 70 ev) m/z: 242(M+), 92, 91, 79, 77, 65, 63, 51.
2.2.2. (E)-2-(benzyloxy)-1-methoxy-4-(2-nitrovinyl)benzene (2)
A solution of compound 1 (10.0 g, 41 mmol), nitromethane (7 mL, 129 mmol) and NH4OAc (8.0 g, 104 mmol) in AcOH (125 mL) was refluxed for 4 h. After cooling, the mixture was diluted with H2O (100 mL) and extracted with CH2Cl2 (3 × 100 mL). The organic solution was washed with brine (2 × 100 mL) and H2O (2 × 100 mL), dried with anhydrous Na2SO4, and evaporated to dryness. Yellow needles were obtained from EtOH corresponding to (E)-2-(benzyloxy)-1-methoxy-4-(2-nitrovinyl)benzene (2) (9.6 g, 80.5%): m.p. 127~128°C (lit.14 m.p. 125~126°C); 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 13.6 Hz, 1H), 7.33~7.46 (m, 6H) 7.18 (dd, J = 2.0, 8.36 Hz, 1H), 7.03 (d, J = 2.0 Hz, 1H), 6.93 (d, J = 8.36 Hz, 1H), 5.17 (s, 2H), 3.95 (s, 3H); MS (EI, 70 ev) m/z: 285 (M+), 92, 91, 77, 65, 63, 51.
2.2.3. 2-(3-(benzyloxy)-4-methoxyphenyl)ethanamine (3)
A solution of compound 2 (4.0 g, 14.0 mmol) in 14 mL of anhydrous THF was added dropwise to a well-stirred suspension of LiAlH4 (2.0 g, 52.8 mmol) in 50 mL of anhydrous THF and was refluxed for 6 h. After the solution was cooled, the excess reagent was destroyed by dropwise addition of EtOAc and 15% aqueous NaOH. After partial evaporation of the filtered portion, the aqueous solution was extracted with CH2Cl2 (3 × 30 mL), and the organic solution was washed with brine (2 × 20 mL) and H2O (2 × 20 mL), dried with anhydrous Na2SO4, and evaporated to dryness, and then 2-(3-(benzyloxy)-4-methoxyphenyl)ethanamine (3) (3.0 g, 84.9%) was obtained as an oil. 1H NMR (400 MHz, CDCl3) δ 6.73~7.44 (m, 8H), 5.12 (s, 2H), 3.85 (s, 3H), 2.85 (t, J = 6.7 Hz, 2H), 2.62 (t, J = 6.7 Hz, 2H), 2.20 (br s, 2H); MS (EI, 70 ev) m/z: 257 (M+), 229, 228, 167, 137, 92, 91, 65.
2.2.4. N-(3-(benzyloxy)-4-methoxyphenethy)acetamide (4a)
A solution of 0.4 mL of acetyl chloride (5.6 mmol) in 5 mL anhydrous CH2Cl2 was added dropwise at 0°C to a solution of compound 3 (1.0 g, 3.88 mmol) and Et3N (1.7 mL, 12.26 mmol) in 20 mL anhydrous CH2Cl2, with stirring at 0°C for 2 h. After the mixture was stirred, 2.5% aqueous HCl was added and the organic solution was washed with brine (2 × 10 mL) and H2O (2 × 10 mL), dried with anhydrous Na2SO4, evaporated to dryness, and pale-yellow solid was obtained. Crude product was crystallized with EtOAc to afford N-(3-benzyloxy-4-methoxyphenylethyl)acetamide (0.94 g, 81.9%) as white crystals. m.p. 106~108°C (lit.15 m.p. 122~123°C); 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 7.2 Hz, 2H), 7.36 (t, J = 7.2 Hz, 2H), 7.30 (d, J = 7.2 Hz, 1H), 6.84 (d, J = 8.8 Hz, 1H), 6.74 (d, J = 6.8 Hz, 2H), 5.14 (s, 2 H), 3.87 (s, 3H), 3.43 (q, J = 6.8, 12.8 Hz, 2H), 2.70 (t, J = 6.8 Hz, 2H), 1.88 (s, 3H).
2.2.5. 6-(benzyloxy)-7-methoxy-1-methyl-3,4-dihydroiso-quinoline (5a)
A solution of 0.9 mL of POCl3 (9.8 mmol) in 6 mL anhydrous CH2Cl2 was added dropwise at 40°C to a solution of compound 4a (0.4 g, 1.06 mmol) in 10 mL anhydrous CH2Cl2, with stirring at 40°C for 3 h, then was poured into ice-water mixture, 2.5% aqueous NaOH was added to make pH about 12, the aqueous solution was extracted with CH2Cl2 (3 × 20 mL), and the organic solution was washed with brine (2 × 10 mL) and H2O (2 × 10 mL), dried with anhydrous Na2SO4, evaporated to dryness and solid was obtained. The crude product was purified with a silica gel column (Petroleum : EtOAc(v/v) = 3 : 1, 200~300 H) to afford 6-(benzyloxy)-7-methoxy-1-methyl-3,4-dihydroisoquinoline (5a) (0.24 g, 80%) as brick red crystals. m.p. 95~96°C; 1H NMR (500 MHz, CDCl3) δ 7.30~7.44 (m, 5H), 7.01 (s, 1H), 6.7 (s, 1H), 5.17 (s, 2H), 3.90 (s, 3H), 3.61 (t, J = 7.2 Hz, 2H), 2.57 (m, J = 1.3, 7.5 Hz, 2H), 2.35 (t, J = 1.3 Hz, 3H).
2.2.6. 8-benzyloxy-9-methoxy-2-(2,4,5-trimethoxyphenyl)-5,6-dihydropyrrolo[2,1-a]isoquinoline (I)
To a solution of 0.52 g compound 5a (1.85 mmol) in 15 mL anhydrous CH3CN was added 0.45 g 2-bromo-1-(2,4,5-trimethoxy-phenyl)-ethanone (1.85 mmol). The reaction mixture was stirred at 85°C for 10 h, then 0.38 g anhydrous K2CO3 (2.75 mmol) was added and continued to stir for another 10 h. After that the mixture was poured into 15 mL brine and extracted with CH2Cl2 (3 × 15 mL), the combined organic layers were dried with anhydrous Na2SO4, evaporated to dry, and brown oil was obtained. The crude product was purified with a silicagelcolumn (Petroleum : EtOAc(v/v) = 2 : 1, 200~300 H) to afford 8-benzyl-9-methoxy-2-(2,4,5-trimethoxyphenyl)-5,6-dihydropyrrolo[2,1-a]isoquinoline (I) (0.65 g, 74.7%) as offwhite sheet solid. m.p. 128°C; IR (KBr) ν: 2993, 2934, 2830, 1614, 1568, 1529, 1508, 1453,1427, 1365, 1336, 1274, 1166, 1130, 1035, 848, 810, 784, 738, 695 cm−1; 1H NMR (500MHz, CDCl3) δ 7.29–7.46 (m, 5H), 7.12 (d, J = 1.6 Hz, 1H), 7.11 (s, 1H), 7.10 (s, 1H), 6.73 (s, 1H), 6.69 (d, J = 1.6 Hz, 1H), 6.60 (s, 1H), 5.14 (s, 2H), 4.05 (t, J = 6.6 Hz, 2H), 3.94 (s, 3H), 3.91 (s, 3H), 3.91 (s, 3H), 3.88 (s, 3H), 2.95 (t, J = 6.6 Hz, 2H); 13C NMR (500 MHz, CDCl3) δ: 29.61, 44.91, 56.96, 56.97, 57.14, 57.43, 72.14, 99.34, 102.13, 107.32, 112.83, 115.18, 117.55, 120.83, 121.02, 123.60, 123.78, 128.03, 128.03, 128.50, 129.21, 129.21, 130.31, 137.98, 144.06, 147.25, 148.13, 149.82, 151.07; MS (LC-MS) m/z: 472 (M+1)+, 367, 318, 273; HRMS (ESI-Q-TOF) calcd for C29H29NO5 [M+1]+ 472.4856, found 472.4819.
2.2.7. N-(3,4-dimethoxyphenethyl)acetamide (4b)
A solution of 7.6 mL of acetyl chloride (0.11 mol) in 10 mL anhydrous CH2Cl2 was added dropwise at 0°C to a solution of compound 3b (10 mL, 0.059 mol) and Et3N (32.8 mL, 0.23 mol) in 25 mL anhydrous CH2Cl2, with stirring at 0°C for 2 h. After the mixture was stirred, 2.5% aqueous HCl was added and the organic solution was washed with brine (2 × 30 mL) and H2O (2 × 20 mL), dried with anhydrous Na2SO4, evaporated to dryness, and yellow solid was obtained. Crude product was crystallized with EtOAc to afford N-(3,4-dimethoxyphenethyl) acetamide (4b) (11.8 g, 90.7%) as yellow crystals. m.p. 85~86°C (lit.16 m.p. 94°C); IR (KBr) ν: 1642.54 cm−1 (–C=O), 3301.49 cm−1 (–NH–).
2.2.8. 6,7-dimethoxy-1-methyl-3,4-dihydroisoquinoline (5b)
A solution of 9.8 mL of POCl3 (0.1 mol) in 40 mL anhydrous CH2Cl2 was added dropwise at 40°C to a solution of compound 4b (3.0 g, 13.4 mmol) in 30 mL anhydrous CH2Cl2, with stirring at 40°C for 3 h, then was poured into ice-water mixture; 2.5% aqueous NaOH was added to make pH about 12, the aqueous solution was extracted with CH2Cl2 (3 × 60 mL), and the organic solution was washed with brine (2 × 50 mL) and H2O (2 × 50 mL), dried with anhydrous Na2SO4, evaporated to dryness, and solid was obtained. The crude product was purified with a silica gel column (Petroleum : EtOAc(v/v) = 1 : 1, 200~300 H) to afford 6,7-dimethoxy-1-methyl-3,4-dihydroisoquinoline (5b) (2.3 g, 83.6%) as brick red crystals. m.p. 98~99°C (lit.17 m.p. 85~96°C); 1H NMR (500 MHz, CDCl3) δ 6.99 (s, 1H), 6.89 (s, 1H), 3.92 (s, 3H), 3.91 (s, 3H), 3.63 (m, J = 1.4, 7.5 Hz, 2H), 2.63 (t, J = 7.5 Hz, 2H), 2.36 (t, J = 1.4 Hz, 3H).
2.2.9. 8,9-dimethoxy-2-(2,4,5-trimethoxyphenyl)-5,6-dihydro[2, 1-a]isoquinoline (II)
To a solution of 1.5 g compound 5b (7.32 mmol) in 20 mL anhydrous CH3CN was added 1.78 g 2-bromo-1-(2,4,5-trimethoxy-phenyl)-ethanone (7.34 mmol). The reaction mixture was stirred at 85°C for 10 h, then 1.52 g anhydrous K2CO3 (11.0 mmol) was added and continued to stir for another 10 h. After that the mixture was poured into 30 mL brine and extracted with CH2Cl2 (3 × 30 mL), the combined organic layers were dried with anhydrous Na2SO4, evaporated to dryness, and brown oil was obtained. The crude product was purified with a silica gel column (Petroleum : EtOAc(v/v) = 2 : 1, 200~300 H) to afford 8,9-dimethoxy-2-(2,4,5-trimethoxyphenyl)-5,6-dihydro[2,1-a]isoquinoline (II) (0.65 g, 72.6%) as gray solid. m.p. 137~138°C; IR (KBr) ν: 2993, 2934, 2836, 1608, 1560, 1530, 1508, 1484, 1397, 1272, 1212, 1126, 1036, 808, 776 cm−1; 1H NMR (500 MHz, CDCl3) δ: 3.01 (t, J = 6.6 Hz, 2H), 3.87 (s, 3H), 3.88 (s, 3H), 3.91 (s, 3H), 3.92 (s, 3H), 3.95 (s, 3H), 4.07 (t, J = 6.6 Hz, 2H), 6.60 (s, 1H), 6.69 (d, J = 1.7 Hz, 1H), 6.70 (s, 1H), 7.08 (s, 1H), 7.12 (s, 1H), 7.13 (d, J = 1.7 Hz, 1H); 13C NMR (500 MHz, CDCl3) δ: 29.07, 44.28, 56.08, 56.15, 56.30, 56.49, 56.78, 98.68, 106.08, 101.36, 111.51, 112.19, 116.90, 120.12, 120.36, 122.43, 122.93, 129.70, 143.40, 147.38, 147.47, 148.38, 150.42; DEPT 135 (500 MHz, CDCl3) δ: two –CH2 (29.06, 44.28), five –CH3 (56.07, 56.14, 56.29,56.48,56.76), six –CH (98.62, 101.34, 106.05, 111.47, 112.15, 120.11); MS (LC-MS) m/z: 396 (M+1)+, 371, 276; HRMS (ESI-Q-TOF) calcd for C23H25NO5 [M+1]+ 396.4852, found 396.4884.
3. Results
The target compounds I and II had been synthesized by our route and their structures were determined by interpretation of spectral data. The 1H NMR and 13C NMR spectra of them were assigned as indicated in Figures 1, 2, 3, and 4.
Figure 1.
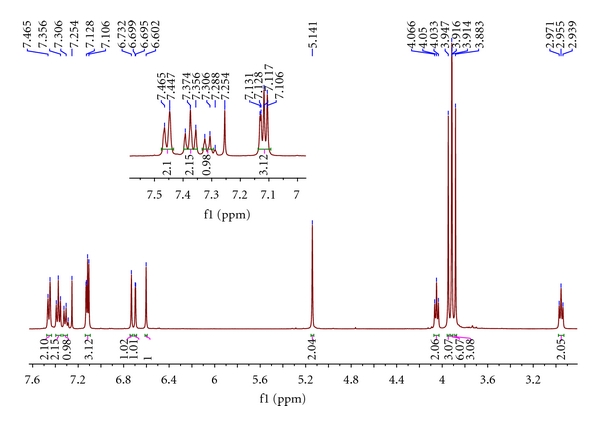
1H NMR spectrum of the I. Inserted figure is the magnification of the part of 7.00–7.50 of chemical shift.
Figure 2.
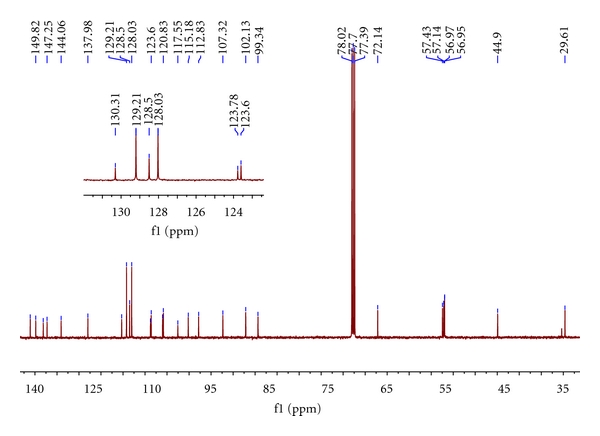
13C NMR spectrum of the I. Inserted figure is the magnification of the part of 120.00–135.00 of chemical shift.
Figure 3.
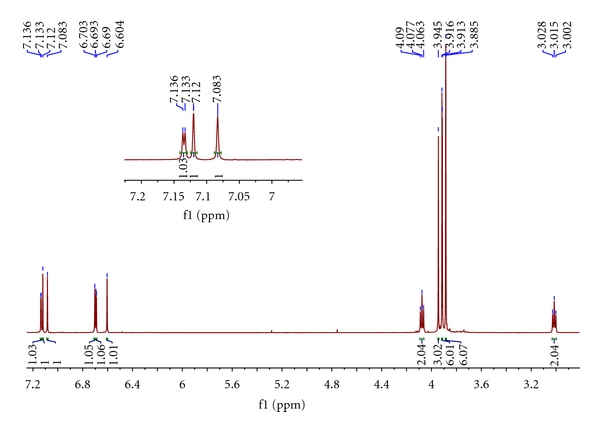
1H NMR spectrum of the II. Inserted figure is the magnification of the part of 7.00–7.20 of chemical shift.
Figure 4.
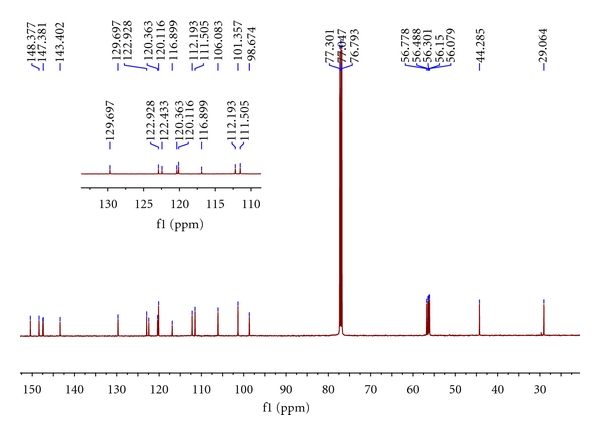
13C NMR spectrum of the II. Inserted figure is the magnification of the part of 110.00–130.00 of chemical shift.
An initial 1H-NMR spectrum of I (in CDCl3) revealed four –OMe–H signals at 3.94 (s, 3H), 3.91 (s, 3H), 3.91 (s, 3H), 3.88 (s, 3H). These peaks are the featured signals of the –OMe–. 2.95 and 4.05 doublets (J = 6.6 Hz) indicate –CH2N– and –CH2– moieties connected with it in the isoquinoline ring. It can be seen that the distinguishing feature of Ar–CH2O 5.14 (s, 2H) is shown in Figure 1. There are several groups of signals in the aromatic region; they are 7.12 (d, J = 1.6 Hz, 1H), 7.11 (s, 1H), 7.10 (s, 1H), 6.73 (s, 1H), 6.69 (d, J = 1.6 Hz, 1H), and 6.60 (s, 1H), respectively. Among them, 7.12 (d, J = 1.6 Hz, 1H) and 6.69 (d, J = 1.6 Hz, 1H) are the signals in the pyrrole ring; this can be estimated from the peak type. Since Ar–H in the Ar–CH2O are influenced by other protons more slightly, they will overlap together and show the multiplet in the spectra. So 7.29–7.46 (m, 5H) is the signal of Ar–H in the Ar–CH2O. A molecular formula of C29H29NO5, resulted from HR-MS data of I. The 13C NMR spectrum of I displayed twenty-seven signals, which represented all twenty-nine C-atoms, eighteen of which were assignable to three aromatic-C moieties and accounted for sixteen spectral signals. Of the remaining eleven signals, four were from OMe (56.96, 56.97, 57.14, 57.43 ppm), and seven were from isoquinoline and pyrrole ring C-atoms.
NMR data of II (see Figures 3 and 4) indicated a C23H25 framework, which HR-MS analysis expanded to a molecular formula of C23H25NO5. The simplest assumed relationship between the two isoquinoline, I as an BnO-substituted II, was reinforced by characterization of the NMR data, which exhibited many similar signals. Specifically, too many shifts of H and C resonances are very similar to each other which proved the basic framework between I and II. The NMR signals which distinguished I from II were those of three aromatic protons appropriate for Ar–H (7.29–7.46 ppm, m, 5H) and –CH2– in the Ar–CH2O. The remaining distinguishing feature was the number of –OMe– signal in 13C NMR at 56-57 ppm.
4. Discussions
I and II from 1-methyl-3,4-dihydroisoquinoline and 2,4,5-trimethoxy-α-halogen-acetophenone were obtained with high yields under mild conditions for the first time. This novel method, as the key reaction step, provides a general and highly efficient method for the preparation of 5,6-dihydropyrrolo[2,1-a]isoquinolines. We envisaged that the 5,6-dihydropyrrolo[2,1-a]isoquinolines could be constructed by the formation of quaternary ammonium salt, and subsequent lactonization in the presence of anhydrous K2CO3. The negative carbon ion of 1-methyl-3,4-dihydroisoquinoline is also active in the Knorr reaction. Both 2,4,5-trimethoxy-α-bromoacetophenone and 2,4,5-trimethoxy-α-chloracetophenone were employed. We found that the yield of the former is about 5% higher than the later. Therefore, 2,4,5-trimethoxy-α-bromoacetophenone is used in the synthesis of I and II.
5. Conclusion
Based on the facile synthetic route depicted in Scheme 1, two novel scaffolds for synthesis of lamellarin analogues 8-benzyloxy-9-methoxy-2-(2,4,5-trimethoxyphenyl)-5,6-dihydropyrrolo[2,1-a] isoquinoline (I) and 8,9-dimethoxy-2-(2,4,5-trimethoxyphenyl)-5,6-dihydro[2,1-a]isoquinoline (II) were obtained under mild condition. These two compounds are characterized by 1H NMR, 13C NMR, IR spectrum, and melting points. The products are stable and may be expected to exhibit biological activities to some extend.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (no. 20372013) The outstanding young talent fund from the Dalian science and technology bureau (2009J22DW038). The authors thank Professor Yecheng You Dalian University and Professor John Joule University of Manchester (UK) for helpful suggestion on this paper.
References
- 1.Andersen RJ, John Faulkner D, Cun-heng H, Van Duyne GD, Clardy J. Metabolites of the marine prosobranch mollusc Lamellaria sp. Journal of the American Chemical Society. 1985;107(19):5492–5495. [Google Scholar]
- 2.Reddy SM, Srinivasulu M, Satyanarayana N, Kondapi AK, Venkateswarlu Y. New potent cytotoxic lamellarin alkaloids from Indian ascidian Didemnum obscurum. Tetrahedron. 2005;61(39):9242–9247. [Google Scholar]
- 3.Ishibashi F, Tanabe S, Oda T, Iwao M. Synthesis and structure-activity relationship study of lamellarin derivatives. Journal of Natural Products. 2002;65(4):500–504. doi: 10.1021/np0104525. [DOI] [PubMed] [Google Scholar]
- 4.Kluza J, Gallego MA, Loyens A, et al. Cancer cell mitochondria are direct proapoptotic targets for the marine antitumor drug lamellarin D. Cancer Research. 2006;66(6):3177–3187. doi: 10.1158/0008-5472.CAN-05-1929. [DOI] [PubMed] [Google Scholar]
- 5.Facompré M, Tardy C, Bal-Mahieu C, et al. Lamellarin D: a novel potent inhibitor of topoisomerase I. Cancer Research. 2003;63(21):7392–7399. [PubMed] [Google Scholar]
- 6.Dias N, Vezin H, Lansiaux A. DNA binders and related subjects. Topics in Current Chemistry. 2005;253:89–109. [Google Scholar]
- 7.Vanhuyse M, Kluza J, Tardy C, et al. Lamellarin D: a novel pro-apoptotic agent from marine origin insensitive to P-glycoprotein-mediated drug efflux. Cancer Letters. 2005;221(2):165–175. doi: 10.1016/j.canlet.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 8.Ploypradith P, Petchmanee T, Sahakitpichan P, Litvinas ND, Ruchirawat S. Total synthesis of natural and unnatural lamellarins with saturated and unsaturated D-rings. Journal of Organic Chemistry. 2006;71(25):9440–9448. doi: 10.1021/jo061810h. [DOI] [PubMed] [Google Scholar]
- 9.Nyerges M, Tőke L. 1,5-Electrocyclisation of azomethine ylides leading to pyrrolo[2,1-a] isoquinolines - Concise construction of the lamellarin skeleton. Tetrahedron Letters. 2005;46(44):7531–7534. [Google Scholar]
- 10.Bermejo A, Andreu I, Suvire F, et al. Syntheses and antitumor targeting G1 phase of the cell cycle of benzoyldihydroisoquinolines and related 1-substituted isoquinolines. Journal of Medicinal Chemistry. 2002;45(23):5058–5068. doi: 10.1021/jm020831a. [DOI] [PubMed] [Google Scholar]
- 11.Cabedo N, Protais P, Cassels BK, Cortes D. Synthesis and dopamine receptor selectivity of the benzyltetrahydroisoquinoline, (R)-(+)-nor-roefractine. Journal of Natural Products. 1998;61(6):709–712. doi: 10.1021/np980008a. [DOI] [PubMed] [Google Scholar]
- 12.Batra S, Sabnis YA, Rosenthal PJ, Avery MA. Structure-based approach to falcipain-2 inhibitors: synthesis and biological evaluation of 1,6,7-Trisubstituted dihydroisoquinolines and isoquinolines. Bioorganic and Medicinal Chemistry. 2003;11(10):2293–2299. doi: 10.1016/s0968-0896(03)00117-2. [DOI] [PubMed] [Google Scholar]