

Abstract
Purpose of review
The purpose of this review is to summarize recent findings implicating complement as an important regulator of T cell immune responses. We then provide perspective for how these newly described mechanisms apply to allograft injury and how they could ultimately influence therapy.
Recent findings
In addition to known effects of serum complement as an effector arm of antibody-initiated injury, T cells and APCs produce complement proteins and upregulate complement receptors following cognate interactions. The locally released and activated, immune cell-derived complement signals predominantly through C3a and C5a binding to their receptors expressed on both partners to induce immune cell activation and differentiation. Complement deficiency or blockade limits T cell mediated autoimmunity and transplant rejection while removal of the complement regulatory protein decay accelerating factor can enhance T cell immunity and accelerate graft rejection.
Summary
Emerging data indicate that immune cell-derived complement physiologically regulates immune cell survival and proliferation, modulating the strength and phenotype of adaptive T cell immune responses involved in transplant rejection. The recognition of the diversity through which complement participates in allograft injury supports the need for continued design and testing of complement inhibitors in human transplant recipients.
Keywords: T cells, allograft rejection, complement, costimulation
Introduction
Complement is traditionally considered a component of the innate immune system required for host defense against invading pathogens (1). The complement cascade also has an established role as a pathogenic effector pathway in transplant rejection. Antibody-initiated, classical pathway activation of serum complement is thought to underlie some forms of vascular allograft injury (2, 3). In support of this, C6 deficient animals are resistant to antibody-mediated rejection (4), C4d deposition is used to diagnose human antibody-mediated vascular rejection (2, 5), and a recent report indicates that a blocking anti-C5 mAb can limit antibody-mediated kidney transplant injury in humans (6).
Several unexpected observations, including the finding that C3 deficient mouse kidneys are accepted by wild type allogeneic hosts with normal serum complement activity (7), have resulted in paradigm-shifting insights into how the complement cascade can influence tissue inflammation and adaptive immunity. Emerging evidence from several research groups indicates that complement produced and activated by cells of the allograft modulates ischemia reperfusion injury (8), and can contribute to the development of renal fibrosis (9). In this review we will focus our discussion on another novel concept, that T cells and antigen presenting cells produce complement components and express complement receptors and that locally activated complement activation products are key regulators of T cell immune responses. We will then provide perspective for how these newly described mechanisms apply to allograft injury and how they could ultimately influence therapy.
Overview of the complement cascade
Complement activation is initiated through the classical, alternative or mannose binding lectin (MBL) pathway which converge at the production of the C3 convertases, central amplification steps within the cascade (Figure 1). Subsequent cleavage of C3 and then C5 initiates formation of the membrane attack complex and also yields soluble and surface bound split products, including C3a, C3b, iC3b, C3dg and C5a, which modulate inflammation via a) direct lysis, b) serving as chemoattractants and activators of innate immune cells (e.g. macrophages) and c) functioning as opsonins, among others (1).
Figure 1.
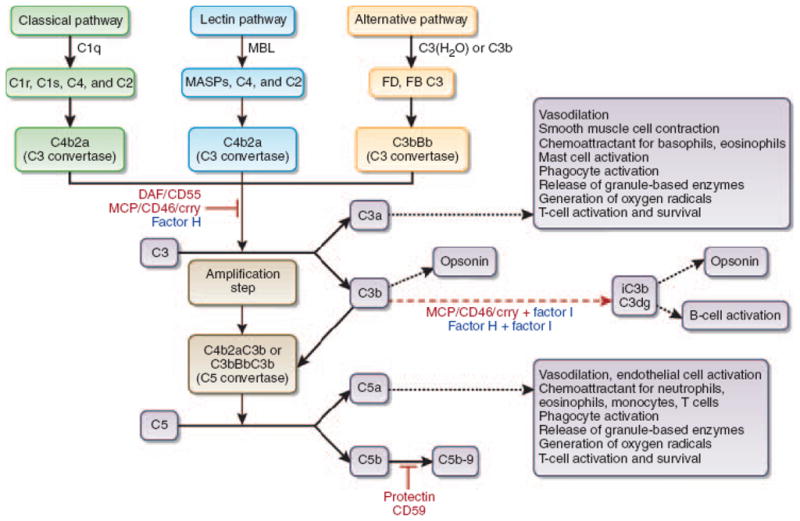
Schematic overview of the complement cascade illustrating the three activation pathways (classical, mannose-binding lectin (MBL), and alternative), the common pathway, the membrane attack complex (C5b-9), and the site of action of the cell surface-expressed complement regulators (red) and the soluble complement regulator Factor H (blue). Representative functions of select cleavage products are listed in boxes.
Reproduced with permission from: Vieyra MB and Heeger PS; Novel aspects of complement in kidney injury; Kidney Int. 2010 Mar;77(6):495–9. Epub 2009 Dec 16.
Current concepts are that complement activation must be regulated in vivo to prevent bystander damage to host cells (1). This regulation is accomplished through secretion and expression of soluble and membrane-bound complement regulatory proteins. Factor H is one of several soluble regulators and decay accelerating factor (DAF or CD55) is one of several membrane bound complement regulatory proteins (others include membrane cofactor protein, CD46 and protectin, CD59, Figure 1). Mechanistically, DAF accelerates the decay of cell-surface assembled, C3 convertases, limiting downstream complement activation and restricting production of the aforementioned cleavage products (10). An important concept is that DAF only functions intrinsically, that is, it only limits complement activation on the cell surface upon which it is expressed.
Complement as a modulator of adaptive immunity including T cell immunity
Interactions between complement and adaptive immunity were initially described in the 1970s when Pepys observed that complement depleted mice failed to mount potent antibody responses (11). Subsequent mechanistic studies have shown that C3dg, a cleavage product of C3b, binds to the B cell-expressed complement receptor 2 (CR2, CD21) and through this interaction, lowers the threshold for B cell activation (1, 12). The effect of complement on B cell immune responses raised the possibility that complement/C3dg might be used as an adjuvant to enhance the efficacy of vaccines aimed at inducing protective pathogen-specific antibodies.
Work from several groups published over the last decade has firmly demonstrated a previously unanticipated role for complement as a regulator of T cell immunity (Figure 2). Our joint group made the novel observation that during cognate interactions between T cells and APCs (macrophages and DCs), both partners upregulate and secrete alternative pathway components C3, fB and fD as well as C5, and upregulate surface expression of C3a receptor (C3aR) and C5a receptor (C5aR) (13, 14). These changes are primarily induced through costimulatory molecule signaling via CD28/CD80/CD86 and CD154/CD40 (14). A transient, physiological downregulation of cell surface expressed DAF permits local complement activation, yielding C3a and C5a, which bind to their receptors, C5aR and C3aR on both the T cell and the APC (13, 14). Signaling via these G-protein coupled receptors in T cells activates phosphoinositide 3-kinase gamma (PI3Kγ), and induces phosphorylation of the central intracellular signaling molecule AKT (see Figure 2)(14, 15). AKT phosphorylation upregulates the anti-apoptotic gene Bcl2 and downregulates surface expression of the pro-apoptotic molecule Fas (15). Together these signals enhance T cell proliferation and diminish T cell apoptosis, explaining the complement-mediated expansion of the effector T cell repertoire following antigenic stimulation (15). Intriguingly, the evidence also indicates that C3aR and C5aR signaling is required for T cell homeostasis, as T cells deficient in both receptors spontaneously undergo accelerated cell death in vitro and in vivo (14)
Figure 2.
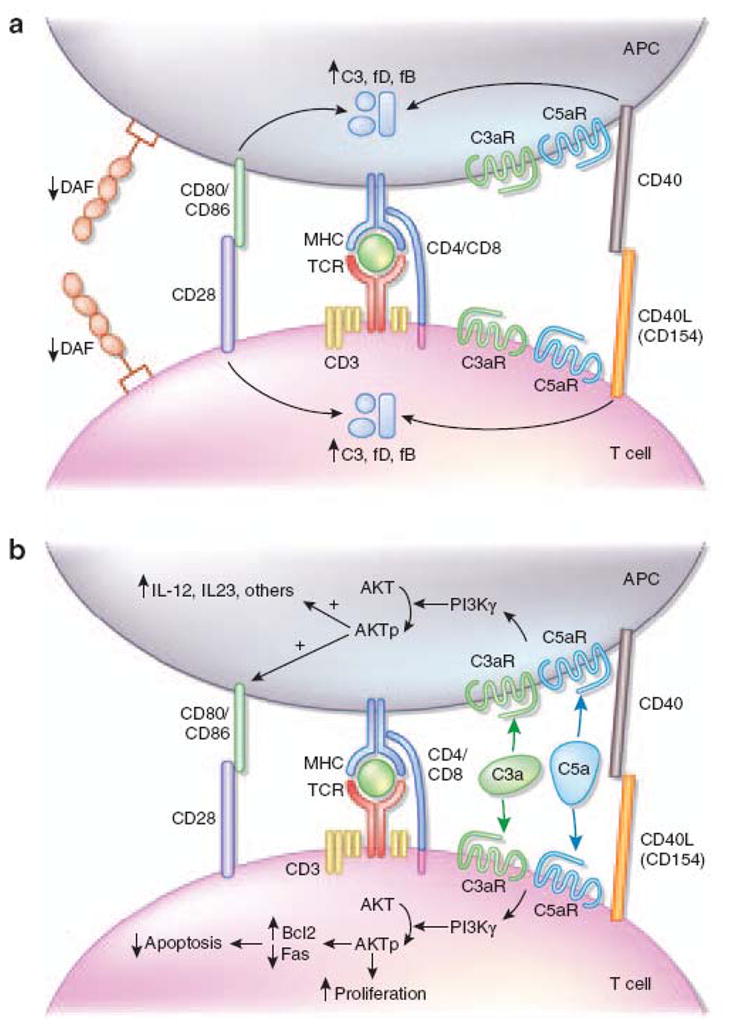
Schematic depiction of how complement modulates T-cell immunity. Cognate T-cell–APC interactions result in the upregulation and release of alternative pathway complement components by both partners and in the downregulation of (a) cell-surface DAF, which permits local complement activation resulting in the production of (b) C3a and C5a. These split products bind to their G-protein-coupled receptors expressed on T cells, signaling through PI3Kγ and AKT to induce proliferation and inhibit apoptosis, while simultaneously activating APCs to upregulate B7 and innate cytokine (for example, IL-12) production. APC, antigen presenting cell; DAF, decay-accelerating factor; IL-12, interleukin-12; PI3Kγ, phosphoinositide 3 -kinase-γ.
Reproduced with permission from: Vieyra MB and Heeger PS; Novel aspects of complement in kidney injury; Kidney Int. 2010 Mar;77(6):495–9. Epub 2009 Dec 16.
The immune cell-derived and locally produced C3a and C5a also bind to C3aR/C5aR on APCs, including dendritic cells and macrophages (Figure 2)(14, 16–18). C5aR/C3aR ligation activates the APCs via PI3Kγ/AKT and inhibition of cAMP/phosphokinase A, together activating NFκB (17, 18). This in turn causes induction and release of innate cytokines (e.g. IL-12, IL-23) and upregulation of APC costimulatory molecules (e.g., CD80, CD86) further amplifying the immune response and modulating the phenotype toward IFNγ-producing Th1 immunity (13–18).
Important in vitro findings supporting these conclusions derived from our group and from the Sacks/Zhou group are that C5aR−/−, C3aR−/−, C5aR−/−/C3aR−/− and C3−/− APCs produce less IL-12, express lower levels of CD80 and are weaker T cell stimulators than WT APCs, while DAF−/− DCs and macrophages (in which restraint on local complement activation is diminished) produce more IL-12 and induce stronger T cell responses than WT (14, 16–18). Moreover, regardless of the phenotype of the APC, T cells deficient in C3aR and C5aR signaling (genetically deficient or blocked) respond poorly to WT and DAF−/− APCs, and undergo accelerated cell death (14, 15).
Studies published in 2009 from another research group identified a different mechanism through which complement modulates T cell immunity (19). These investigators, using DCs from C1q−/− mice and rare C1q-deficient humans, showed that DC-derived C1q can bind in an autocrine fashion to C1q receptors on the DCs and in concert with CD40 signaling, phosphorylate ERK-1/2 and p38 protein kinases, increasing IL-12 production and CD80/86 expression, and as a result enhance T cell activation and differentiation.
Using a bone marrow chimera strategy, we documented that bone marrow (BM) cell-derived, as opposed to systemic/serum-derived, complement and DAF are true regulators of T cell immunity in vivo. Chimeric mice with C3−/− BM-derived cells did not respond to alloantigenic stimuli despite having normal serum complement, while C3 deficient chimeras with WT (C3+) BM exhibited normal T cell alloreactivity (15, 20). Analogously, BM chimeras produced using C5aR deficient donors or recipients confirmed that T cell immunity is dependent on C5aR expression on BM-derived cells (15, 20).
Complement and DAF regulate both pathogenic and protective T cell immunity
The effects of immune cell-derived complement are relevant to multiple disease models. C3−/− mice exhibit enhanced susceptibility to viral infection (21). Conversely, DAF−/− mice are better protected and produce stronger T cell responses to lymphocytic choriomeningitis virus infection than WT controls (22). C3aR/C5aR deficient animals are highly susceptible to herpes keratitis and to toxoplasma gondii infection, in the latter case, producing little IL-12 and weak T cell immunity required for protection from this pathogen (14).
Complement regulates T cell autoimmunity as well. Experimental allergic encephalomyelitis (EAE) is one model of T cell mediated autoimmunity that mimics aspects of human multiple sclerosis. In response to immunization with myelin oligodendrocyte glycoprotein, DAF−/− mice develop more severe paralysis than wild type animals, which is associated with stronger autoreactive T cell immunity, enhanced IL-17 production, and diffuse T cell epitope spreading (14, 23, 24). These effects are C5aR- and C3aR-dependent as mice deficient in either or both of these G-protein coupled receptors develop weaker T cell responses and are resistant to EAE, regardless of DAF expression (14). Additional studies published in the last 2 years indicate that complement activation drives autoreactive pathogenic T cells in a model of autoimmune focal and segmental glomerulosclerosis (FSGS) in DAF−/− mice (25), and that serum-derived C5a interacting with immune cell-expressed C5aR is an essential mediator in an IL-17-dependent model of autoimmune arthritis (26). Finally, our research group reported in 2010 that immune cell-derived C3 is required for the induction of T cell dependent, autoimmune diabetes in mice (27).
Complement and T cell immunity following transplantation
Complement dependent effects on alloreactive T cell immunity regulate the phenotypic expression of immune-mediated transplant injury in animal models. In addition to the aforementioned observation that wild type mice do not reject allogeneic C3 deficient kidneys (7), wild type mice reject DAF−/− heart allografts (enhances local complement activation) with accelerated kinetics compared to wild type grafts (20). The accelerated rejection of DAF−/− heart transplants is associated with augmented anti-donor T cell reactivity and is notable in animals devoid of B cells, confirming that local complement activation accelerates graft rejection through a T cell-dependent mechanism. Moreover, the effect is complement dependent because heart grafts deficient in both DAF and C3 exhibit prolonged survival and stimulate weak T cell responses.
Using a bone marrow chimera strategy we determined that immune cell-derived and/or donor graft-derived complement, but not serum complement, regulate expansion of both alloreactive CD4+ and CD8+ T cells following transplantation (20). In other work, our joint group showed that DAF deficiency accelerates rejection and enhances T cell alloimmunity in murine models of both corneal and skin transplantation (13, 28). Results presented at the American Transplant Congress in San Diego, May 2010, further revealed that immune cell-derived complement is involved in the CD4 T cell help required for CD8 cell mediated allograft rejection (29)
Other studies published within the last year indicate that local complement production influences effector CD8 T cell responses to allogeneic vascular endothelial cells (EC) (30). Following stimulation with IFNγ, TNFα, and IL-1, murine EC produce alternative pathway complement components which activate locally yielding C5a (30). Experiments performed using in vitro culture systems and in vivo heart transplantation models showed that this EC-produced and locally activated complement regulates T cell expansion and function. Consistent with the aforementioned in vitro studies, the effects of EC-derived complement are in part transmitted through C5aR signalling on T cells, as C5aR deficiency or blockade abrogates responsiveness (30).
Studies published by others showed that C5a/C5aR interactions are pathogenic mediators of T cell dependent kidney transplant rejection in rodents. C5aR blockade prolonged kidney transplant survival in rodents, a result associated with an abrogation of intragraft mononuclear cell infiltration and a diminution in T cell alloreactivity (31). Other studies presented at the American Transplant Congress 2009 revealed that a blocking anti-C5 mAb synergizes with CTLA4-Ig to prevent T cell priming and prolong heart transplant survival in mice (32). Finally, recently published work indicates that genetic deficiency of C5aR in the donor and in the recipient limits T cell alloreactivity and results in prolonged murine kidney allograft survival (33). Together these observations are consistent with the interpretation that complement is a physiologically important regulator of alloreactive T cell immunity.
Therapeutic approaches/data in humans
Selected reports suggest that immune cell-derived and/or graft-derived complement contributes to human transplant rejection. The quantity of RNA message for alternative pathway complement components and complement receptors, including C5aR and C3aR, is higher in human transplant tissue with histologic evidence of rejection compared to non-injured control tissue (31, 34). Gene expression profiling of human kidney transplants reveals higher expression of several complement genes in deceased donor grafts with longer ischemic times, and interestingly, the complement gene upregulation correlates inversely with early and late renal function (35). In vitro studies presented in abstract form at the XIII international complement workshop (NY, Aug 2010) showed that analogous to the murine findings, human DCs produce complement and C5aR and C3aR signaling seems to be important in DC activation and function (36).
In another report, donor kidney expression of a specific polymorphic variant of C3 is associated with worse posttransplant outcomes (37). The precise mechanism through which this mutation alters allograft injury in human transplant recipients remains unclear and an independent study of a disparate and larger patient population could not verify these initial findings (38).
Conclusion
While the complement system was originally discovered as a serum component that “complemented” antibodies in the killing of bacteria, it is now apparent that complement has a multitude of other functions. Understanding complexities of complement’s contributions to transplant injury requires an understanding of the various activation pathways (e.g., classical pathway activation in antibody-mediated injury, alternative pathway activation as pathogenic in renal ischemia reperfusion, MBL (lectin) dependent in GI and cardiac ischemia (see accompanying article in this issue by Berger and Daha) and T cell activation) as well as comprehension of the expression and function of complement regulatory proteins and the source of the complement (serum or non-liver-derived). Emerging data indicate that immune cell-derived complement activation physiologically regulates immune cell survival and proliferation, modulating the strength and phenotype of adaptive T cell immune responses involved in alloimmune transplant rejection.
Consideration of the cellular source and local function of complement has potential therapeutic implications. Antibodies capable of blocking serum complement activation, including an anti-C5 antibody may be most beneficial to decrease post-transplant graft injury caused by alloantibody-initiated complement activation. Small molecule receptor inhibitors, which are in development for human use, may better penetrate tissues to restrain downstream consequences of the released cleavage products (e.g. C3a and C5a) and thereby restrain complement’s influence over immune cells. The recognition of the diversity through which complement participates in allograft injury supports the need for continued design and testing of complement inhibitors in human transplant recipients.
Acknowledgments
This work was supported by NIH grants AI43578 and AI071185 to PSH. HR is a recipient of a fellowship grant from the National Kidney Foundation.
Abreviations
- APC
antigen presenting cell
- BM
bone marrow
- C3aR
C3a receptor
- C5aR
C5a receptor
- DAF
decay accelerating factor
- DC
dendritic cell
- EAE
experimental allergic encephalomyelitis
- fB
complement factor B
- fD
complement factor D
- mAb
monoclonal antibody
- MBL
mannose binding lectin
- PI3Kγ
phosphoinositide 3-kinase gamma
References
- 1*.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–797. doi: 10.1038/ni.1923. This is an outstanding, in depth review of the complement cascade highlighting recent developments in all aspect of the field. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baldwin WM, Ota H, Rodriguez ER. Complement in transplant rejection: diagnostic and mechanistic considerations. Springer Semin Immunopathol. 2003;25(2):181–197. doi: 10.1007/s00281-003-0133-3. [DOI] [PubMed] [Google Scholar]
- 3.Wehner J, Morrell CN, Reynolds T, Rodriguez ER, Baldwin WM., 3rd Antibody and complement in transplant vasculopathy. Circulation research. 2007;100(2):191–203. doi: 10.1161/01.RES.0000255032.33661.88. [DOI] [PubMed] [Google Scholar]
- 4.Qian Z, Wasowska BA, Behrens E, Cangello DL, Brody JR, Kadkol SS, et al. C6 produced by macrophages contributes to cardiac allograft rejection. The American journal of pathology. 1999;155(4):1293–1302. doi: 10.1016/S0002-9440(10)65231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins AB, Schneeberger EE, Pascual MA, Saidman SL, Williams WW, Tolkoff-Rubin N, et al. Complement activation in acute humoral renal allograft rejection: diagnostic significance of C4d deposits in peritubular capillaries. J Am Soc Nephrol. 1999;10(10):2208–2214. doi: 10.1681/ASN.V10102208. [DOI] [PubMed] [Google Scholar]
- 6.Stegall M, Tayyab D, Lynn C, Justin B, Patrick D, JM G. Terminal Complement Inhibition Decreases Early Acute Humoral Rejection in Sensitized Renal Transplant Recipients. Am J Transplant. 2010;10(s4):39. [Google Scholar]
- 7.Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nature medicine. 2002;8(6):582–587. doi: 10.1038/nm0602-582. [DOI] [PubMed] [Google Scholar]
- 8.Sheerin NS, Risley P, Abe K, Tang Z, Wong W, Lin T, et al. Synthesis of complement protein C3 in the kidney is an important mediator of local tissue injury. FASEB J. 2008;22(4):1065–1072. doi: 10.1096/fj.07-8719com. [DOI] [PubMed] [Google Scholar]
- 9.Tang Z, Lu B, Hatch E, Sacks SH, Sheerin NS. C3a mediates epithelial-to-mesenchymal transition in proteinuric nephropathy. J Am Soc Nephrol. 2009;20(3):593–603. doi: 10.1681/ASN.2008040434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medof ME, Kinoshita T, Nussenzweig V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J Exp Med. 1984;160(5):1558–1578. doi: 10.1084/jem.160.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pepys MB. Role of complement in the induction of immunological responses. Transplant Rev. 1976;32:93–120. doi: 10.1111/j.1600-065x.1976.tb00230.x. [DOI] [PubMed] [Google Scholar]
- 12.Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science (New York, NY. 1996;271(5247):348–350. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- 13.Heeger PS, Lalli PN, Lin F, Valujskikh A, Liu J, Muqim N, et al. Decay-accelerating factor modulates induction of T cell immunity. J Exp Med. 2005;201(10):1523–1530. doi: 10.1084/jem.20041967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28(3):425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112(5):1759–1766. doi: 10.1182/blood-2008-04-151068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lalli PN, Strainic MG, Lin F, Medof ME, Heeger PS. Decay accelerating factor can control T cell differentiation into IFN-gamma-producing effector cells via regulating local C5a-induced IL-12 production. J Immunol. 2007;179(9):5793–5802. doi: 10.4049/jimmunol.179.9.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li K, Anderson KJ, Peng Q, Noble A, Lu B, Kelly AP, et al. Cyclic AMP plays a critical role in C3a-receptor-mediated regulation of dendritic cells in antigen uptake and T-cell stimulation. Blood. 2008;112(13):5084–5094. doi: 10.1182/blood-2008-05-156646. [DOI] [PubMed] [Google Scholar]
- 18*.Peng Q, Li K, Wang N, Li Q, Asgari E, Lu B, et al. Dendritic cell function in allostimulation is modulated by C5aR signaling. J Immunol. 2009;183(10):6058–6068. doi: 10.4049/jimmunol.0804186. In this paper, the authors confirm and extend previous work performed by this research group and others showing that C5a/C5aR signaling modulates DC function. C5a enhances DC immunogenicity by upregulating costimulatory molecules and increasing IL-12 production, among other effects. The signaling pathways are described. [DOI] [PubMed] [Google Scholar]
- 19**.Baruah P, Dumitriu IE, Malik TH, Cook HT, Dyson J, Scott D, et al. C1q enhances IFN-gamma production by antigen-specific T cells via the CD40 costimulatory pathway on dendritic cells. Blood. 2009;113(15):3485–3493. doi: 10.1182/blood-2008-06-164392. In this paper the authors define a novel role for DC produced C1q as a modulator of DC function. Colocalization between C1q and CD40 upon cognate T cell APC interactions is noted. The effects were shown to be independent of toll-like receptor signaling. [DOI] [PubMed] [Google Scholar]
- 20.Pavlov V, Raedler H, Yuan S, Leisman S, Kwan WH, Lalli PN, et al. Donor deficiency of decay-accelerating factor accelerates murine T cell-mediated cardiac allograft rejection. J Immunol. 2008;181(7):4580–4589. doi: 10.4049/jimmunol.181.7.4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suresh M, Molina H, Salvato MS, Mastellos D, Lambris JD, Sandor M. Complement component 3 is required for optimal expansion of CD8 T cells during a systemic viral infection. J Immunol. 2003;170(2):788–794. doi: 10.4049/jimmunol.170.2.788. [DOI] [PubMed] [Google Scholar]
- 22.Fang C, Miwa T, Shen H, Song WC. Complement-dependent enhancement of CD8+ T cell immunity to lymphocytic choriomeningitis virus infection in decay-accelerating factor-deficient mice. J Immunol. 2007;179(5):3178–3186. doi: 10.4049/jimmunol.179.5.3178. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, Lin F, Strainic MG, An F, Miller RH, Altuntas CZ, et al. IFN-gamma and IL-17 production in experimental autoimmune encephalomyelitis depends on local APC-T cell complement production. J Immunol. 2008;180(9):5882–5889. doi: 10.4049/jimmunol.180.9.5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Miwa T, Hilliard B, Chen Y, Lambris JD, Wells AD, et al. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med. 2005;201(4):567–577. doi: 10.1084/jem.20040863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25*.Bao L, Haas M, Pippin J, Wang Y, Miwa T, Chang A, et al. Focal and segmental glomerulosclerosis induced in mice lacking decay-accelerating factor in T cells. The Journal of clinical investigation. 2009;119(5):1264–1274. doi: 10.1172/JCI36000. While not directly relevant to transplantation, this paper highlights the importance of complement regulation on T cell function. T cells from DAF−/− mice preferentially induced a model of glomerulonephritis that mimics many aspects of human focal and segmental sclerosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26*.Hashimoto M, Hirota K, Yoshitomi H, Maeda S, Teradaira S, Akizuki S, et al. Complement drives Th17 cell differentiation and triggers autoimmune arthritis. J Exp Med. 2010;207(6):1135–1143. doi: 10.1084/jem.20092301. The authors extend previously reported work in other disease models such as EAE (see ref 23) by demonstrating that C5a is an important inducer of pathogenic IL-17 producing autoreactive T cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin M, Yin N, Murphy B, Medof ME, Segerer S, Heeger PS, et al. Immune Cell Derived C3 is Required for Autoimmune Diabetes Induced by Multiple Low Doses of Streptozotocin. Diabetes. doi: 10.2337/db10-0044. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28*.Esposito A, Suedekum B, Liu J, An F, Lass J, Strainic MG, et al. Decay Accelerating Factor is Essential for Successful Corneal Engraftment. Am J Transplant. 2010;10(4):527–534. doi: 10.1111/j.1600-6143.2009.02961.x. In the absence of the complement regulatory molecule DAF, minor antigen disparate corneal grafts, which are normally accepted by recipients, undergo rapid rejection. The findings not only highlight the role of complement as a regulator of T cell alloimmunity but demonstrate that the brittle state of ocular tolerance is also modulated by local complement activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vieyra M, Leisman S, Kwan W, Heeger P. Enhanced Immune Cell-Derived Complement Activation Bypasses the Need for CD4 Help for Rejecting Murine Heart Transplants. Am J Transplant. 2010;10(s4):158. [Google Scholar]
- 30**.Raedler H, Yang M, Lalli PN, Medof ME, Heeger PS. Primed CD8 T-Cell Responses to Allogeneic Endothelial Cells Are Controlled by Local Complement Activation. Am J Transplant. 2009 doi: 10.1111/j.1600-6143.2009.02723.x. Endothelial cells (ECs) are known to activate alloreactive CD8 T cells as they enter a vascularized allografts. Herein the authors show that EC-derived complement plays a key role in this process. Complement component deficiency or blockade prevents murine ECs from activating T cells, both in vitro and in vivo. The findings add new mechanistic insight into the basis of T cell/EC interactions and highlight the need for additional studies focusing on complement blockade to prevent T cell mediated allograft injury. [DOI] [PubMed] [Google Scholar]
- 31.Gueler F, Rong S, Gwinner W, Mengel M, Brocker V, Schon S, et al. Complement 5a Receptor Inhibition Improves Renal Allograft Survival. J Am Soc Nephrol. 2008 doi: 10.1681/ASN.2007111267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leisman S, Rother R, Hunter J, Johnson K, Heeger P, Leisman Staci A, Rother Russell, Hunter Jeffrey, Johnson Krista, Heeger Peter S. Anti-C5 mAb Plus Low Dose CTLA4 Ig Inhibit Alloreactive T Cells and Prolong Heart Graft Survival in Mice. Am J Transplant. 2009;9(s2):209. [Google Scholar]
- 33*.Li Q, Peng Q, Xing G, Li K, Wang N, Farrar CA, et al. Deficiency of C5aR prolongs renal allograft survival. J Am Soc Nephrol. 2010;21(8):1344–1353. doi: 10.1681/ASN.2009090977. Murine kidney transplantation is significantly prolonged in the absence of C5aR in the donor and in the recipient. The enhanced survival is associated with marked reduction in pathology and weaker alloreactive T cell immunity, underscoring the concept that complement regulates alloreactive T cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keslar K, Rodriguez ER, Tan CD, Starling RC, Heeger PS. Complement gene expression in human cardiac allograft biopsies as a correlate of histologic grade of injury. Transplantation. 2008;86(9):1319–1321. doi: 10.1097/TP.0b013e3181889831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naesens M, Li L, Ying L, Sansanwal P, Sigdel TK, Hsieh SC, et al. Expression of complement components differs between kidney allografts from living and deceased donors. J Am Soc Nephrol. 2009;20(8):1839–1851. doi: 10.1681/ASN.2008111145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li K, Fazekasova H, Gomes C, Wang N, Sagoo P, Peng Q, et al. Expression of complement components, receptors and regulators by human dendritic cells and the role of C3a and C5a in modulation of dendritic cell function. Molecular Immunology. 2010;47(13):2232. doi: 10.1016/j.molimm.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown KM, Kondeatis E, Vaughan RW, Kon SP, Farmer CK, Taylor JD, et al. Influence of donor C3 allotype on late renal-transplantation outcome. The New England journal of medicine. 2006;354(19):2014–2023. doi: 10.1056/NEJMoa052825. [DOI] [PubMed] [Google Scholar]
- 38*.Varagunam M, Yaqoob MM, Dohler B, Opelz G. C3 polymorphisms and allograft outcome in renal transplantation. The New England journal of medicine. 2009;360(9):874–880. doi: 10.1056/NEJMoa0801861. In contrast to previously published work (see ref 38), this large study did not detect a relationship between C3 allotype and kidney transplant outcome. Differences in patient population and specifics of study design could account for the disparate results. Importantly, it is not yet fully understood how the allotypic differences in the C3 molecule impact C3 function. [DOI] [PubMed] [Google Scholar]