

Abstract
This work describes the integration of expanded bed adsorption (EBA) and adsorptive protein refolding operations used to recover purified and biologically active human basic fibroblast growth factor from inclusion bodies expressed in E. coli. Insoluble overexpressed human basic fibroblast growth factor has been purified on CM Hyper Z matrix by expanded bed adsorption after isolation and solubilization in 8 M urea. The adsorption was made in expanded bed without clarification steps such as centrifugation. Column refolding was done by elimination of urea and elution with NaCl. The human basic fibroblast growth factor was obtained as a highly purified soluble monomer form with similar behavior in circular dichroism and fluorescence spectroscopy as native protein. A total of 92.52% of the available human basic fibroblast growth factor was recovered as biologically active and purified protein using the mentioned purification and refolding process. This resulted in the first procedure describing high-throughput purification and refolding of human basic fibroblast growth factor in one step and is likely to have the greatest benefit for proteins that tend to aggregate when refolded by dilution.
1. Introduction
Production of therapeutic proteins in inclusion bodies is useful due to the efficacy of insoluble expression such as high product yield and protection against degradation by proteases [1]. The recovery of biologically active protein from such inclusion bodies requires refolding protocols.
In general, the methods used for inclusion body solubilization result in a soluble protein that is biologically inactive. The solublized proteins do not have native conformation and must be transferred into conditions that allow the formation of the native structure. During this period, the correct folding pathway competes, often in disadvantage with misfolding and aggregation of the target protein. Protein refolding involves intramolecular interactions and follows first-order kinetics [2].
Refolding yields commonly decrease with increasing initial concentrations of the unfolded protein independent of the refolding method applied [3]. Aggregates are formed by nonnative intramolecular hydrophobic interactions between protein folding intermediates, which have not yet buried their hydrophobic amino acid stretches. Therefore, prevention of hydrophobic intermolecular interaction during the first steps of refolding is crucial to allow successful renaturation at high protein concentrations [4].
A very efficient strategy to prevent aggregation is to minimize the risk of intermolecular interactions by adsorbing the denatured protein molecules to a solid support, thus, effectively separating the individual protein molecules from each other during refolding. The first studies on the refolding of denatured proteins after adsorption to a solid matrix surface were carried out many years ago. Thus, Epstein and Anfinsen [5] adsorbed trypsin and ribonuclease to carboxymethyl cellulose for the purpose of studying the reversible reduction of their disulfide bonds. Studies on the refolding of chymotrypsinogen and trypsin after covalent attachment to agarose gel have been performed [6], resulting in 50–70% refolding yield. In 1979, Mozhaev et al. [7] covalently immobilized trypsin for the same reason. Following the development of new protein adsorbent media during the last couple of decades, various chromatographic methods have been developed to improve refolding yield. Noncovalent (reversible) adsorption of denatured proteins to ion-exchange media was used by Creighton [8] for the purpose of refolding.
The need to use large quantities of hbFGF in formulation study in our lab and its high market cost led us to develop its refolding and purification on a large scale.
Heparin affinity chromatography is widely utilized for the purification of hbFGF but it has not been possible so far to obtain homogeneous protein preparations in a single-step process. Heparin affinity chromatography is commonly combined with a cation-exchange step, maybe followed from subsequent purification steps, such as copper chelate or hydrophobic interaction or gel permeation chromatography [9, 10].
These traditional techniques are limited with recombinant proteins by intrinsic problem of aggregation [11]. Indeed, most of the recombinant proteins are overexpressed as insoluble inclusion bodies or soluble small aggregates. The challenge is to convert the inactive and insoluble inclusion bodies and small protein aggregates into soluble, correctly folded biologically active products [12]. Mannen et al. [13] showed for the first time an expanded bed protein refolding using a solid phase artificial chaperone. They used the artificial chaperone-assisted refolding method and the expanded bed technique providing a novel industrial scale protein refolding system. Cho et al. [11] have also proposed an expanded bed adsorption-mediated refolding process of a fusion protein. Exploiting the advantages of expanded bed adsorption chromatography, which is an alternative to traditional clarification and the first chromatography step [14–16], they could provide a higher yield with much less aggregation and, thus, reducing the number of the renaturation process steps.
The recombinant hbFGF has been overexpressed as inclusion bodies to increase the quantity of recovered and to facilitate purification. The cell homogenate including inclusion bodies and small soluble aggregates was dissolved by urea and then the unfolded recombinant hbFGF was allowed to adsorb on the chromatographic support. Here, on-column purification and refolding procedure was based on an ion exchange bed technology and was done in one step. The incentive of this approach was to obtain a one step purification of high quantity of hbFGF suitable for automation.
2. Materials and Methods
2.1. hbFGF Expression and Extraction
Cloning and expression of rhbFGF was described by Alibolandi et al. [17]. Briefly, the cell pellets of 1 L LB culture of induced transformants were collected and resuspended in 30 mL cold PBS, pH 7.0. Then, cells were lysed by six passes through high-pressure homogenizer (APV Manton Gaulin).
2.2. Solubilization of Inclusion Bodies
The homogenate (containing soluble and insoluble proteins) was diluted with 270 mL of 8 M urea, PBS pH 7.0 and agitated 8 h at 10°C. Then, the homogenate was directly used for expanded bed cation exchange adsorption.
2.3. Chromatographic Procedure
2.3.1. Study Degree of Expansion
Firstly, expanded bed adsorption of two matrices (CM sepharose Streamline and CM Hyper Z) has been compared. Two buffers were evaluated, PBS, pH 7.0 buffer and 8 M urea, PBS, pH 7.0 buffer. The flow rate was also compared for twofold degree of expansion. The degree of expansion was calculated from the ratio of expanded bed height (L) to sedimented bed height (L0). To study the expansion versus flow rate, increments of 1 mL/min of the flow rate were used from 1 mL/min until the flow rate necessary to obtain a twofold degree of expansion. In addition, the influence of high concentrations of urea and sample on the expansion degree of both matrices was studied. The matrix allowing the higher flow rate was chosen for the subsequent purification step.
2.3.2. Purification of Insoluble hbFGF
Cation exchange expanded bed adsorption was performed with CM Hyper Z (60 mL, 10.4 cm sedimented gel) packed in the Streamline 25 column. The column was linked to a Biopilot workstation (Amersham Pharmacia Biotech). Equilibration/expansion was performed with 8 M urea, PBS, pH 7.0 buffer at 40 mL/min to obtain a twofold degree of expansion. The homogenate was applied at 40 mL/min, followed by washing with the equilibration buffer until UV baseline was reached. Then, the pump was turned off and the bed sedimented. Next, the adaptor was moved down towards the sedimented bed surface. After a run of two volumes (sedimented gel) of equilibration buffer, refolding was performed with 10 column volumes of PBS, pH 7.0 buffer at a flow velocity of 40 mL/min using a downward flow in the sedimented bed mode. Then, elution was performed in two steps, 0.6 M NaCl, PBS pH 7.0 for the elution of hbFGF and 1 M NaCl, PBS, pH 7 for the elution of contaminants and run at 40 mL/min.
2.4. Analytical Procedures
2.4.1. Sodium Dodecyl Sulphate-Polyacrylamide Gel Electrophoresis and Immunoblot Analysis
Analysis of the fractions containing hbFGF was performed by electrophoresis on 12% SDS-polyacrylamide gels under reducing conditions according to Laemmli method [18].
The concentration of hbFGF was calculated by determining the total protein concentration using the Bradford method [19] and by measuring the percentage of hbFGF by gel scanning (Image Station 440 CF, Kodak, Rochester, NY, USA). In addition, the precise amount of hbFGF in fractions was measured by ELISA kit (R&D, USA).
Western blotting analysis was also applied to detect the hbFGF in all fractions. All samples were size fractionized by 12% SDS-PAGE and then transferred to a 0.2 μm pore size nitrocellulose membrane (Schleicher & Schuell, USA). Rabbit anti-hbFGF anti serum (Sigma, USA) and horseradish peroxidase conjugated goat-antirabbit antibody (Sigma, USA) were used as primary and secondary antibody. The immunoreactions were visualized using 0.5 mg/mL diaminobenzidine HCl (Merck, USA) and 0.01% H2O2 (Merck, USA).
2.4.2. Circular Dichroism (CD)
CD spectroscopy was used to determine the refolded protein secondary and tertiary structures using JASCO J715 spectropolarimeter and a quartz cuvette of 0.1 cm path length. The protein concentration of about 0.5 mg/mL was used for each CD measurement. The CD spectra were corrected for buffer contributions.
2.4.3. Fluorescence Spectroscopy
Determination of protein fluorescence spectrum was performed with a Hitachi F-4010 fluorescence spectrophotometer using a quartz cuvette with 1 cm path length. Excitation and emission slits were set as 10 nm. Fluorescence spectra were then recorded with an excitation of 295 nm and recording the emission spectra from 200 to 500 nm. The samples were incubated for 3 h at room temperature before fluorescence measurements were recorded.
3. Results and Discussion
3.1. Comparison of Matrices
Before purification, expanded bed adsorption of two matrices has been compared: CM sepharose and CM Hyper Z Streamline. Two buffers were investigated, PBS, pH 7.0 buffer and 8 M urea, PBS, pH 7.0 buffer. The flow rate was measured for twofold degree of expansion.
For a twofold degree of expansion with PBS pH 7, the CM Hyper Z matrix allowed a 40 mL/min flow rate which is twice that of CM sepharose streamline. Figure 1 shows that 8 M urea had less effect on CM Hyper Z than CM sepharose Streamline. The influence was similar with cellular protein extract in presence of 8 M urea. Therefore, for purification we employed the CM Hyper Z matrix, which allowed a high flow rate and gave suitable performance in the presence of 8 M urea. Moreover, in a previous study, it was showed that CM sepharose matrix was efficient to purify soluble hbFGF [17].
Figure 1.
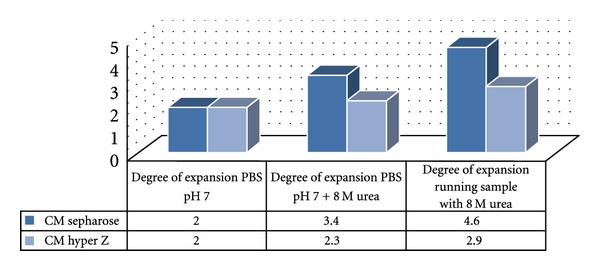
Influence of fluid on expansion degree with CM sepharose and CM hyper Z matrices. Column 1: PBS pH 7; column 2: PBS pH 7 + 8 M urea; column 3: cell lysate in PBS pH 7 + 8 M urea.
Equilibration/expansion was performed with PBS, pH 7.0 buffer at 40 mL/min upward flow to obtained twofold degree of expansion. Then, equilibration was performed with 8 M urea, PBS pH 7.0 buffer, giving a 2.3-fold degree of expansion. The inclusion bodies solubilized in 8 M urea (300 mL) were applied at 40 mL/min and gave a 2.9-fold degree of expansion.
3.2. Purification Step and Analysis of Purified Refolded hbFGF
Injection of the sample was followed by washing with 8 M urea, PBS pH 7 buffer until UV baseline was reached. Then, the gel bed was sedimented and a run of two volumes (sedimented gel) of equilibration buffer was performed. Urea was eliminated with 10 column volumes of PBS, pH 7.0 buffer at a flow rate of 40 mL/min using a downward flow in the sedimented bed mode.
Next, the elution was performed, 0.4 M NaCl, PBS pH 7.0 for the elution of hbFGF and 0.8 M NaCl, PBS pH 7 for the elution of contaminants and was run at 40 mL/min (Figure 2). All fractions were analyzed by SDS-page. The purification results are shown in Table 1. The cell lysate containing 378 mg of target protein was applied onto the column, 664.3 mg proteins passed into the flow through, and the hbFGF estimated by ELISA was about 23.3 mg. Peak 1 contains 349.72 mg hbFGF with 92.52% of recovery. The recovery and purification value are higher than those obtained in cation exchange purification of soluble hbFGF [17].
Figure 2.
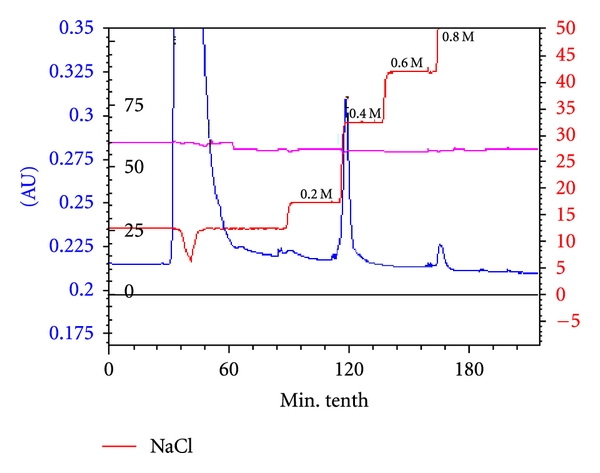
Purification of insoluble hbFGF with cation exchange expanded bed adsorption. Column: CM Hyper Z (60 mL of gel). Sample: unclarified cell lysate in 8 M urea, PBS pH 7. Equilibration buffer: 8 M urea, PBS pH 7. Refolding buffer: PBS pH 7. Elution buffer: 0.2–2 M NaCl, 20 mM sodium bicarbonate pH 9. Detection at 280 nm; flow rate: 40 mL/min. Flow through: no retained fraction; peak 1: 0.4 M NaCl eluted fraction; peak 2: 0.8 M NaCl eluted fraction.
Table 1.
Purification of hbFGF with the CM hyper Z matrix.
| Volume | Protein | Total protein | hbFGF | Total hbFGF | Recovery | Purification fold | |
|---|---|---|---|---|---|---|---|
| (mL) | (mg/mL) | (mg) | (mg/mL) | (mg) | % | ||
| Denatured cell lysate | 300 | 4.2 | 1260 | 1.26 | 378 | ||
| Flow through | 910 | 0.73 | 664.3 | 0.02 | 23.3 | 6.16 | 0.116 |
| Peak 1 | 183 | 2 | 366 | 1.91 | 349.72 | 92.52 | 3.51 |
| Peak 2 | 162 | 0.1 | 16.2 | 0.03 | 4.86 | 1.285 | 1.11 |
However, purification with heparin affinity required an additional step to obtain a single band on SDS-PAGE [20]. The production of hbFGF as inclusion bodies allowed us to reduce the volume culture for the same quantity of protein and then the time of process. Besides the reduction of the number of purification steps, fast removal of proteolytic enzymes and a shortening of the total process time may help to prevent losses in mass and quality of recombinant hbFGF due to degradation and precipitation.
Moreover, the use of cation exchange using CM hyper Z allowed us to avoid further step for purification. All fractions were analyzed by SDS-PAGE (Figure 3). The lane containing the flow through shows a low level of hbFGF. It seems that this hbFGF corresponds to insufficient refolded hbFGF. Indeed, without urea treatment, all the hbFGF would have passed in the flow through (data not shown). The native hbFGF from previous purification is used as reference native hbFGF [21]. The molecular mass of refolded hbFGF seemed to correspond to that of the native hbFGF. The folding of refolded hbFGF obtained was evaluated by fluorescence spectroscopy and circular dichroism analysis. The same spectral properties were obtained between refolded and native hbFGF (Figure 4). Quantitative analysis of the CD spectra also indicates that refolded hbFGF is predominantly in beta sheet and other conformations with very little alpha-helix fraction. These results are in agreement with earlier observations [22].
Figure 3.

(a) 12% SDS-PAGE analysis of fractions from purification of hbFGF. M: Molecular weight marker; lane 1: cell lysate in 8 M urea, PBS pH 7; lane 2: flow through; lane 3: peak 1; lane 4: peak 2. (B) Western blotting of cell lysate and fractions from purification of hbFGF. Lane 1: cell lysate; lane 2: flow through; lane 3: peak 1; lane 4: peak 2.
Figure 4.
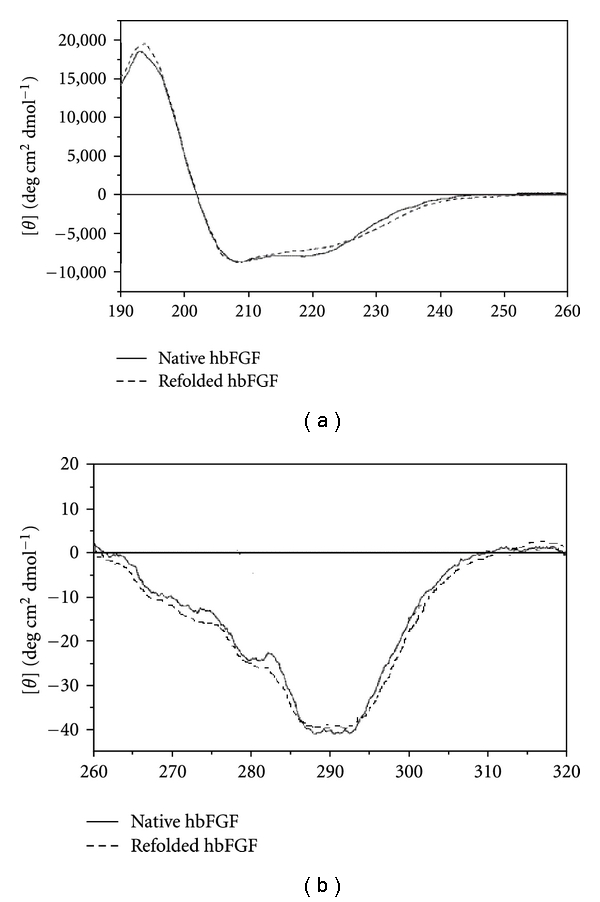
Circular dichroism (CD) spectra of refolded and native hbFGF (a) far-UV spectra for native and refolded hbFGF; (b) near-UV CD spectra for native and refolded hbFGF.
It is well established that fluorescence intensity obtained at an excitation wavelength of 295 nm correlates with the exposure of hydrophobic tryptophan residues on the surface of the polypeptide [23] and hence the extent of protein unfolding. hbFGF carrys 8 tryptophan residues [24]. Fluorescence emission of refolded hbFGF exhibited an emission characteristic that was comparable to native hbFGF. Here, fluorescence emission of purified refolded hbFGF exhibited a very low emission characteristic that was comparable to native hbFGF (Figure 5).
Figure 5.
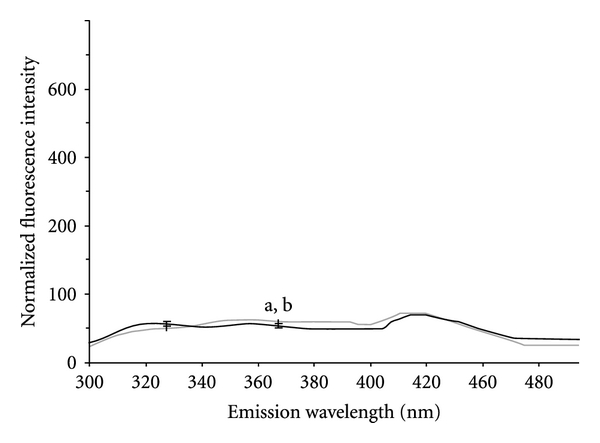
Fluorescence spectra profile of refolded and native hbFGF.
Obtained results from this study indicate the good performance of this technique in purification and refolding of hbFGF.
It has been noted that high expanded bed adsorption is also efficient in recovering active proteins from the unclarified cell culture [25–29].
This technique was applied to purified proteins from different sources like yeast [30], bacterial cells [31, 32], and cell cultures [33, 34].
On the other hand, expanded bed adsorption was found an easy technique to scale-up and automation due to the reduction of the number of purification steps [35].
Here, high density of CM Hyper Z beads allows efficient processing of cell lysate at very high viscosity (urea 8 M) and high flow rate (40 mL/min). Insoluble and soluble (monomer and small aggregates) hbFGF could be purified after solubilization in 8 M urea and adsorption in expanded bed mode to avoid centrifugation. After elimination of urea on the column and elution with NaCl, the hbFGF was obtained as a highly purified soluble form with similar behavior in fluorescence spectroscopy, circular dichroism, and electrophoresis as native hbFGF. The results of one-step protein purification and on-column refolding of hbFGF with cation exchange expanded bed chromatography demonstrate that it is a promising process allowing automation.
References
- 1.Clark EB. Protein refolding for industrial processes. Current Opinion in Biotechnology. 2001;12(2):202–207. doi: 10.1016/s0958-1669(00)00200-7. [DOI] [PubMed] [Google Scholar]
- 2.Wetlaufer DB, Xie Y. Control of aggregation in protein refolding: a variety of surfactants promote renaturation of carbonic anhydrase II. Protein Science. 1995;4(8):1535–1543. doi: 10.1002/pro.5560040811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta P, Hall CK, Voegler AC. Effect of denaturant and protein concentrations upon protein refolding and aggregation: a simple lattice model. Protein Science. 1998;7(12):2642–2652. doi: 10.1002/pro.5560071218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vallejo LF, Rinas U. Strategies for the recovery of active proteins through refolding of bacterial inclusion body proteins. Microbial Cell Factories. 2004;3, article 11 doi: 10.1186/1475-2859-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Epstein CJ, Anfinsen CB. The reversible reduction of disulfide bonds in trypsin and ribonuclease coupled to carboxymethyl cellulose. The Journal of Biological Chemistry. 1962;237:2175–2179. [PubMed] [Google Scholar]
- 6.Sinha NK, Light A. Refolding of reduced, denatured trypsinogen and trypsin immobilized on agarose beads. Journal of Biological Chemistry. 1975;250(22):8624–8629. [PubMed] [Google Scholar]
- 7.Mozhaev VV, Martinek K, Berezin IV. Effect of immobilization on protein (trypsin) folding. Molekulyarnaya Biologiya. 1979;13(1):73–80. [PubMed] [Google Scholar]
- 8.Creighton TE. Folding of proteins adsorbed reversibly to ionexchange resins. In: Oxender DL, editor. UCLA Symposia on Molecular and Cellular Biology, New Series. Vol. 39. New York, NY, USA: Alan R. Liss; 1986. pp. 249–257. [Google Scholar]
- 9.Lobb RR, Harper JW, Fett JW. Purification of heparin-binding growth factors. Analytical Biochemistry. 1986;154(1):1–14. doi: 10.1016/0003-2697(86)90487-2. [DOI] [PubMed] [Google Scholar]
- 10.Shing Y. Biaffinity chromatography of fibroblast growth factors. Methods in Enzymology. 1991;198:91–95. doi: 10.1016/0076-6879(91)98011-t. [DOI] [PubMed] [Google Scholar]
- 11.Cho TH, Ahn SJ, Lee EK. Refolding of protein inclusion bodies directly from E. coli homogenate using expanded bed adsorption chromatography. Bioseparation. 2001;10(4-5):189–196. doi: 10.1023/a:1016305603569. [DOI] [PubMed] [Google Scholar]
- 12.Li M, Su ZG, Janson JC. In vitro protein refolding by chromatographic procedures. Protein Expression and Purification. 2004;33(1):1–10. doi: 10.1016/j.pep.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 13.Mannen T, Yamaguchi S, Honda J, Sugimoto S, Nagamune T. Expanded-bed protein refolding using a solid-phase artificial chaperone. Journal of Bioscience and Bioengineering. 2001;91(4):403–408. doi: 10.1263/jbb.91.403. [DOI] [PubMed] [Google Scholar]
- 14.Draeger NM, Chase HA. Liquid fluidized bed adsorption of protein in the presence of cells. Bioseparation. 1991;2(2):67–80. [PubMed] [Google Scholar]
- 15.Chase HA, Draeger NM. Affinity purification of proteins using expanded beds. Journal of Chromatography. 1992;597(1-2):129–145. doi: 10.1016/0021-9673(92)80103-2. [DOI] [PubMed] [Google Scholar]
- 16.Johansson HJ, Jagersten C, Shiloach J. Large scale recovery and purification of periplasmic recombinant protein from E. coli using expanded bed adsorption chromatography followed by new ion exchange media. Journal of Biotechnology. 1996;48(1-2):9–14. doi: 10.1016/0168-1656(96)01390-9. [DOI] [PubMed] [Google Scholar]
- 17.Alibolandi M, Mirzahoseini H, Abad MA, Azami movahed M. High level expression of human basic fibroblast growth factor in Escherichia coli: evaluating the effect of the GC content and rare codons within the first 13 codons. African Journal of Biotechnology. 2010;9(16):2456–2462. [Google Scholar]
- 18.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 19.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72(1-2):248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 20.Garke G, Deckwer WD, Anspach FB. Preparative two-step purification of recombinant human basic fibroblast growth factor from high-cell-density cultivation of Eschericia coli. Journal of Chromatography B. 2000;737(1-2):25–38. doi: 10.1016/s0378-4347(99)00394-1. [DOI] [PubMed] [Google Scholar]
- 21.Alibolandi M, Mirzahoseini H, Nehi FM, Tabatabaian G, Amini H, Sardari S. Improving recombinant protein solubility in Escherichia coli: identification of best chaperone combination which assists folding of human basic fibroblast growth factor. African Journal of Biotechnology. 2010;9(47):8100–8109. [Google Scholar]
- 22.Prestrelski SJ, Arakawa T, Kenney WC, Byler DM. The secondary structure of two recombinant human growth factors, platelet-derived growth factor and basic fibroblast growth factor, as determined by Fourier-transform infrared spectroscopy. Archives of Biochemistry and Biophysics. 1991;285(1):111–115. doi: 10.1016/0003-9861(91)90335-g. [DOI] [PubMed] [Google Scholar]
- 23.Ladokhin AS, Jayasinghe S, White SH. How to measure and analyze tryptophan fluorescence in membranes properly, and why bother? Analytical Biochemistry. 2000;285(2):235–245. doi: 10.1006/abio.2000.4773. [DOI] [PubMed] [Google Scholar]
- 24.Dubey VK, Lee J, Blaber M. Redesigning symmetry-related mini-core regions of FGF-1 to increase primary structure symmetry: thermodynamic and functional consequences of structural symmetry. Protein Science. 2005;14(9):2315–2323. doi: 10.1110/ps.051494405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hjorth R, Kampe S, Carlsson M. Analysis of some operating parameters of novel adsorbents for recovery of proteins in expanded beds. Bioseparation. 1995;5(4):217–223. [PubMed] [Google Scholar]
- 26.Finette GMS, Mao QM, Hearn MTW. Examination of protein adsorption in fluidized bed and packed bed columns at different temperatures using frontal chromatographic methods. Biotechnology and Bioengineering. 1997;58(1):35–46. doi: 10.1002/(sici)1097-0290(19980405)58:1<35::aid-bit4>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 27.Wang G. Isolation and purification of phycoerythrin from red alga Gracilaria verrucosa by expanded-bed-adsorption and ion-exchange chromatogaphy. Chromatographia. 2002;56(7-8):509–513. [Google Scholar]
- 28.Bermejo R, Felipe MA, Talavera EM, Alvarez-Pez JM. Expanded bed adsorption chromatography for recovery of phycocyanins from the microalga Spirulina platensis. Chromatographia. 2006;63(1-2):59–66. [Google Scholar]
- 29.Bermejo R, Acién FG, Ibáñez MJ, Fernández JM, Molina E, Alvarez-Pez JM. Preparative purification of B-phycoerythrin from the microalga Porphyridium cruentum by expanded-bed adsorption chromatography. Journal of Chromatography B. 2003;790(1-2):317–325. doi: 10.1016/s1570-0232(03)00168-5. [DOI] [PubMed] [Google Scholar]
- 30.Raymond F, Rolland D, Gauthier M, Jolivet M. Purification of a recombinant protein expressed in yeast: optimization of analytical and preparative chromatography. Journal of Chromatography B. 1998;706(1):113–121. doi: 10.1016/s0378-4347(97)00512-4. [DOI] [PubMed] [Google Scholar]
- 31.Maurizi G, Cioccio VD, Macchia G, et al. Purification of human recombinant interleukin 1 receptor antagonist proteins upon Bacillus subtilis sporulation. Protein Expression and Purification. 1997;9(2):219–227. doi: 10.1006/prep.1996.0681. [DOI] [PubMed] [Google Scholar]
- 32.Willoughby N, Martin P, Titchener-Hooker N. Extreme scale-down of expanded bed adsorption: purification of an antibody fragment directly from recombinant E. coli culture. Biotechnology and Bioengineering. 2004;87(5):641–647. doi: 10.1002/bit.20173. [DOI] [PubMed] [Google Scholar]
- 33.Batt BC, Yabannavar VM, Singh V. Expanded bed adsorption process for protein recovery from whole mammalian cell culture broth. Bioseparation. 1995;5(1):41–52. [PubMed] [Google Scholar]
- 34.Thömmes J, Bader A, Halfar M, Karau A, Kula MR. Isolation of monoclonal antibodies from cell containing hybridoma broth using a protein A coated adsorbent in expanded beds. Journal of Chromatography A. 1996;752(1-2):111–122. doi: 10.1016/s0021-9673(96)00504-3. [DOI] [PubMed] [Google Scholar]
- 35.Frej AKB, Johansson HJ, Johansson S, Leijon P. Expanded bed adsorption at production scale: scale-up verification, process example and sanitization of column and adsorbent. Bioprocess Engineering. 1997;16(2):57–63. [Google Scholar]