

Abstract
The DNA-relaxing enzyme topoisomerase I (Top1) can be inhibited by heterocyclic compounds such as indolocarbazoles and indenoisoquinolines. Carbohydrate and hydroxyl-containing side chains are essential for the biological activity of indolocarbazoles. The current study investigated how similar functionalities could be “translated” to the indenoisoquinoline system and how stereochemistry and hydrogen bonding affect biological activity. Herein is described the preparation and assay of indenoisoquinolines substituted with short-chain alcohols, diols, and carbohydrates. Several compounds (including those derived from sugars) display potent Top1 poisoning and antiproliferative activities. The Top1 poisoning activity of diol-substituted indenoisoquinolines is dependent upon stereochemistry. Although the effect is striking, molecular modeling and docking studies do not indicate any reason for the difference in activity due to similar calculated interactions between the ligand and Top1-DNA complex and ambiguity about the binding mode. A stereochemical dependence was also observed for carbohydrate-derived indenoisoquinolines. Although similar trends were observed in other classes of Top1 inhibitors, the exact nature of this effect has yet to be elucidated.
Introduction
Topoisomerase I (Top1) is an enzyme that relaxes supercoiled DNA. Relaxed DNA is required for many cellular processes such as DNA replication, transcription, and repair.1–4 Top1 relaxes DNA through a cycle of cleavage and religation steps involving the active site residue Tyr723. This residue attacks the phosphodiester backbone, breaking the single strand and forming a covalent “cleavage complex” in which the unbroken strand undergoes “controlled rotation” and relaxes the DNA. After relaxation, the scissile strand is religated and the enzyme is released. As inhibition of Top1 can lead to cell death,5–7 several Top1 inhibitors have been developed as a targeted approach for anti-cancer therapy. Camptothecin (1)8 and its clinically used analogues, topotecan (2) and irinotecan (3)9,10 (Figure 1), were found to inhibit Top1 activity by intercalating into the cleavage complex and preventing the religation step of the catalytic cycle. As a result, advancing replication forks collide with the cleavage complex, resulting in double-stranded DNA breaks and apoptosis.5–7 Compounds that inhibit the religation reaction are commonly known as “Top1 poisons”.4–7
Figure 1.

Representative Top 1 Poisons
Although these inhibitors possess potent antitumor activity, issues regarding solubility and bioactivity, dose-limiting toxicity,9–11 and most importantly, the instability of the hydroxy lactone and associated pharmacokinetic liabilities,12,13 led to the development of therapeutic alternatives. A COMPARE analysis14,15 performed on the cytotoxicity profile of synthetic indenoisoquinoline 4 showed many similarities to the cytotoxicity profile of camptothecin, indicating that compound 4 may exert its action through inhibition of Top1.15,16 Indeed, indenoisoquinolines such as 4 and MJ-III-65 (5)17 inhibit the religation reaction by an intercalative mechanism like camptothecin,16,18 and Top1-linked DNA single strand breaks have been detected by immunocomplex assays, alkaline elution after proteinase digestion, and as histone γH2AX foci in various human cancer cell lines.14,19,20 Furthermore, the DNA single-strand breaks resulting from treatment of MCF7 cells with the indenoisoquinoline 4 reversed more slowly than those induced by camptothecin.14 This is expected to offer a potential clinical advantage for indenoisoquinolines vs. the camptothecins because the rapid reversibility of Top1-DNA-camptothecin cleavage complexes necessitates long infusion times in order to ensure maximum clinical benefit.21 Additionally, indenoisoquinolines are chemically stable, and many compounds in this class possess high antiproliferative activity. After a series of comprehensive structure-activity relationship (SAR) studies,22–25 two compounds, indotecan (6) and indimitecan (7) were eventually promoted into Phase I clinical trials through the National Cancer Institute.16,20 Cleavage complex trapping by both compounds 6 and 7 in human colon cancer HT29 cells results in histone γH2AX phosphorylation, which can be used as a pharmacodynamic biomarker to monitor drug effects in cancer patients.26
Indolocarbazoles, a series of bis-indole alkaloids isolated predominantly from various bacterial species, also demonstrate promising antitumor activity through inhibition of Top1. One of the first indolocarbazoles shown to exhibit antiproliferative activity through selective inhibition of Top1 was rebeccamycin (8).27 Since then, many potent indolocarbazoles have been synthesized28–31 and edotecarin (9) recently finished Phase III clinical trials.10,32
Structurally, the indolocarbazoles bear a carbohydrate substituent on an indole nitrogen, and like the lactam side chain of the indenoisoquinolines,22 the carbohydrate substituent present in the indolocarbazoles is required for effective Top1 inhibition.28,29 A comparison of the crystal structures33 of the Top1 ternary complexes containing the indolocarbazole SA315F (10)34 and indenoisoquinoline MJ238 (11, Figure 2a) reveals that the carbohydrate substituent of the indolocarbazole and the lactam side chain of the indenoisoquinoline both extend out into the DNA major groove region. This relationship is shown in Figure 2b. The aromatic cores of both compounds face Arg364 (with which they likely interact). The side chains sit in close proximity to Asn352 and occupy similar spatial areas, where, accounting for flexibility, they could easily hydrogen bond with this residue, water, or flanking base pairs.
Figure 2.

Structures of compounds 10 and 11 (2a) and a crystal structure alignment showing both ligands in ternary complex (2b). Compound 10 is colored green and 11 is colored magenta; possible interactions with Arg364 (minor groove) and Asn352 (major groove; from individual crystal structures 1SC7 and 1SEU; colored to match the corresponding ligand) are shown. The scissile DNA strand (flanking base pairs removed) is shown at the top of the figure and colored by element; corresponding nonscissile strand is on the bottom. Distances (in Å) are measured from heavy-atom to heavy atom and the diagram is programmed for wall-eyed (relaxed) viewing.
In addition to the similar binding orientation, both the indenoisoquinoline and the indolocarbazole SARs have demonstrated that the presence of hydrogen bonding groups (i.e., hydroxyl) on their respective side chains correlates with an increase in bioactivity.23,24,30 Together, these findings led to the hypothesis that a potent indenoisoquinoline could be developed by strategically combining carbohydrate side chains (such as those of the indolocarbazoles) with the indenoisoquinoline core. In essence, two elements proven to be effective for their respective inhibitor classes could be “mixed-and-matched.”
Additionally, in an initial study, two indenoisoquinolines bearing propanediol substituents (Figure 3) indicated that anti-Top1 activity appeared to be dependent upon stereochemistry. The (S)-isomer 12a possesses high Top1 inhibitory activity, whereas the (R)-isomer 12b is largely inactive (and the racemate 12c’s activity is between that of the enantiopure forms). This result provided a rationale to investigate other short-chain alcohol substituents with hope of adding stereochemical and orientation-based effects to the indenoisoquinoline SAR.
Figure 3.

Stereochemistry affects the bioactivity of indenoisoquinolines. Top1 inhibitory activity is expressed as relative to 1 μM camptothecin: 0, no inhibitory activity; +, between 20 and 50% activity; ++, between 50 and 75% activity; +++, between 75% and 95% activity; ++++, equipotent.
The present communication details the design, synthesis, and evaluation of several series of novel indenoisoquinolines, including those substituted with amines derived from aldohexoses and aldopentoses and those bearing shorter chiral side chains. These strategies aim to combine active elements from different classes of Top1 inhibitors and elucidate the roles that stereochemistry, orientation, and hydrogen bonding play on biological activity.
Rationale and Chemistry
Aldopentose and Aldohexose Substituents
Previously, installation of indenoisoquinoline side chains involved condensation of the indenoisochromenone lactone (a vinylogous anhydride, e.g. compound 16, Scheme 1) with a primary amine.35 Therefore, the preparation of carbohydrate-derived indenoisoquinolines required the conversion of the aldose into an aminodeoxyalditol. Initially, a synthesis was envisioned that proceeded through reductive benzylamination of the aldehyde and cleavage of the benzyl group. Disappointingly, deprotection did not proceed as planned, and the first step was irreproducible and either yielded mixtures of products or failed entirely for substrates such as D-xylose (13d, Table 1) and D-glucose (13c). Although it was also speculated that protection of the hydroxyl groups of the monosaccharide would be necessary to carry out the required transformations, Winestock and Plaut’s method for preparation of aminodeoxyalditols was eventually employed without recourse to protecting groups.36 This method is shown below for D-arabinose (15a, Scheme 1) and proved successful for all sugars investigated.
Scheme 1 a.

aReagents and conditions: (a) i. H2NOH·HCl, NaOMe, EtOH, r.t. ii. 13a, 70 °C; (b) Pt(IV)O2, H2 (40 psi), AcOH, r.t; (c) MeOH or CHCl3 (MeOH for example shown), reflux.
Table 1.
Aldopentose and Aldohexose-Based Substituents.
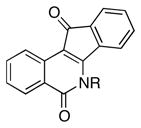 | |||
|---|---|---|---|
| Aldose | Oxime | Amine | Indenoisoquinoline R group |
 13a |
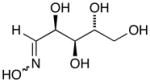 14a |
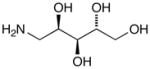 15a |
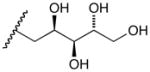 17a |
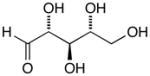 13b |
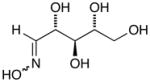 14b |
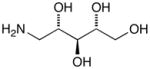 15b |
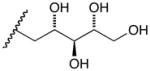 17b |
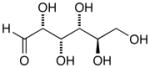 13c |
 14c |
 15c |
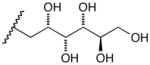 17c |
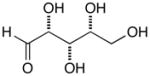 13d |
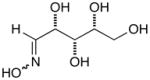 14d |
 15d |
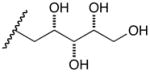 17d |
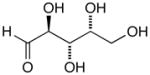 13e |
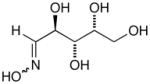 14e |
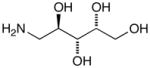 15e |
 17e |
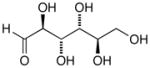 13f |
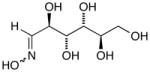 14f |
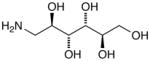 15f |
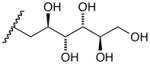 17f |
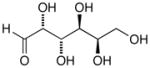 13g |
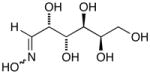 14g |
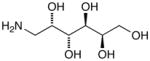 15g |
 17g |
 13h |
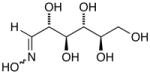 14h |
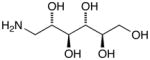 15h |
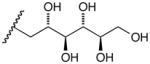 17h |
D-Arabinose (13a) was treated with hydroxylamine hydrochloride in the presence of sodium methoxide to afford the corresponding oxime 14a as a mixture of (E)- and (Z)- diastereomers.37,38 The mixture was hydrogenated in the presence of catalytic Pt(IV)O2 to afford D-arabitylamine (15a)36 in quantitative yield following ion-exchange chromatography. Likewise, D-ribose (13b), D-glucose (13c), D-xylose (13d), D-lyxose (13e), D-mannose (13f), D-galactose (13g) and D-allose (13h) were converted into their respective oximes (or diastereomeric mixtures of oximes) 14b–h (structures given in Table 1). These oximes were reduced to amines 15b–h, respectively. To prepare indenoisoquinolines, lactone 1639 was condensed with excess aminodeoxyalditol in refluxing MeOH or CHCl3 (also shown for arabinose in Scheme 1). The indenoisoquinolines 17a–h were obtained in moderate yields.
Previous studies from our laboratories revealed that nitrated indenoisoquinolines have much higher bioactivity than unsubstituted (and other substituted) indenoisoquiolines.25,40 To evaluate the hypothesis that nitration would improve potency with carbohydrate substituents present, an indenoisoquinoline (19) was prepared from D-glucamine (15c) and the nitrated lactone 18,25,40 as shown in Scheme 2.
Scheme 2 a.

aReagents and Conditions: (a) MeOH, reflux.
Three-Carbon Alcohol and Diol Substituents
Indenoisoquinolines 12a, 12b, and 12c were prepared by condensation of lactone 16 with commercially available aminoalcohols 20a, 20b, and 20c to afford the (S), (R), and racemic compounds, respectively (Scheme 3). As the stereochemistry of the 2′-hydroxyl group appeared to play a significant role in the inhibition of Top1 (Figure 3) and there is no crystal structure of these compounds in ternary complex with DNA and Top1 available, additional compounds were synthesized in order to elucidate the function (and possible stereochemical SAR) of the 2′ substituent.
Scheme 3 a.

aReagents and Conditions: (a) CHCl3 or MeOH, reflux.
It was hypothesized that the 2′-hydroxyl group may form a crucial hydrogen bond in the ternary complex. To test this hypothesis, the 2′-hydroxyl group was first replaced with a methyl group, abolishing the H-bonding potential at this position. The racemic amino alcohol 21c, along with the enantiopure alcohols 21a and 21b (all commercially available), were condensed with lactone 16 to give the corresponding indenoisoquinolines 22a–c (Scheme 3).
Another analogue, 25 (Scheme 4), possesses a 2′-keto group instead of the 2′-hydroxyl, switching the substituent’s properties from H-bond donor/acceptor to solely H-bond acceptor. Failure to selectively oxidize the 2′-hydroxyl of 12c led to the approach depicted in Scheme 4. 1-Amino-2,3-propanediol (20c) was condensed with lactone 16 to yield the racemic indenoisoquinoline 12c. Selective protection of the primary alcohol (as the silyl ether 23) was followed by oxidation of the secondary alcohol using Ley’s conditions41 to yield indenoisoquinoline 24. Acidic cleavage of the protecting group afforded the ketone analogue 25.
Scheme 4 a.

aReagents and conditions: (a) CHCl3, reflux; (b) TBDPSCl, Et3N, 4-DMAP, CH2Cl2; (c) NMO, TPAP, CH2Cl2, r.t.; (d) methanolic HCl, r.t.
We next sought to determine the exact function of the primary alcohol. If the primary alcohol participates in hydrogen bonding in the ternary complex (as those of indolocarbazoles are proposed to do33), there are two potential explanations for the activity of a diol such as 12a. A hydrogen-bonding interaction in the complex involving the primary alcohol could place the secondary alcohol in a position for additional interactions, or the reverse could occur: the stereochemistry of the secondary alcohol could orient the primary alcohol for more favorable binding via intramolecular interactions. Analogues that allowed for investigation of these possible explanations were thus prepared.
Scheme 5 depicts the synthesis of “truncated” diol indenoisoquinolines 27a–c and the amino analogue 27d. Lactone 16 was condensed with commercially available alcohols 26a–d to afford the respective indenoisoquinolines 27a–d. Compounds 27a–c eliminated the 3′ hydrogen-bonding group entirely (a similar strategy as that used to elucidate the function of the 2′-hydroxyl), whereas in compound 27d the hydroxyl group was replaced with an amino group.
Scheme 5 a.

aReagents and conditions: (a) CHCl3, reflux.
Additionally, two ring-substituted diol analogues were prepared using the same rationale for the synthesis of 19 (vide supra). Dimethoxy- and nitro-substituted lactones (28 and 18, respectively)25,40 were condensed with (S)-diol 20a (which conferred good activity upon the unsubstituted lactone core, see Figure 3) to yield the corresponding analogues 29 and 30 (Scheme 6).
Scheme 6 a.

aReagents and conditions: (a) CHCl3, reflux.
Biological Results and Discussion
The indenoisoquinoline analogues were tested for antiproliferative activity in the National Cancer Institute’s Developmental Therapeutics Assay against cell lines derived from approximately 60 different human tumors.42,43 After an initial one-dose screening assay, (at 10−5 molar), selected compounds were tested at five concentrations ranging from 10−8 to 10−4 M. Results are reported in Table 2 as GI50 values for selected cell lines, and overall antiproliferative potency is quantified as a mean-graph midpoint (MGM). This value can be considered an average GI50 across all cell lines tested; compounds whose GI50 values fall outside the concentration range tested (10−8 to 10−4 M) are assigned GI50 values of either 10−8 M or 10−4 M, respectively. Additionally, the values for growth percent, a measure of inhibitory activity in the initial 10−5 molar assay, are given for most compounds, including those not selected for the five-concentration screen. For comparison, Top1 and antiproliferative activity data for camptothecin (1),17 lead indenoisoquinolines 4 and 5,23,44 and clinical candidates 6 and 723,45 are included.
Table 2.
Antiproliferative Potencies and Topoisomerase I Inhibitory Activities of Substituted Indenoisoquinolines and Relevant Compounds.
| Compd | Cytotoxicity (GI50 in μM)a |
MGMb | Growth Percentc | Top1 Cleavaged | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lung | Colon | CNS | Melanoma | Ovarian | Renal | Prostate | Breast | |||||
| HOP-62 | HCT-116 | SF-539 | UACC-62 | OVCAR-3 | SN12C | DU-145 | MCF7 | |||||
| 117 | 0.01 | 0.03 | 0.01 | 0.01 | 0.22 | 0.02 | 0.01 | 0.01 | 0.0405 ± 0.0187f | -d | ++++ | |
| 417,44 | 1.3 | 35 | 41 | 4.2 | 73 | 68 | 37 | 1.58 | 20.0 ± 14 | - | ++ | |
| 517,44 | 0.02 | 0.10 | 0.04 | 0.03 | 0.5 | <0.01 | <0.01 | <0.01 | 0.11 | - | ++++ | |
| 623,45 | 1.78 | 1.15 | 0.04 | 0.03 | 74.1 | 0.813 | 0.155 | 0.37 | 4.64 ± 1.25 | - | ++++ | |
| 723,45 | <0.01 | <0.01 | 0.037 | <0.01 | 0.085 | <0.01 | <0.01 | 0.01 | 0.079 ± 0.023 | - | ++++ | |
| 12a | 5.01 | - | 7.41 | 5.37 | 14.5 | 6.92 | 5.75 | 4.90 | 8.71 | - | +++ | |
| 12b | 3.79 | 4.47 | 5.72 | 4.36 | 16.1 | 3.77 | 2.34 | 3.04 | 6.24 ± 0.22 | - | 0/+ | |
| 12c | 7.76 | 4.36 | 5.89 | 7.58 | 18.6 | 2.95 | 2.88 | 3.55 | 5.13 | - | ++ | |
| 17a | 8.71 | 5.62 | 7.59 | 7.41 | >100 | 20.0 | 16.6 | 4.57 | 19.9 | 58.44 | ++ | |
| 17b | 2.95 | 3.39 | 2.14 | 3.23 | 13.5 | 2.40 | 1.32 | 1.38 | 3.31 | 17.28 | 0 | |
| 17c | - | - | - | - | - | - | - | - | - | 97.16e | ++ | |
| 17d | 3.23 | 3.39 | 7.94 | 2.88 | 24.0 | 2.24 | 2.75 | 1.74 | 4.07 | 34.91 | ++ | |
| 17e | 6.46 | 3.39 | 11.2 | 6.31 | 13.2 | 5.01 | 3.98 | 1.90 | 7.76 | 50.62 | +++ | |
| 17f | - | - | - | - | - | - | - | - | 69.35 | + | ||
| 17g | - | - | - | - | - | - | - | - | 67.99 | + | ||
| 17h | 3.23 | 4.17 | 5.75 | 2.95 | 13.8 | 3.39 | 3.31 | 1.78 | 5.75 | 49.31 | - | |
| 19 | 7.35 | 5.08 | 2.61 | 2.98 | 4.26 | 17.7 | 4.76 | 0.88 | 4.60 ± 0.53 | 58.05 | ++++ | |
| 22a | - | - | - | - | - | - | - | - | - | ++ | ||
| 22b | - | - | - | - | - | - | - | - | 78.60 | + | ||
| 22c | 17.6 | 15.9 | 14.4 | 11.7 | 25.7 | 37.1 | 23.7 | 34.4 | 15.6 ± 2.15 | - | + | |
| 25 | 40.7 | 30.9 | 20.0 | 58.9 | 5.23 | 81.3 | 17.8 | 32.3 | 20.9 | 72.36 | + | |
| 27a | 3.39 | 5.52 | 6.31 | 10.2 | 21.4 | 9.33 | 8.32 | 3.23 | 10.2 | 55.23 | +++ | |
| 27b | - | - | - | - | - | - | - | - | 54.88 | +++ | ||
| 27c | 3.46 | 5.75 | 6.76 | 6.16 | 16.2 | 3.98 | 4.36 | 3.80 | 7.9 | 50.24 | + | |
| 27d | 0.276 | 0.263 | 0.344 | 0.318 | 1.32 | 0.206 | 0.130 | 0.198 | 0.348 ± 0.109 | -23.49 | ++ | |
| 29 | 0.336 | 0.230 | 0.214 | 0.144 | 0.287 | 0.501 | 0.419 | 0.097 | 0.401 ± 0.055 | - | +++ | |
| 30 | 0.089 | 0.025 | 0.157 | 0.098 | 0.309 | 0.241 | 0.040 | 0.016 | 0.156 ± 0.061 | - | ++++ | |
The cytotoxicity GI50 values are the concentrations corresponding to 50% growth inhibition.
Mean graph midpoint for growth inhibition of all human cancer cell lines successfully tested, ranging from 10−8 to 10−4 molar.
Percentage of cell growth in a one-dose assay at 10 μM. This data has recently been incorporated into the NCI protocol (implemented ca. 2006), thus, growth percent is only available for select compounds.
Compound-induced DNA cleavage due to Top1 inhibition is graded by the following rubric relative to 1 uM camptothecin: 0, no inhibitory activity; +, between 20 and 50% activity; ++, between 50 and 75% activity; +++, between 75% and 95% activity; ++++, equipotent. The 0/+ ranking is between 0 and +.
Some compounds were not selected for further testing, refer to text for details.
For MGM GI50 values in which a standard error appears, the GI50 values for individual cell lines are the average of two determinations; values without standard error are from one determination. The values for 1, 4, 5, 6, and 7 are from many determinations.
Top1 inhibition was measured by a compound’s ability to induce enzyme-linked DNA breakage and is graded by the following semiquantitative scale relative to 1 μM camptothecin: 0, no inhibitory activity; +, between 20 and 50% activity; ++, between 50 and 75% activity; +++, between 75% and 95% activity; ++++, equipotent. The 0/+ score is defined as between 0 and +. Many compounds in this series can act as Top1 poisons, and a representative example of Top1-linked DNA cleavage by these indenoisoquinolines can be observed in Figure 4. As can be observed, the cleavage patterns resemble indenoisoquinoline 4, with compound 30 displaying the highest potency.
Figure 4.

Top1-mediated DNA cleavage induced by indenoisoquinolines 29, 30, 17a, and 17c. Lane 1: DNA alone; lane 2: DNA plus Top1; lanes 3–20: DNA plus Top1 and indenoisoquinolines as indicated above gel. Numbers and arrows on the left indicate cleavage site positions.
As the data for the short-chain alcohol and diol substituents could, in part, be used to explain the results of aldopentose and hexose substitution, these results will be discussed first. As noted earlier, the Top1 inhibitory activity of compounds 12a and 12b differs drastically (+++ and 0/+, respectively). An attempt to explain this difference was made through analysis of hypothetical binding modes using molecular modeling and docking. Both compounds were docked with GOLD using a defined active site centroid in a mutant, solvated Top1-DNA complex.33,46 The ternary complexes containing the highest-ranked ligand poses were then minimized and visualized with SYBYL 8.1 (see experimental section for full details).
Although our docking protocol was extensively validated (see experimental section for full details), the top ligand poses were dependent upon the assigned charge sets. Nonetheless, several docking runs indicated that a mixture of two dominant poses for compound 12a were returned regardless of the charge set used, and these poses had similar GOLD fitness scores (between 72–75). A “normal” mode was returned as the top pose when MMFF94s47 charges were assigned to the ligand. In this mode, the position of the aromatic core is effectively identical to that in crystallized indenoisoquinoline-Top1-DNA ternary complexes in which the polycyclic system of the inhibitor is similar to that of 12a.33 The ketone hydrogen bonds with Arg364 and the polar side chain projects into the major groove. This mode is depicted in Figure 5a. When Gasteiger-Hückel charges were assigned instead, a “flipped” binding mode was returned as the top-ranked pose (although normal modes were also returned as alternates). Interestingly, flipped binding modes were not returned in the top five poses during the docking validation runs with camptothecin (1), topotecan (2), and indenoisoquinoline 11, regardless of the ligand, charges, or force field used. In this binding mode, the ligand is flipped 180° along the long axis of the indenoisoquinoline and 180° relative to the short axis, positioning the lactam carbonyl in a location to hydrogen bond with Arg364 (Figure 5b). The side chain is thus calculated to project into an open area of the minor groove. Although there is currently no literature evidence available to support this binding mode for indenoisoquinolines (ab initio calculations indicate normal modes are more favorable46), X-ray crystallography indicates that norindenoisoquinolines, which share a similar aromatic core, bind in a flipped mode, and ab initio calculations indicate that the flipped mode of norindenoisoquinolines is more favorable.48,49 Moreover, the GOLD fitness scores for 12a binding in the “normal” mode (72.98) and the “flipped mode” (74.30) were similar, indicating a small difference in binding energies between the two modes. On the other hand, the prior ab initio calculations carried out at the MP2 level, which indicate a preference for binding in the “normal mode”, are likely to be more accurate because they take dispersion into consideration.46 They also agree the X-ray crystallography studies that show compound 11 binding in the “normal mode”.33
Figure 5.

Hypothetical models of the ternary complexes with Top1, DNA, and diols 12a and 12b. All views are from the major groove, scissile-strand side. The normal binding mode of compound 12a (green ligand) is shown in 5a. The flipped binding mode of compound 12a (cyan ligand) is shown in 5b. The normal (only) binding mode of compound 12b (red ligand) is shown in Figure 5c. All other structures are colored by element, water molecules are shown as red spheres and relevant substructures are labeled. Hydrogen bonds and polar contacts are shown and labeled. Distances (in Å) are from heavy atom to heavy atom. The diagrams are programmed for wall-eyed (relaxed) viewing.
Overall, the molecular modeling results indicate that both binding modes are theoretically possible and should at least be considered. In both cases, the aromatic core of the indenoisoquinoline is well intercalated into the complex without any visible steric hindrance, projecting the diol side chain beyond the flanking base pairs where it is free to interact with the surrounding structures. In both binding modes, polar contacts and water-mediated H-bonds are observed. For compound 12a, the ketone of the aromatic core is predicted to hydrogen-bond with Arg364 (Figure 5a). Additional contacts (including water-mediated H-bonds) are formed with Met428, Asn352, and two flanking base pairs. Similar networks of water are reported to stabilize the side chains in similar models of polar indenoisoquinolines and aromathecins.50 In the flipped binding mode (Figure 5b), most major-groove contacts are absent, but the diol side chain forms two H-bonds in the minor groove: with the flanking base pair Tgp11, and the amino acid Asp533. The lactam carbonyl also binds to Arg364. For compound 12b, only the normal binding mode was returned by GOLD regardless of assigned charge set (Figure 5c).
All things considered, it is difficult to determine the cause of a stereochemical effect on the basis of these studies alone, which are complicated by the possibility of an alternate binding mode. Similar interactions within the ternary complex are observed, and there is no observable difference between the normal binding modes of the polycyclic systems of compounds 12a and 12b. Compound 12b, in normal mode, also forms H-bonds (including water-mediated) with two flanking base pairs, Arg364, and Tyr426.
Interestingly, the cytotoxic effects exerted by these two compounds, and many others in this series, are similar despite differing Top1 inhibitory activities. This indicates that Top1 inhibition is not the sole harbinger of antiproliferative potency. The lack of correlation between Top1 inhibition and cytotoxic activity is not surprising since it has been observed before for indenoisoquinolines,17 indolocarbazoles28 and other classes of Top1 inhibitors substituted with similar side chains.51 The effect could be due to factors including differences in solubility, activity at another target, or cellular ADME properties.
In the all models and modes, the 2′-hydroxyl of the diols forms a hydrogen bond, and as predicted from these models, indenoisoquinolines 22a–c exhibit a loss of Top1 inhibition relative to compounds 12a and 27a–b, regardless of stereochemistry. The lower activity of 22a–c could be due to the loss of hydrogen bonding between the 2′-hydroxyl and surrounding structures. Additionally, the ketone analogue 25 possesses a diminished ability to inhibit Top1, indicating that the role of the secondary hydroxyl of 12a could be to serve as a hydrogen-bond donor.
Indenoisoquinolines 27a and 27b, which lack a primary hydroxyl group, demonstrate no reduction in Top1 inhibition when compared to the reduction observed for indenoisoquinolines lacking the secondary hydroxyl. These results indicate that the primary alcohol may not be as important as the 2′-hydroxyl substituent, despite the contacts it forms in the models. Unlike compounds 12a–12c, however, the stereochemistry of the secondary alcohol has little effect on Top1 inhibition (cf. compounds 27a and 27b, it is unknown why the racemate 27c has lower activity). This result, along with the observed absence of a stereochemical effect for 22a and 22b, supports the hypothesis that the presence of both the primary and secondary alcohol is required for the differences in activity observed initially in 12a and 12b.
Compound 27d exists in the racemic form and its Top1 inhibitory activity is comparable to the activity of racemate 12c, indicating that the primary amine does not improve affinity in the ternary complex. Likewise, the hydroxyl-to-amine switch did not prove effective at enhancing Top1 inhibition for indolocarbazoles.52 A difference between these two racemates is observed in their cytotoxicities, however. Indenoisoquinoline 12c exerts an MGM GI50 of 5.13 μM whereas indenoisoquinoline 27d is much more potent with an MGM GI50 of 0.348 μM. It is unknown exactly how the amine exerts this effect, but one possibility is that the protonated primary amine aids in targeting the drug to negatively charged DNA52–54 or enters cells via an amino acid or polyamine transport system.24
As predicted, the dimethoxy- and nitro-analogues of compound 12a proved to be more cytotoxic than the parent compound (29, 0.401 μM; 30, 0.156 μM) while maintaining Top1 inhibition. The increase in anti-Top1 potency of 30 is proposed to be associated with hydrogen bonding between the nitro substituent and enzyme residues. Additionally, the strong electron-withdrawing nature of the nitro substituent may increase the π-stacking interaction between the indenoisoquinoline aromatic core and flanking DNA bases by enabling charge-transfer complex formation and increasing charge complementarity between the intercalator and the neighboring bases. Both of these hypotheses were previously proposed for active nitrated indenoisoquinolines bearing similar substitution patterns.25,40
Indenoisoquinolines 17a–h, derived from aldopentose and aldohexose sugars, were also assayed. Although these indenoisoquinolines demonstrated a fairly small range of MGM GI50 values (3 μM to 20 μM), Top1 inhibitory activity varied significantly. The compounds derived from D-arabinose (17a), D-glucose (17c), D-xylose (17d) and D-lyxose (17e) demonstrate the most potent Top1 inhibitory activity (++/+++), indicating that carbohydrate substituents can be successfully combined with the indenoisoquinoline system. Interestingly, arabityl substitution produced indolocarbazoles with significant DNA-targeting activity as well.55 The remaining compounds [those derived from ribose (17b), mannose (17f), and galactose (17g)] possess only weak inhibitory activity (0 or +), demonstrating a strong stereochemical dependence.
Nonetheless, it is a formidable task to disentangle the full effects that sugar-substitution may have on bioactivity. Unlike in previous studies performed with indolocarbazoles,30 the presence of additional hydrogen bond donors does not linearly increase activity (compare compounds 17e and 17f), indicating that the orientation of the hydrogen-bond donors is the major determinant.
Due to the limitation of predictive reliability of the molecular models and the possibility of multiple binding modes, an analogous study with hypothetical models of carbohydrate-derived indenoisoquinolines was not performed. It is likely that these structures make more polar contacts than compounds 12a and 12b, and it is well established that increasing the degree of ligand flexibility (such as in these carbohydrate side chains) further erodes the reliability of docking.56 It is likely that the differences in Top1 inhibitory activity are due to a complex relationship between orientation (e.g. possible interaction between adjacent hydroxyls directed by stereochemistry, as proposed for 12a and 12b), intramolecular hydrogen bonding, and side-chain flexibility. Perhaps the relative stereochemistry of the hydroxyl groups may serve to create an “active” or more sterically favored conformation in some cases.
Although it is difficult to definitively explain the stereochemical determinants of the SAR, it is worth noting that similar trends have been observed for other Top1 inhibitors. In a study performed by Facompre et al.,57 it was discovered that the stereochemistry of the glycosidic bond (i.e., α vs β) between the carbohydrate and the indole moieties of indolocarbazoles has an effect on biological activity,57–59 and the stereochemistry of the sugar itself may be responsible for DNA-sequence recognition and binding.60 Differences in Top1 poisoning activity (but interestingly, not cytotoxicity) between indolocarbazoles substituted with different cyclic sugars and disaccharides have also been observed28,57–59,61 and it has been proposed that there are no “universal” Top1 SAR trends for carbohydrate substituents.57 Stereochemical effects have also been observed in topoisomerase-poisoning saccharide analogues of anthracyclines.62 Even camptothecin (1) exhibits a dramatic relationship between stereochemistry and biological activity: natural camptothecin exists with the 20-(S)-configuration, and inversion of the stereochemistry at this position attenuates the Top1 inhibitory activity greatly.63,64
Conclusions
Several series of alcohol, diol, and carbohydrate-substituted indenoisoquinolines were prepared to evaluate the hypotheses that (1) carbohydrate moieties could be successfully “translated” from the indolocarbazole system to the indenoisoquinoline system, and (2) that changing the stereochemistry of these and shorter chiral substituents could in turn cause disparate biological effects from which valuable SAR data could be gleaned. Promisingly, indenoisoquinolines derived from aldopentoses and aldohexose demonstrated notable activity across a panel of cancer cell lines. Several of these compounds also displayed potent Top1 inhibitory activity when compared to camptothecin. A stereochemical effect was observed, but a convincing explanation of it cannot be offered at the present time.
Similar effects were observed for the indenoisoquinolines substituted with three-carbon alcohols and diols. The stereochemistry of the 2′-hydroxyl group is important for Top1 inhibitory activity, but only in the presence of an adjacent primary alcohol.
The activity of the indenoisoquinolines can be increased by ring substitution and by replacement of a primary alcohol with an amino group. Together, these biological trends aid in the development of a comprehensive indenoisoquinoline SAR and could provide guidance for further optimization. Improvement of our two indenoisoquinolines 6 and 7 that are presently in phase 1 clinical trials is a significant objective.
Experimental Section
General
Solvents and reagents were purchased from commercial vendors and were used without any further purification. Melting points were determined using capillary tubes with a Mel-Temp apparatus and are uncorrected. Infrared spectra were obtained using KBr pellets or on salt plates using CHCl3 as the solvent. IR spectra were recorded using a Perkin-Elmer 1600 series or Spectrum One FTIR spectrometer. 1H NMR spectra were recorded at 300 MHz using a Bruker ARX300 spectrometer with a QNP probe. Mass spectral analyses were performed at the Purdue University Campus-Wide Mass Spectrometry Center. ESIMS was performed using a FinniganMAT LCQ Classic mass spectrometer system. EI/CIMS was performed using a Hewlett-Packard Engine or GCQ FinniganMAT mass spectrometer system. Combustion microanalyses were performed at the Purdue University Microanalysis Laboratory using a Perkin-Elmer Series II CHNS/O model 2400 analyzer. All reported values are within 0.4% of the calculated values; purity of biologically important compounds is ≥95%. Analytical thin-layer chromatography was carried out on Baker-flex silica gel IB2-F plates and compounds were visualized with short wavelength UV light and KMnO4 staining. Silica gel flash chromatography was performed using 230–400 mesh silica gel and ion-exchange chromatography was performed using Dowex 50W-X8-100 resin activated with H2O. Lactones 16, 18, and 28 were prepared according to literature procedures.25,35,40
General Procedure for Preparation of Sugar Oximes (14a–h)36,37
Hydroxylamine hydrochloride (0.976–10.8 g, 14.0–155 mmol) was diluted with absolute EtOH (6–65 mL) or an equivalent amount of dry MeOH. Two drops of phenolphthalein (1% solution in EtOH) were added. A solution of sodium methoxide (10.0–133 mmol) or an equivalent amount of sodium ethoxide (in the case of 13g) in EtOH or MeOH (6–90 mL) was added slowly to the suspension, upon which a white precipitate formed. Addition of base was halted when the mixture stayed pink for approximately one minute (or the pH was titrated back to this point by addition of minimal hydroxylamine hydrochloride). The mixture was stirred for several minutes and filtered to remove salts. The filtrate was warmed to 70 °C, and an aldopentose or aldohexose (sugars 13a–h, 1.0–12.0 g, 5.56–66.0 mmol) was added in small portions. The mixture was stirred at 70 °C until the sugar had completely dissolved (10 min-4 h; typical time, 20 min) and cooled to room temperature. The oximes either precipitated from solution (compounds 14a, 14b, and 14f–h) or were isolated as semi-solids or syrups after concentration and drying in vacuo (compounds 14c–e).
D-Arabinose Oxime (14a)38
By the general procedure, hydroxylamine hydrochloride (5.90 g, 84.9 mmol), NaOMe (4.54 g, 84.0 mmol) and D-arabinose (13a, 5.45 g, 36.3 mmol) afforded the title compound [a mixture of 70% (E)-oxime and 30% (Z)-oxime] as a white microcrystalline solid (4.54 g, 76%): mp 128–136 °C (lit38 mp 135–136 °C). 1H NMR (D2O) δ 7.55 (dd, J = 6.1, 1.8 Hz, 1 H), 4.47 (dd, J = 5.9, 2.7 Hz, 1 H), 3.80–3.60 (m, 5 H). Some of the hydroxyl groups are not visible due to exchange with residual water. Small resonances at 6.91 (dd, J = 5.6, 1.8 Hz, 1 H) and 5.07 (dd, J = 5.6, 2.0 Hz, 1 H) ppm [corresponding to the (Z)-oxime] are visible.
D-Ribose Oxime (14b)38
By the general procedure, hydroxylamine hydrochloride (5.90 g, 84.9 mmol), NaOMe (~4.75 g, 88.0 mmol) and D-ribose (13b, 5.45 g, 36.3 mmol) afforded the title compound [>90% (Z)-oxime] as a white microcrystalline solid (4.13 g, 69%) after filtration; additional product crystallized from the mother liquor upon standing: mp 137–140 °C (lit38 mp 138–139 °C). 1H NMR (D2O) δ 6.86 (d, J = 6.3 Hz, 1 H), 5.05 (dd, J = 6.2, 3.7 Hz, 1 H), 3.80-3.54 (m, 5 H). Some of the hydroxyl groups are not visible due to exchange with residual water. Very small resonances at 7.50 (d, J = 6.9 Hz, 1 H) and 4.40 (m, 1 H) ppm [corresponding to the (E)-oxime] are visible.
D-Glucose Oxime (14c)
By the general procedure, hydroxylamine hydrochloride (10.8 g, 155 mmol), NaOMe (7.2 g, 133 mmol) and D-glucose (13c, 12.0 g, 66.6 mmol) afforded the crude product (13.0 g, 100% with some residual solvent) as a colorless semisolid. Resonances corresponding to the carbonyl protons of (E)- and (Z)-oximes were visible (along with some unreacted aldehyde) and the crude product was used as such without further purification.
D-Xylose Oxime (14d)
By the general procedure, hydroxylamine (2.82 g, 40.6 mmol), NaOMe (2.19 g, 40.6 mmol) and D-xylose (13d, 2.50 g, 16.6 mmol) afforded the title compound [80% (E)-oxime] as a colorless, clear syrup (2.93 g, 100% with residual solvent) after drying in vacuo. 1H NMR (D2O) δ 7.50 (d, J = 6.8 Hz, 1 H), 4.35 (d, J = 5.7 Hz, 1 H), 3.75-3.56 (m, 5 H). Some of the hydroxyl groups are not visible due to exchange with residual water. Resonances at 6.86 (d, J = 5.6 Hz, 1 H) and 4.95 (m, 1 H) ppm [corresponding to the (Z)-oxime] are visible; ESIMS m/z (rel. intensity) 188 (MNa+, 100).
D-Lyxose Oxime (14e)38
By the general procedure, hydroxylamine hydrochloride (1.58 g, 22.8 mmol), NaOMe (1.23 g, 22.8 mmol) and D-lyxose (13e, 1.40 g, 9.32 mmol) afforded the title compound [85% (E)-oxime] as a pale-yellow semisolid (1.61 g, 100% with residual solvent) after drying in vacuo. 1H NMR (D2O) δ 7.51 (d, J = 7.0 Hz, 1 H), 4.25 (t, J = 7.4 Hz, 1 H), 3.84-3.76 (m, 1 H), 3.70-3.55 (m, 3 H). Some of the hydroxyl groups are not visible due to exchange with residual water. Resonances at 6.90 (d, J = 6.8 Hz, 1 H) and 4.93 (m, 1 H) ppm [corresponding to the (Z)-oxime] are visible.
D-Mannose Oxime (14f)65
By the general procedure, hydroxylamine hydrochloride (2.24 g, 32.2 mmol), NaOMe (1.50 g, 27.8 mmol), and D-mannose (13f, 2.50 g, 14.0 mmol) afforded the title compound [>90% (E)-oxime] as a white solid (2.23 g, 82%): mp 173–176 °C [lit65 mp 183–185 °C (dec)]. 1H NMR (CD3OD) δ 7.44 (d, J = 6.9 Hz, 1 H), 4.23 (dd, J = 8.2, 6.9 Hz, 1 H), 3.86-3.77 (m, 3 H), 3.73-3.61 (m, 2 H). Some of the hydroxyl groups are not visible due to exchange with residual water. A small resonance at ~6.80 ppm [corresponding to the (Z)-oxime] is visible; CIMS m/z (rel. intensity) 196 (MH+, 84), 178 [(MH+ – H2O), 14], 103 [(MH+-3H2O – C3H3), 100].
D-Galactose Oxime (14g)66
By the general procedure, hydroxylamine hydrochloride (0.95 g, 13.7 mmol), NaOEt (0.64 g, 9.45 mmol), and D-galactose (13g, 1.0 g, 5.56 mmol) afforded the title compound [a mixture of 50% (E)- and 50% (Z)-oxime] as a white solid (1.01 g, 94 %): mp 172–174 °C (lit66 mp 176 °C). 1H NMR (CD3OD) (E)-oxime: δ 7.50 (d, J = 6.6, 1 H), 5.17 (dd, J = 5.5, 1.7 Hz, 1 H), 3.88-3.83 (m, 1 H), 3.72-3.60 (m, 5 H); (Z)-oxime: 6.80 (d, J = 5.5 Hz, 1 H), 4.48 (dd, J = 6.6, 1.5 Hz, 1 H), 3.88-3.83 (m, 1 H), 3.72-3.60 (m, 5 H). Some of the hydroxyl groups are not visible due to exchange with residual water; ESIMS m/z (rel. intensity) 218 (MNa+, 51), 195 (MH+, 100).
D-Allose Oxime (14h)
By the general procedure, hydroxylamine hydrochloride (0.973 g, 14.0 mmol), NaOMe (0.541 g, 10.0 mmol) and D-allose (13h, 0.900 g, 5.00 mmol) afforded the title compound [>95% (Z)-oxime] as a white solid (0.668 g, 68%): mp 140–142 °C. 1H NMR (CD3OD) δ 6.77 (d, J = 6.2 Hz, 1 H), 5.08 (dd, J = 6.2, 3.6 Hz, 1 H), 3.88 (dd, J = 7.6, 3.6 Hz, 1 H), 3.82-3.71 (m, 2 H), 3.66-3.60 (m, 2 H). The hydroxyl groups are not visible due to exchange with residual water. Very small resonances at ~7.40 and ~4.40 ppm [corresponding to the (E)-oxime] are visible; ESIMS m/z (rel. intensity) 412 (2MNa+, 100), 218 (MNa+, 65), 195 (MH+, 4).
General Procedure for Oxime Reduction (To Yield Amines 15a–h)36
The sugar oxime (0.6–10 g, 3.06–51.2 mmol) was diluted with glacial AcOH (10–65 mL), and Pt(IV)O2 (0.06–0.600 g, generally around 1% w/w was sufficient) was added. The mixture was hydrogenated with shaking on a Parr apparatus at 35–45 psi, for between 18 h and 3.5 days, or until the mixture was clear. The mixture was then filtered to remove the catalyst, concentrated, dissolved in H2O (80–250 mL) and loaded onto an ion-exchange column packed in H2O. The column was washed with H2O (80–200 mL) after loading, and then the desired aminodeoxyalditol was eluted using NH4OH (3 N, between 80–250 mL). The alkaline fraction was concentrated to yield the crude aminodeoxyalditols (15a–h), which were used without further purification to synthesize indenoisoquinolines.
D-Arabitylamine (15a)
From 14a (3.00 g, 18.2 mmol), the general procedure afforded the desired product as a dark yellow syrup (2.77 g, 100% with some residual water). 1H NMR (CD3OD) δ 3.80-3.50 (m, 6 H), 3.90-2.72 (m, 2 H). Some of the hydroxyl groups and the primary amine are not visible due to exchange with residual water.
D-Ribitylamine (15b)
From 14b (3.00 g, 18.2 mmol), the general procedure afforded the desired product as a light brown syrup (2.78 g, 100% with some residual water). 1H NMR (CD3OD) δ 3.77-3.52 (m, 5 H), 2.90-2.73 (m, 2 H). Hydroxyl groups and the primary amine are not visible due to exchange with residual water.
D-Glucamine (15c)67
From crude 14c (10.0 g, 51.2 mmol), the general procedure afforded the desired product as a dark brown oil (6.37 g, 70%). 1H NMR resonances were consistent with crude D-glucamine and this material was used without further purification to prepare 17c and 19.
D-Xylitylamine (15d)
From 14d (2.93 g, 17.7 mmol), the general procedure afforded the desired product as a dark yellow syrup (2.16 g, 81%). 1H NMR (D2O) δ 3.75-3.50 (m, 5 H), 2.76-2.61 (m, 2 H). Hydroxyl groups and the primary amine are not visible due to exchange with the solvent; ESIMS m/z (rel. intensity) 152 (MH+, 100).
D-Lyxitylamine (15e)
From 14e (1.61 g, 9.70 mmol), the general procedure afforded the desired product as a brown syrup (1.27 g, 87%). 1H NMR (CD3OD) δ 3.87 (t, J = 5.7 Hz, 1 H), 3.65-3.59 (m, 3 H), 3.44 (d, J = 7.0 Hz, 1 H), 2.97 (dd, J = 13.2, 3.5 Hz, 1 H), 2.91 (dd, J = 13.1, 7.1 Hz, 1 H). Hydroxyl groups and the primary amine are not visible due to exchange with residual water; ESIMS m/z (rel. intensity) 286 [(2MH+ – NH3)+, 100], 152 (MH+, 18).
D-Mannitylamine (15f)67
From 14f (2.00 g, 12.0 mmol), the general procedure afforded the desired product as a dark-brown oil (1.35 g, 97%). 1H NMR (CD3OD) δ 3.86-3.59 (m, 6 H), 3.29-3.22 (m 1 H), 2.99 (dd, J = 12.2, 8.0 Hz, 1 H). Hydroxyl groups and the primary amine are not visible due to exchange with residual water; ESIMS m/z (rel. intensity) 182 (MH+, 86).
D-Galactitylamine (15g)
From 14g (1.01 g, 5.17 mmol), the general procedure afforded the desired product as a dark-brown oil (0.95 g, 100% with some residual water). 1H NMR (CD3OD) δ 4.11-4.00 (m, 1 H), 3.80 (dt, J = 1.3, 6.4 Hz, 1 H) 3.64-3.51 (m, 5 H), 3.10-2.97 (m, 2 H). Some hydroxyl groups and the primary amine are not visible due to exchange with residual water; CIMS m/z (rel. intensity) 182 (MH+, 100).
D-Allitylamine (15h)
From 14h (0.600 g, 3.01 mmol), the general procedure afforded the desired product as a dark-brown oil (0.475 g, 86%). 1H NMR (CD3OD) δ 4.10-3.98 (m, 1 H), 3.86-3.53 (m, 5 H), 3.30-2.90 (m, 2 H). Hydroxyl groups and the primary amine are not visible due to exchange with residual water; ESIMS m/z (rel. intensity) 346 [(2MH+ – NH3)+, 100], 182 (MH+, 2).
General Procedure for Preparation of Indenoisoquinolines 12a–c, 17a–h, 19, 22a–c, 25, 27a–d, 29, and 30
A solution of the appropriate lactone, 16 (0.040–0.254 g, 0.14–1.02 mmol), 18 (0.040–0.215 g, 0.136–0.733 mmol), or 28 (0.100 g, 0.324 mmol) and the primary amine (0.05–0.36 g, 0.33–2.20 mmol, 2.0–3.0 equiv.) in either MeOH (15–125 mL) or CHCl3 (20–70 mL) was heated to reflux. After 2–36 h (typical time: 18 h), the reaction mixture was cooled to room temperature. In cases where precipitation resulted, the precipitate was collected by filtration and washed with CHCl3 (50–300 mL). In cases where the product was soluble in the reaction solvent, the reaction mixture was concentrated and the residue was dissolved in CHCl3 (20–70 mL). The solution was washed with H2O (3 × 10–30 mL), sat. NaCl (in some cases), dried over anhydrous sodium sulfate, and concentrated to afford the desired compounds after chromatography (as described for individual compounds).
(2′S)-5,6-Dihydro-6-[(2′,3′-dihydroxypropyl)]-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (12a)
From lactone 16 (0.150 g, 0.604 mmol) and alcohol 20a (0.110 g, 1.21 mmol) in MeOH (50 mL), the general procedure afforded the title compound as a yellow-orange solid (0.159 g, 82%) after purification by flash column chromatography (SiO2, eluting with a gradient of CHCl3 to 5% MeOH in CHCl3): mp 200–205 °C. IR (KBr) 3400, 1705, 1655, 1611, 1503 cm−1; 1H NMR (DMSO-d6) δ 8.61 (d, J = 8.1 Hz, 1 H), 8.23 (d, J = 8.1 Hz, 1 H), 8.07 (d, J = 7.4 Hz, 1 H), 7.85 (m, 1 H), 7.56-7.46 (m, 4 H), 5.14 (d, J = 5.1 Hz, 1 H), 4.99 (t, J = 5.6 Hz, 1 H), 4.60-4.50 (m, 2 H), 4.00-3.90 (m, 1 H), 3.60 (t, J = 5.5 Hz, 2 H); ESIMS m/z (rel intensity) 322 (MH+, 73), 304 [(MH+ – H2O)+, 100]. Anal. Calcd for C19H15NO4: C, 71.02; H, 4.71; N, 4.36. Found: C, 70.73; H, 4.60; N, 4.31.
(2′R)-5,6-Dihydro-6-[(2′,3′-dihydroxypropyl)]-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (12b)
From lactone 16 (0.100 g, 0.403 mmol) and alcohol 20b (0.073 g, 0.806 mmol) in MeOH (50 mL), the general procedure afforded the title compound as a yellow-orange solid (0.096 g, 74%) after washing with EtOAc-hexanes: mp 200–205 °C. IR (KBr) 3349, 1704, 1641, 1611, 1503, 1425, 1316, 1045, 879, 757 cm−1; 1H NMR (DMSO-d6) δ 8.61 (d, J = 7.8 Hz, 1 H), 8.22 (d, J = 8.1 Hz, 1 H), 8.07 (d, J = 7.6 Hz, 1 H), 7.84 (m, 1 H), 7.56-7.43 (m, 4 H), 5.14 (d, J = 5.1 Hz, 1 H), 4.99 (t, J = 5.1 Hz, 1 H), 4.60-4.47 (m, 2 H), 4.00-3.90 (m, 1 H), 3.60 (t, J = 5.3 Hz, 2 H); ESIMS m/z (rel intensity) 321 (MH+, 73), 304 [(MH+ – H2O)+, 100]. Anal. Calcd for C19H15NO4: C, 71.02; H, 4.71; N, 4.36. Found: C, 70.86; H, 4.66; N, 4.34.
(2′RS)-5,6-Dihydro-6-[(2′,3′-dihydroxypropyl)]-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (12c)
From lactone 16 (0.140 g, 0.604 mmol) and alcohol 20c (0.110 g, 1.21 mmol) in MeOH (50 mL), the general procedure afforded the title compound as a yellow-orange solid (0.150 g, 77%) after purification by flash column chromatography (SiO2, eluting with a gradient of CHCl3 to 5% MeOH in CHCl3): mp 209–212 °C. IR (KBr) 3400, 1705, 1655, 1611, 1503 cm−1; 1H NMR (DMSO-d6) δ 8.61 (d, J = 8.1 Hz, 1 H), 8.23 (m, 1 H), 8.07 (d, J = 7.4 Hz, 1 H), 7.85 (m, 1 H), 7.56-7.46 (m, 4 H), 5.14 (d, J = 5.1 Hz, 1 H), 4.99 (d, J = 5.6 Hz, 1 H), 4.57 (m, 2 H), 4.00 (m, 1 H), 3.60 (t, J = 5.5 Hz, 2 H); ESIMS m/z (rel intensity) 322 (MH+, 48), 304 [(MH+ – H2O)+, 100]. Anal. Calcd for C19H15NO4·0.2 H2O: C, 70.23; H, 4.78; N, 4.31. Found: C, 69.85; H, 4.38; N, 4.66.
(2′R, 3′S, 4′R)-5,6-Dihydro-6-(2′, 3′, 4′, 5′-tetrahydroxypentyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (17a)
From lactone 16 (0.061 g, 0.241 mmol) and amine 15a (0.050 g, 0.331 mmol) in MeOH (20 mL), the general procedure afforded the title compound as a red solid (0.067 g, 72%) that was used without further purification: mp 231–233 °C. IR (KBr) 3281, 2925, 1705, 1658, 1501, 1457, 1378 cm−1; 1H NMR (DMSO-d6) δ 8.61 (d, J = 8.1 Hz, 1 H), 8.20 (t, J = 7.4 Hz, 1 H), 7.81 (t, J = 7.4 Hz, 1 H), 7.54-7.44 (m, 4 H), 4.92 (d, J = 7.5 Hz, 1 H), 4.84 (d, J = 5.9 Hz, 1 H), 4.62-4.41 (m, 4 H), 4.32 (m, 1 H), 3.66 (t, J = 5.8 Hz, 1 H), 3.54-3.34 (m, 4 H); ESIMS m/z (rel intensity) 381 [(M – H+)−, 100]. Anal. Calcd for C21H19NO6·0.5 H2O: C, 64.61; H, 5.16; N, 3.39. Found: C, 64.27; H, 5.10; N, 3.39.
(2′S,3′S,4′R)-5,6-Dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (17b)
From lactone 16 (0.254 g, 1.02 mmol) and amine 15b (0.316 g, 2.05) in MeOH (125 mL), the general procedure afforded the title compound as an orange solid (0.295 g, 75%) after flash column chromatography (SiO2, eluting with a gradient of CHCl3 to 20% MeOH in CHCl3): mp 218–221 °C. IR (KBr) 3476, 3380, 1691, 1656, 1504, 1069, and 761 cm−1; 1H NMR (DMSO-d6) δ 8.60 (d, J = 7.8 Hz, 1 H), 8.21-8.18 (m, 2 H), 7.83 (m, 1 H), 7.54-7.42 (m, 4 H), 5.19 (d, J = 5.0 Hz, 1 H), 4.98 (d, J = 5.5 Hz, 1 H), 4.77 (d, J = 4.5 Hz, 1 H), 4.68-4.62 (m, 3 H), 4.49 (t, J = 5.4 Hz, 1 H), 4.22-4.10 (m, 1 H), 3.67-3.61 (m, 3 H), 3.48-3.45 (m, 1 H); ESIMS m/z (rel intensity) 382 (MH+, 89), 364 [(MH+ – H2O)+, 100]. Anal. Calcd for C21H19NO6·0.39 H2O: C, 64.94; H, 5.13; N, 3.61. Found: C, 64.56; H, 4.73; N, 4.00.
(2′S,3′R,4′R,5′R)- 5,6-Dihydro-6-(2′, 3′, 4′, 5′, 6′-pentahydroxyhexyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (17c)
From lactone 16 (0.200 g, 0.806 mmol) and amine 15c (0.363 g, 2.0 mmol) in MeOH (60 mL), the general procedure afforded the title compound as an orange solid (0.067 g, 20%) after flash column chromatography (SiO2, eluting with a gradient of CHCl3 to 20% MeOH in CHCl3): mp 240–244 °C. IR (KBr) 3430, 2085, 1627, 1549, 1504, 1424, 1316, 1269, 1179 cm−1. 1H NMR (DMSO-d6) δ 8.61 (d, J = 8.1 Hz, 1 H), 8.22 (t, J = 8.3 Hz, 2 H), 7.81 (t, J = 7.0 Hz, 1 H), 7.54-7.44 (m, 4 H), 5.13 (d, J = 4.9 Hz, 1 H), 4.85 (d, J = 6.4 Hz, 1 H), 4.70-4.60 (m, 1 H), 4.60-4.54 (m, 3 H), 4.43 (t, J = 5.4 Hz, 1 H), 4.20-4.10 (br m, 1 H), 3.90-3.80 (br m, 1 H), 3.64-3.40 (m, 4 H); negative ion ESIMS m/z (rel. intensity) 821 [2(M – H+)−, + H+, 100], 410 [(M – H+)−, 92]. Anal. Calcd for C22H21NO7·1 H2O: C, 61.53; H, 5.40; N, 3.26. Found: C, 61.78; H, 5.42; N, 3.47.
(2′S,3′R,4′R)-5,6-Dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (17d)
From lactone 16 (0.100 g, 0.403 mmol) and amine 15d (0.251 g, 1.66 mmol) in MeOH (60 mL), the general procedure afforded the title compound as a red solid (0.028 g, 18%) that was used without further purification: mp 220–224 °C. IR (film) 3374, 1701, 1625, 1549, 1506, 1032, 884, 757 cm−1; 1H NMR (DMSO-d6) δ 8.63 (d, J = 7.8 Hz, 1 H), 8.25-8.22 (m, 2 H), 7.85 (t, J = 6.9 Hz, 1 H), 7.57-7.44 (m, 4 H), 5.05 (d, J = 5.1 Hz, 1 H), 4.87 (d, J = 6.2 Hz, 1 H), 4.79-4.53 (m, 4 H), 4.06–4.16 (br m, 1 H), 3.40–3.56 (m, 5 H); EIMS m/z (rel. intensity) 247 [(M – C5H10O4)+, 100], 381 (M+, 5). Anal. Calcd for C21H19NO6·1.0 H2O: C, 63.15; H, 5.30; N, 3.51. Found: C, 63.01; H, 5.03; N, 3.54.
(2′R,3′R,4′R)-5,6-Dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (17e)
From lactone 16 (0.112 g, 0.450 mmol) and amine 15e (0.136 g, 0.900 mmol) in MeOH (30 mL), the general procedure afforded the title compound as an orange solid (0.024 g, 5%) after flash column chromatography (SiO2, eluting with a gradient of CHCl3 to 20% MeOH in CHCl3): mp 183–185 °C. IR (KBr) 3351, 2924, 1756, 1738, 1704, 1655, 1609, 1548, 1504, 1429, 1316, 1267, 1197 cm−1; 1H NMR (DMSO-d6) δ 8.61 (d, J = 8.1 Hz, 1 H), 8.22 (d, J = 8.1 Hz, 1 H), 8.08 (d, J = 7.6 Hz, 1 H), 7.83 (t, J = 7.6, 1 H), 7.58-7.43 (m, 4 H), 5.08 (d, J = 5.7 Hz, 1 H), 4.82 (d, J = 6.9 Hz, 1 H), 4.72-4.53 (m, 3 H), 4.36 (d, J = 6.6 Hz, 1 H), 4.10-4.09 (br m, 1 H), 3.70-3.42 (m, 4 H); CIMS m/z (rel. intensity) 382 (MH+, 50), 364 [(MH+ – H2O)+, 100]. Anal. Calcd for C21H19NO6·1.1 H2O: C, 62.95; H, 5.32; N, 3.50. Found: C, 62.56; H, 4.91; N, 3.32.
(2′R, 3′R, 4′R, 5′R)- 5,6-Dihydro-6-(2′, 3′, 4′, 5′, 6′-pentahydroxyhexyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (17f)
From lactone 16 (0.137 g, 0.550 mmol) and amine 15f (0.250 g, 1.38 mmol) in MeOH (45 mL), the general procedure afforded the desired product as an orange solid (0.097 g, 43%) after washing with CHCl3 (~500 mL): mp 244–246 °C. IR (KBr) 3450, 3302, 3235, 3071, 2969, 1713, 1654, 1634, 1610, 1549, 1504, 1414, 1317, 1200, 1075, 1012 cm−1; 1H NMR (DMSO-d6) δ 8.61 (d, J = 8.0 Hz, 1 H), 8.22 (d, J = 7.8 Hz, 1 H), 8.05 (d, J = 7.5 Hz, 1 H), 7.83 (t, J = 7.6, 1 H) 7.56-7.43 (m, 4 H), 5.02 (d, J = 5.7 Hz, 1 H), 4.80 (d, J = 6.9 Hz, 1 H), 4.75-4.45 (m, 2 H), 4.51 (d, J = 5.0 Hz, 1 H), 4.41 (t, J = 5.6 Hz, 1 H), 4.24 (d, J = 7.5 Hz, 1 H), 4.10-4.00 (br m, 1 H), 3.83 (t, J = 7.6 Hz, 1 H), 3.64-3.39 (m, 4 H); CIMS m/z (rel. intensity) 248 [(MH+ – C6H12O5), 100], 412 (MH+, 15). Anal. Calcd for C22H21NO7·0.8 H2O: C, 62.05; H, 5.35; N, 3.29. Found: C, 61.73; H, 5.08; N, 3.19.
(2′S,3′R,4′S,5′R)-5,6-Dihydro-6-(2′,3′,4′,5′-tetrahydroxypentyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (17g)
From lactone 16 (0.319 g, 1.29 mmol) and amine 15g (0.797 g, 4.40 mmol), in MeOH (90 mL), the general procedure afforded the desired product as an orange solid (0.041 g, 8%): mp 261–265 °C. IR (KBr) 3419, 2951, 1971, 1697, 1610, 1573, 1547, 1501, 1458, 1421, 1320, 1268 cm−1; 1H NMR (DMSO-d6) δ 8.62 (d, J = 8.1 Hz, 1 H), 8.22 (d, J = 8.1 Hz, 2 H), 7.85 (t, J = 7.2 Hz, 1 H), 7.44–7.58 (m, 4 H), 4.88 (d, J = 3.9 Hz, 1 H), 4.78 (d, J = 6.1 Hz, 1 H), 4.66-4.62 (m, 1 H), 4.53-4.46 (m, 2 H), 4.37-4.33 (m, 1 H), 4.26 (d, J = 6.5 Hz, 1 H), 4.15-4.13 (m, 1 H), 3.81 (q, J = 6.2 Hz, 1 H), 3.58-3.42 (m, 4 H); EIMS m/z (rel. intensity) 247 [(M – C6H12O5)+, 100], 411 (M+, 2). Anal. Calcd for C22H21NO7: C, 64.23; H, 5.14; N, 3.40. Found: C, 63.91; H, 5.09; N, 3.43.
(2′S,3′S,4′R,5′R)-5,6-Dihydro-6-(2′,3′,4′,5′,6′-pentahydroxyhexyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (17h)
From lactone 16 (0.110 g, 0.440 mmol) and amine 15h (0.200 g, 1.10 mmol) in MeOH (30 mL), the general procedure afforded the title compound as an orange solid (0.045 g, 25%) after washing with H2O (10 mL) and CHCl3 (~200 mL): mp 201–205 °C. IR (KBr) 3373, 1703, 1639, 1019, 1548, 1504, 1425, 1316, 1263, 1062, 758 cm−1; 1H NMR (DMSO-d6) δ 8.61 (d, J = 8.1 Hz, 1 H), 8.27 (d, J = 7.3 Hz, 1 H), 8.22 (d, J = 8.0 Hz, 1 H), 7.84 (t, J = 8.2 Hz, 1 H) 7.55-7.42 (m, 4 H), 5.27 (d, J = 5.1 Hz, 1 H), 4.97 (d, J = 5.3 Hz, 1 H), 4.84 (d, J = 4.4 Hz, 1 H), 4.67 (d, J = 5.8 Hz, 3 H), 4.47 (t, J = 5.6 Hz, 1 H), 4.27-4.20 (m, 1 H), 3.85-3.80 (m, 1 H), 3.67-3.42 (m, 4 H); negative ion ESIMS m/z (rel. intensity) 410 [(M – H+)−, 36]. Anal. Calcd for C22H21NO7·1 H2O: C, 61.53; H, 5.40; N, 3.26. Found: C, 61.30; H, 5.25; N, 3.46.
(2′S,3′R,4′R,5′R)-5,6-Dihydro-6-(2′,3′,4′,5′,6′-pentahydroxyhexyl)-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (19)
From lactone 18 (0.040 g, 0.136 mmol) and amine 15c (0.062 g, 0.341 mmol) in MeOH (15 mL), the general procedure afforded the title compound as an orange solid (0.021 g, 34%) after concentrating, re-suspending in CHCl3 (20 mL), filtering, and washing with CHCl3 (~30 mL) and ether (~30 mL): mp 265–267 °C. IR (KBr) 3401, 2934, 1713, 1649, 1615, 1559, 1505, 1426, 1334, 1076 cm−1; 1H NMR (DMSO-d6) δ 8.92 (d, J = 2.4 Hz, 1 H), 8.78 (d, J = 8.8 Hz, 1 H), 8.61-8.50 (m, 2 H), 7.64-7.56 (m, 4 H), 5.20 (d, J = 5.0 Hz, 1 H), 4.94 (d, J = 6.5 Hz, 1 H), 4.90-4.80 (m, 1 H), 4.62-4.58 (m, 3 H), 4.45 (t, J = 5.0 Hz, 1 H), 4.20-4.10 (m, 1 H), 3.90-3.80 (m, 1 H), 3.65-3.34 (m, 4 H); negative ion ESIMS m/z (rel. intensity) 455 [(M – H+)−, 24], 291 [(M – H+) – C6H12O5,]−, 100. Anal. Calcd for C22H21NO7: C, 57.90 H, 4.42; N, 6.14. Found: C, 57.66; H, 4.66; N, 6.48.
(1′S)-5,6-Dihydro-6-(1′-hydroxy-2′-methylethyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (22a)
From lactone 16 (0.100 g, 0.403 mmol) and alcohol 21a (0.108 g, 1.21 mmol) in CHCl3 (30 mL), the general procedure afforded the desired product as a dark red solid (0.086 g, 71%) after extraction and washing with ether: mp 215–217 °C. IR (KBr) 3061, 2980, 2937, 2890, 1658, 1575, 1457, 1418, 1262, 1171, 1017 cm−1; 1H NMR (CDCl3) δ 8.67 (d, J = 8.1 Hz, 1 H), 8.27 (d, J = 8.1 Hz, 1 H), 7.75 (t, J = 7.0 Hz, 1 H), 7.65–7.58 (m, 2 H), 7.49-7.37 (m, 3 H), 5.14-5.08 (m, 1 H), 4.42-4.36 (m, 1 H), 4.18-4.12 (m, 1 H), 3.68 (dd, J = 8.1, 3.7 Hz, 1 H), 1.77 (d, J = 7.0, 3 H); negative ion ESIMS m/z (rel. intensity) 288 [(MH+ – H2O, 100], 306 (MH+, 12). Anal. Calcd for C19H15NO3: C, 74.74; H, 4.95; N, 4.59. Found: C, 74.45; H, 4.78; N, 4.57.
(1′R)-5,6-Dihydro-6-(3′-hydroxy-1′-methylethyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (22b)
From lactone 16 (0.100 g, 0.403 mmol) and alcohol 21b (0.108 g, 1.21 mmol) in CHCl3 (30 mL), the general procedure afforded the desired product as a dark red solid (0.074 g, 60%) after extraction and washing with ether: mp 216–217 °C. IR (KBr) 3390, 2978, 2941, 2891, 1698, 1659, 1610, 1548, 1500, 1421, 1374, 1047 cm−1; 1H NMR (CDCl3) δ 8.67 (d, J = 8.1 Hz, 1 H), 8.26 (d, J = 8.0 Hz, 1 H), 7.71 (td, J = 7.0 Hz, 1.3 Hz, 1 H), 7.65-7.59 (m, 2 H), 7.48-7.39 (m, 3 H), 5.13-5.08 (m, 1 H), 4.46-4.37 (m, 1 H), 4.18-4.11 (m, 1 H) 3.71 (dd, J = 8.0, 3.9 Hz, 1 H), 1.77 (d, J = 7.0 Hz, 3 H); ESIMS m/z (rel. intensity) 288 [(MH+ – H2O)+, 100], 306 (MH+, 84). Anal. Calcd for C19H15NO3·0.8 H2O·0.05 CHCl3: C, 70.25; H, 5.15; N, 4.30. Found: C, 70.12; H, 4.76; N, 4.01.
(1′RS)-5,6-Dihydro-6-(3′-hydroxy-2′-methylethyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (22c)
From lactone 16 (0.100 g, 0.403 mmol) and alcohol 21c (0.108 g, 1.21 mmol) in CHCl3 (30 mL), the general procedure afforded the desired product as a dark red solid (0.077 g, 63%) after extraction and washing with ether: mp 225–227 °C. IR (KBr) 3399, 2985, 2881, 1659, 1612, 1423 cm−1; 1H NMR (DMSO-d6) δ 8.59 (d, J = 8.1 Hz, 1 H), 8.18 (d, J = 8.1 Hz, 1 H), 7.89 (d, J = 7.8 Hz, 1 H), 7.81 (td, J = 7.7 Hz, 1.3 Hz, 1 H), 7.61-7.47 (m, 4 H), 5.05 (t, J = 5.8 Hz, 2 H), 4.32-4.28 (m, 1 H), 3.82-3.78 (m, 1 H), 1.64 (d, J = 6.8, 3 H); ESIMS m/z (rel. intensity) 288, [(MH+ – H2O)+, 83], 306 (MH+, 5). Anal. Calcd for C19H15NO3: C, 74.74; H, 4.95; N, 4.59. Found: C, 74.39; H, 5.12; N, 4.43.
(2′RS)-5,6-Dihydro-6-[2′-hydroxy-3′-tert-(butyldiphenylsilyloxy)propyl]-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (23)
Compound 12c (0.350 g, 1.09 mmol) was dissolved in CH2Cl2 (30 mL), and TBDPSCl (0.329 g, 1.20 mmol) was added. Triethylamine (0.121 g, 1.20 mmol) and a catalytic amount of 4-DMAP were added. The solution was allowed to stir at room temperature. After 18 h, the solution was diluted with CH2Cl2 (50 mL), washed with H2O (3 × 30 mL), and dried over Na2SO4. The solution was concentrated under vacuum to yield a red oil. The oil was purified by flash column chromatography (SiO2, up to 50:50 EtOAc–hexanes) to yield the desired product as a red solid (0.220 g, 60%): mp 163–165 °C. IR (film) 3684, 3020, 2400, 1659, 1521, 1427, 1216 cm−1; 1H NMR (DMSO-d6) δ 8.61 (d, J = 8.0 Hz, 1 H), 8.23 (d, J = 7.7 Hz, 1 H), 8.07 (d, J = 7.6 Hz, 1 H), 7.85-7.79 (m 1 H), 7.70-7.66 (m 4 H), 7.56-7.29 (m, 10 H), 5.35 (d, J = 5.2 Hz, 1 H), 4.76-4.58 (m, 2 H), 4.16-4.13 (m, 1 H), 3.83 (d, J = 4.7, 2 H), 1.03 (s, 9 H); ESIMS m/z (rel intensity) 1140 [(2MNa)+, 100], 582 [(MNa)+, 34], 560 (MH+, 4). Anal. Calcd for C35H33NO4Si: C, 75.10; H, 5.94; N, 2.50. Found: C, 75.17; H, 5.91; N, 2.51.
5,6-Dihydro-6-[2′-oxo-3′-tert-(butyldiphenylsilyloxy)propyl]-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (24)
Indenoisoquinoline 23 (0.200 g, 0.358 mmol) was dissolved in CH2Cl2 (65 mL). N-Methylmorpholine-N-oxide (0.083 g, 0.715 mmol) and TPAP (5%, 0.006 g, 0.018 mmol) were added to the solution. The reaction mixture was allowed to stir at room temperature for 4.5 h. The mixture was then diluted with CHCl3 (40 mL) and washed with H2O (5 × 40 mL). The organic layers were dried over Na2SO4 and concentrated under vacuum to yield a brownish-red solid. The solid was flushed through SiO2 with 40:60 EtOAc–hexanes, and the filtrate was concentrated under vacuum to yield the desired product as a brownish-red solid (0.161 g, 81%): mp 175–178 °C. IR (film) 3401, 2091, 1665, 1551, 1503, 1428, 1315, 1112 cm−1; 1H NMR (DMSO-d6) δ 8.61 (d, J = 8.0 Hz, 1 H), 8.23 (d, J = 8.0 Hz, 1 H), 8.04 (d, J = 7.6 Hz, 1 H), 7.87 (t, J = 8.0 Hz, 1 H), 7.70-7.68 (m, 4 H), 7.60-7.39 (m, 11 H), 5.64 (s, 2 H), 4.83 (s, 2 H), 1.03 (s, 9 H); ESIMS m/z (rel. intensity) 558 (MH+, 52). Anal. Calcd for C35H33NO4Si·1.3 H2O: C, 72.34; H, 5.83; N, 2.41. Found: C, 72.29; H, 5.63; N, 2.59.
5,6-Dihydro-6-(3′-hydroxy-2′-oxopropyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (25)
Acetyl chloride (0.07 mL) was added dropwise to MeOH (1.8 mL). The resulting methanolic HCl was cooled to 20 °C, and a solution of ketone 24 (0.030 g, 0.054 mmol) in CH2Cl2 (2 mL) was added. The reaction mixture was allowed to stir for 22 h. The solution was concentrated under vacuum and diluted with CHCl3 (20 mL). The organic layer was washed with H2O (4 × 15 mL), dried over Na2SO4, and concentrated under vacuum to yield an orange solid. The solid was purified by flash column chromatography (SiO2), eluting with a gradient of 10% EtOAc in hexanes to EtOAc, to yield the desired product as an orange powder (0.006 g, 34%): mp 211–213 °C. IR (film) 3435, 3020, 2400, 1729, 1708, 1658, 1550, 1504, 1427, 1215 cm−1; 1H NMR (DMSO-d6) δ 8.62 (d, J = 8.3 Hz, 1 H), 8.22 (d, J = 7.9 Hz, 1 H), 7.90 (t, J = 7.9 Hz, 1 H), 7.58-7.48 (m, 5 H), 5.79 (t, J = 6.0 Hz, 1 H), 5.65 (s, 2 H), 4.48 (d, J = 6.1 Hz, 2 H); CIMS m/z (rel intensity) 248 [(MH+ – C3H4O2)+ 100], 320 (MH+, 65). Anal. Calcd for C19H13NO4·0.4 H2O: C, 69.89; H, 4.26; N, 4.29. Found: C, 69.53; H, 4.01; N, 4.20.
(2′R)-5,6-Dihydro-6-(2′-hydroxypropyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (27a)
From lactone 16 (0.100 g, 0.403 mmol) and alcohol 26a (0.091 g, 1.21 mmol) in CHCl3 (30 mL), the general procedure was followed to afford the title compound as a red solid (0.074 g, 61%) after washing with ether: mp 176–180 °C. IR (KBr) 3484, 2969, 2924, 1706, 1610, 1424 cm−1; 1H NMR (DMSO-d6) δ 8.61 (d, J = 7.8 Hz, 1 H), 8.23 (d, J = 8.1 Hz, 1 H), 7.93 (d, J = 7.5 Hz, 1 H), 7.85-7.80 (m, 1 H), 7.59-7.47 (m, 4 H), 5.11 (d, J = 4.8 Hz, 1 H), 4.51-4.38 (m, 2 H), 4.10-4.00 (m, 1 H), 1.27 (d, J = 6.2 Hz, 3 H); CIMS m/z (rel intensity) 306 (MH+, 100). Anal. Calcd for C19H15NO3·0.3 H2O: C, 73.44 H, 5.06; N, 4.51. Found: C, 73.06; H, 5.22; N, 4.38.
(2′S)-5,6-Dihydro-6-(2′-hydroxypropyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (27b)
From lactone 16 (0.100 g, 0.403 mmol) and alcohol 26b (0.091 g, 1.21 mmol) in CHCl3 (30 mL), the general procedure was followed to afford the title compound as a red solid (0.101 g, 82%) after washing with ether: mp 185–188 °C. IR (KBr) 3484, 3068, 2970, 1708, 1648, 1574, 1503, 1423, 1317, 1266, 1196, 1064 cm–1; 1H NMR (CDCl3) 8.56 (d, J = 8.1 Hz, 1 H), 8.26 (d, J = 8.1 Hz, 1 H), 7.66-7.29 (m, 6 H), 4.60-4.52 (m, 2 H), 4.48-4.41 (m, 1 H), 3.12 (d, J = 5.1 Hz, 1 H), 1.49 (d, J = 6.2 Hz, 3 H); negative ion ESIMS m/z (rel intensity) 306 (MH+, 37), 288 [(MH+ – H2O)+, 100]. Anal. Calcd for C19H15NO3: C, 74.74 H, 4.95; N, 4.59. Found: C, 74.53; H, 5.13; N, 4.43.
(2′RS)-5,6-Dihydro-6-(2′-hydroxypropyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (27c)
From lactone 16 (0.100 g, 0.403 mmol) and alcohol 26c (0.091 g, 1.21 mmol) in CHCl3 (30 mL), the general procedure was followed to afford the title compound as an orange solid (0.108 g, 87%) after washing with ether: mp 191–194 °C. IR (KBr) 3474, 2975, 2917, 1706, 1610, 1424 cm−1; 1H NMR (DMSO-d6) δ 8.60 (d, J = 8.0 Hz, 1 H), 8.22 (dd, J = 8.1, 0.71 Hz, 1 H), 7.91 (d, J = 7.4 Hz, 1 H), 7.84-7.78 (m, 1 H), 7.59-7.44 (m, 4 H), 5.11 (d, J = 4.8 Hz, 1 H), 4.53-4.31 (m, 2 H), 4.10-4.00 (m, 1 H), 1.25 (d, J = 6.3 Hz, 3 H); CIMS m/z (rel intensity) 306 (MH+, 100). Anal. Calcd for C19H15NO3: C, 74.74; H, 4.95; N, 4.59. Found: C, 74.75; H, 5.14; N, 4.40.
(2′RS)-5,6-Dihydro-6-(3′-amino-2′-hydroxypropyl)-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (27d)
From lactone 16 (0.100 g, 0.403 mmol) and alcohol 26d (0.109 g, 1.21 mmol) in CHCl3 (30 mL), the general procedure was followed to afford the title compound as an orange solid (0.107 g, 83%) after washing with ether: mp 185–188 °C. IR (KBr) 3359, 1673, 1547, 1505, 1427 cm−1; 1H NMR (DMSO-d6) δ 8.69 (d, J = 8.0 Hz, 1 H), 8.21 (d, J = 8.1 Hz, 1 H), 8.04 (d, J = 7.9 Hz, 1 H), 7.83 (t, J = 7.2 Hz, 1 H), 7.57-7.41 (m, 4 H), 4.60-4.30 (m, 2 H), 3.90-3.80 (m, 1 H), 2.80-2.65 (m, 2 H); the hydroxyl and primary amino group are not visible due to exchange with residual water; ESIMS m/z (rel intensity) 321 (MH+, 100). Anal. Calcd for C19H15N2O3·1.2H2O: C, 66.73; H, 5.42; N, 8.19. Found: C, 67.04; H, 5.30; N, 7.85.
(2′S)-5,6-Dihydro-6-(2′,3′-dihydroxypropyl)-2,3-dimethoxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (29)
From lactone 28 (0.100 g, 0.324 mmol) and alcohol 20a (0.059 g, 0.628 mmol) in MeOH (30 mL), the general procedure was followed to afford the title compound as a red solid (0.051 g, 41%): mp 220–222 °C. IR (KBr) 3402, 2965, 2928, 1697, 1632, 1479, 1429, 1396, 1263 cm−1; 1H NMR (DMSO-d6) δ 8.01 (s, 1 H), 7.99 (d, J = 7.8 Hz, 1 H), 7.54-7.39 (m, 4 H), 5.11 (d, J = 5.1 Hz, 1 H), 4.97-4.95 (m, 1 H), 4.57-4.51 (m, 2 H), 4.94-4.90 (m, 1 H), 3.96 (s, 3 H), 3.87 (s, 3 H), 3.57-3.54 (m, 2 H); ESIMS m/z (rel intensity) 382 (MH+, 100). Anal. Calcd. for C21H19NO6: C, 66.13; H, 5.02; N, 3.67. Found: C, 65.77; H, 5.10; N, 3.59.
(2′S)-5,6-Dihydro-6-(2′,3′-dihydroxypropyl)-3-nitro-5,11-dioxo-11H-indeno[1,2-c]isoquinoline (30)
From lactone 18 (0.215 g, 0.733 mmol) and alcohol 20a (0.200 g, 2.20 mmol) in MeOH (60 mL), the general procedure was followed to afford the title compound as a yellow solid (0.171 g, 43%): mp 254–256 °C. IR (KBr) 3320, 2946, 1714, 1659, 1613, 1502, 1425, 1333, 1201 cm−1; 1H NMR (DMSO-d6) δ 8.88 (d, J = 2.3 Hz, 1 H), 8.75 (d, J = 9.0 Hz, 1 H), 8.59-8.55 (m, 1 H), 8.18 (d, J = 7.1 Hz, 1 H), 7.64-7.53 (m, 3 H), 5.19 (d, J = 5.0 Hz, 1 H), 5.06 (t, J = 5.0 Hz, 1 H), 4.60-4.50 (m, 2 H), 4.10-4.00 (m, 1 H), 3.64 (t, J = 4.9 Hz, 2 H); negative ion ESIMS m/z (rel intensity) 365 [(M – H+)−, 89]. Anal. Calcd. for C19H14N2O6: C, 62.30; H, 3.85; N, 4.59. Found: C, 61.93; H, 3.86; N, 7.48.
Topoisomerase I-Mediated DNA Cleavage Reactions
Human recombinant Top1 was purified from Baculovirus as previously described.68 DNA cleavage reactions were prepared as previously reported20 (for review see69) with the exception of the DNA substrate. Briefly, a 117-bp DNA oligonucleotide (Integrated DNA Technologies) encompassing the previous identified Top1 cleavage sites identified in the 161-bp fragment from pBluescript SK(−) phagemid DNA was employed. This 117-bp oligonucleotide contains a single 5′-cytosine overhang, which was 3′-end labeled by fill-in reaction with [α-32P]-dGTP in React 2 buffer (50 mM Tris-HCl, pH 8.0, 100 mM MgCl2, 50 mM NaCl) with 0.5 units of DNA polymerase I (Klenow fragment, New England BioLabs). Unincorporated 32P-dGTP was removed using mini Quick Spin DNA columns (Roche, Indianapolis, IN), and the eluate containing the 3′-end-labeled DNA substrate was collected. Approximately 2 nM of radiolabeled DNA substrate was incubated with recombinant Top1 in 20 μL of reaction buffer [10 mM Tris-HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 15 μg/mL BSA] at 25 °C for 20 min in the presence of various concentrations of compounds. The reactions were terminated by adding SDS (0.5% final concentration) followed by the addition of two volumes of loading dye (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromphenol blue). Aliquots of each reaction were subjected to 20% denaturing PAGE. Gels were dried and visualized by using a Phosphoimager and ImageQuant software (Molecular Dynamics). For simplicity, cleavage sites were numbered as previously described in the 161-bp fragment.68
Docking and Modeling Studies
Crystal structure preparation
The crystal structure of a ternary complex containing topoisomerase I, DNA, and topotecan, was downloaded from the Protein Data Bank (PDB ID 1K4T).7 This crystal structure was used due to the presence of co-crystallized water. The open carboxylate form, an atom of Hg, and molecule of PEG were deleted, and hydrogens were added in SYBYL 8.3.
A “mutant” crystal structure, containing the correct cleavage site for an indenoisoquinoline, was prepared by substituting the flanking (−1) A-T pair of this “prepared” structure with a G-C pair.46 The adenine -> guanine mutation was performed using the “Mutate Monomers” function of SYBYL. The thymine -> cytosine mutation was performed by manually changing the atom types. The energies of these two base pairs were subsequently minimized (with all other structures frozen in an aggregate) using the standard Powell method, the MMFF94 force field and MMFF94s charges, a distance-dependant dielectric function, and a 0.05 kcal/mol*Å energy gradient convergence criterion. The ternary complex centroid coordinates for docking were defined using the crystallized ligand as the center of the binding pocket (x = 21.3419, y = −3.9888, z = 28.2163). This ligand was then deleted.
Docking validation
To validate the docking protocol, the crystal structures of camptothecin (PDB ID 1T8I),43 and an indenoisoquinoline (PDB ID 1SC7)43 were downloaded, and their respective ligands were extracted. The ligand from the topotecan structure was extracted as well and used for validations. For topotecan and campothecin, several atom types in the quinoline ring were reset from type C.2 to type C.Ar. For the indenoisoquinoline, the carboxyl group was fixed according to SYBYL atom types. Hydrogens were added to all ligands, and minimization was performed using the MMFF94 force field with MMFF94s charges, using a conjugate gradient method, distance-dependent dielectric function, and converging to 0.01 kcal/mol*Å. Docking was performed with GOLD 3.2 using default parameters and the coordinates defined by the crystal structure as described above. The top-ranked GOLD poses for each ligand were all within 1.5 Å RMSD. The top pose for each ligand was merged into the mutant crystal structure, and minimization was subsequently performed on a sphere with a radius of 6 Å containing the ligand. These structures were allowed to move during the minimization. The surrounding structures were frozen in an aggregate. Minimization were performed using the standard Powell method, the MMFF94 force field and MMFF94s charges, a distance-dependant dielectric function, and a 0.05 kcal/mol*Å energy gradient convergence criterion. These final minimized complexes were then compared to the original structures for camptothecin, topotecan, and the indenoisoquinoline MJ238 by aligning the proteins using the ‘Align Structures by Homology’ tool in SYBYL, using the alpha-carbons as the reference point. The resulting GOLD and crystal structure poses were compared using the smart_rmsd function in GOLD. The correct binding modes were observed in all cases. RMSD values were as follows: topotecan, 0.699 Å, camptothecin, 1.20 Å, indenoisoquinoline, 2.27 Å (likely higher due to the flexible side chain). Virtually identical results were obtained when the validation ligands were constructed de novo in SYBYL.
Modeling of Indenoisoquinolines
Indenoisoquinolines 12a and 12b were constructed in SYBYL. Hydrogens were added, and the ligands were minimized using either the MMFF94 force field with MMFF94 charges, or the Tripos force field with Gasteiger-Huckel charges. Each ligand (two per charge set, four total) was docked into the mutant crystal structure using GOLD 3.2 using default parameters and the coordinates defined by the crystal structure as described above. The top three poses for each ligand were examined, and both the normal (compounds 12a and 12b) and flipped (compound 12a only) ligands were merged into the crystal structure, and the entire complex was subsequently subjected to minimization using a standard Powell method, the MMFF94 force field and MMFF94s charges, a distance-dependant dielectric function, and a 0.05 kcal/mol*Å energy gradient convergence criterion. The ligand overlays in Figure 2 were constructed by aligning the crystal structures of 1SC7 and 1SEU using the ‘Align Structures by Homology’ function with the alpha-carbons as the reference.
Acknowledgments
This work was made possible by the National Institutes of Health (NIH) through support with Research Grant UO1 CA89566, by the Frederick N. Andrews Fellowship sponsored by Purdue University (K.E.P.), and by a Purdue Research Foundation Grant (#203202, M.A.C.). In vitro cytotoxicity testing was conducted through the Developmental Therapeutics Program, DCTD, National Cancer Institute, under contract NO1-CO-56000. This research was also supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. M.A.C. thanks Dr. Karl Wood, Evgeny Kiselev, and Dr. Xiangshu Xiao (Oregon Health and Science University) for valuable discussions.
Abbreviations
- Top1
topoisomerase I
- SAR
structure-activity relationship
- MGM
mean-graph midpoint
- 4-DMAP
4-(N,N-dimethylamino)pyridine
- TBDPSCl
tert-butyldiphenylsilyl chloride
- TPAP
tetra-N-propylammonium perruthenate
- GOLD
Genetic Optimisation for Ligand Docking
- MMFF94
Merck Molecular Force Field
References
- 1.Pommier E. Topoisomerase I Inhibitors: Camptothecins and Beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 2.Stewart L, Redinbo MR, Qiu X, Hol WGJ, Champoux JJ. A Model for the Mechanism of Human Topoisomerase I. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 3.Wang JC. Cellular Roles of DNA Topoisomerases: A Molecular Perspective. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 4.Pommier Y. DNA Topoisomerase I Inhibitors: Chemistry, Biology, and Interfacial Inhibition. Chem Rev. 2009;109:2894–2902. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailly C. Topoisomerase I Poisons and Suppressors as Anticancer Drugs. Curr Med Chem. 2000;7:39–58. doi: 10.2174/0929867003375489. [DOI] [PubMed] [Google Scholar]
- 6.Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin Induces Protein-linked DNA Breaks via Mammalian DNA Topoisomerase I. J Biol Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- 7.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Stewart L. The Mechanism of Topoisomerase I Poisoning by a Camptothecin Analogue. Proc Natl Acad Sci US A. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca acuminata. J Am Chem Soc. 1966;88:3888–3890. [Google Scholar]
- 9.Thomas CJ, Rahier NJ, Hecht SM. Camptothecin: Current Perspectives. Bioorg Med Chem. 2004;12:1585–1604. doi: 10.1016/j.bmc.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 10.Teicher B. Next Generation of Topoisomerase I Inhibitors: Rationale and Biomarker Strategies. Biochem Pharmacol. 2008;75:1262–1271. doi: 10.1016/j.bcp.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Schaeppi U, Fleischm Rw, Cooney DA. Toxicity of Camptothecin (NSC-100880) Cancer Chemother Rep, Part 3. 1974;5:25–36. [PubMed] [Google Scholar]
- 12.Luzzio MJ, Besterman JM, Emerson DL, Evans MG, Lackey K, Leitner PL, McIntyre G, Morton B, Myers PL, Peel M, Sisco JM, Sternbach DD, Tong WQ, Truesdale A, Uehling DE, Vuong A, Yates J. Synthesis and Antitumor-Activity of Novel Water-Soluble Derivatives of Camptothecin as Specific Inhibitors of Topoisomerase-I. J Med Chem. 1995;38:395–401. doi: 10.1021/jm00003a001. [DOI] [PubMed] [Google Scholar]
- 13.Mi ZH, Burke TG. Differential Interactions of Camptothecin Lactone and Carboxylate Forms with Human Blood Components. Biochemistry. 1994;33:10325–10336. doi: 10.1021/bi00200a013. [DOI] [PubMed] [Google Scholar]
- 14.Kohlhagen G, Paull K, Cushman M, Nagafuji P, Pommier Y. Protein-Linked DNA Strand Breaks Induced by NSC 314622, a Novel Noncamptothecin Topoisomerase I Poison. Mol Pharmacol. 1998;54:50–58. doi: 10.1124/mol.54.1.50. [DOI] [PubMed] [Google Scholar]
- 15.Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd MR. Display and Analysis of Patterns of Differential Activity of Drugs Against Human Tumor Cell Lines: Development of Mean Graph and COMPARE Algorithm. J Natl Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 16.Pommier Y, Cushman M. The Indenoisoquinoline Noncamptothecin Topoisomerase I Inhibitors: Update and Perspectives. Mol Cancer Ther. 2009;8:1008–1014. doi: 10.1158/1535-7163.MCT-08-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagarajan M, Morrell A, Fort BC, Meckley MR, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis and Anticancer Activity of Simplified Indenoisoquinoline Topoisomerase I Inhibitors Lacking Substituents on the Aromatic Rings. J Med Chem. 2004;47:5651–5661. doi: 10.1021/jm040025z. [DOI] [PubMed] [Google Scholar]
- 18.Pommier Y, Leo E, Zhang HL, Marchand C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antony S, Kohlhagen G, Agama K, Jayaraman M, Cao S, Durrani FA, Rustum YM, Cushman M, Pommier Y. Cellular Topoisomerase I Inhibition and Antiproliferative Activity by MJ-III-65 (NSC 706744), an Indenoisoquinoline Topoisomerase I Poison. Mol Pharmacol. 2005;67:523–530. doi: 10.1124/mol.104.003889. [DOI] [PubMed] [Google Scholar]
- 20.Antony S, Agama KK, Miao ZH, Takagi K, Wright MH, Robles AI, Varticovski L, Nagarajan M, Morrell A, Cushman M, Pommier Y. Novel Indenoisoquinolines NSC 725776 and NSC 724998 Produce Persistent Topolsomerase I Cleavage Complexes and Overcome Multidrug Resistance. Cancer Res. 2007;67:10397–10405. doi: 10.1158/0008-5472.CAN-07-0938. [DOI] [PubMed] [Google Scholar]
- 21.Pommier Y. Eukaryotic DNA Topoisomerase I: Genome Gate Keeper and Its Intruders, Camptothecins. Semin Oncol. 1996;23:1–10. [PubMed] [Google Scholar]
- 22.Morrell A, Placzek MS, Steffen JD, Antony S, Agama K, Pommier Y, Cushman M. Investigation of the Lactam Side Chain Length Necessary for Optimal Indenoisoquinoline Topoisomerase I Inhibition and Cytotoxicity in Human Cancer Cell Cultures. J Med Chem. 2007;50:2040–2048. doi: 10.1021/jm0613119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagarajan M, Morrell A, Ioanoviciu A, Antony S, Kohlhagen G, Agama K, Hollingshead M, Pommier Y, Cushman M. Synthesis and Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Substituted with Nitrogen Heterocycles. J Med Chem. 2006;49:6283–6289. doi: 10.1021/jm060564z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagarajan M, Xiao X, Antony S, Kohlhagen G, Pommier Y, Cushman M. Design, Synthesis, and Biological Evaluation of Indenoisoquinoline Topoisomerase I Inhibitors Featuring Polyamine Side Chains on the Lactam Nitrogen. J Med Chem. 2003;46:5712–5724. doi: 10.1021/jm030313f. [DOI] [PubMed] [Google Scholar]
- 25.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis of Nitrated Indenoisoquinolines as Topoisomerase I Inhibitors. Bioorg Med Chem Lett. 2004;14:3659–3663. doi: 10.1016/j.bmcl.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Pommier Y, Cushman M. The Indenoisoquinoline Noncamptothecin Topoisomerase I Inhibitors: Update and Perspectives. Mol Cancer Ther. 2009;8:1008–1014. doi: 10.1158/1535-7163.MCT-08-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nettleton DE, Doyle TW, Krishnan B, Matsumoto GK, Clardy J. Isolation and Structure of Rebeccamycin - a New Antitumor Antibiotic from Nocardia Aerocoligenes. Tetrahedron Lett. 1985;26:4011–4014. [Google Scholar]
- 28.Prudhomme M. Rebeccamycin Analogues as Anti-cancer Agents. Eur J Med Chem. 2003;38:123–140. doi: 10.1016/s0223-5234(03)00011-4. [DOI] [PubMed] [Google Scholar]
- 29.Bailly C, Riou JF, Colson P, Houssier C, RodriguesPereira E, Prudhomme M. DNA Ceavage by Topoisomerase I in the Presence of Indolocarbazole Derivatives of Rebeccamycin. Biochemistry. 1997;36:3917–3929. doi: 10.1021/bi9624898. [DOI] [PubMed] [Google Scholar]
- 30.Zhang GS, Shen J, Cheng H, Zhu LZ, Fang LY, Luo SZ, Muller MT, Lee GE, Wei LJ, Du YG, Sun DX, Wang PG. Syntheses and Biological Activities of Rebeccamycin Analogues with Uncommon Sugars. J Med Chem. 2005;48:2600–2611. doi: 10.1021/jm0493764. [DOI] [PubMed] [Google Scholar]
- 31.Long BH, Rose WC, Vyas DM, Matson JA, Forenza S. Discovery of Antitumor Indolocarbazoles: Rebeccamycin, NSC655649 and Fluoroindolocarbazoles. Curr Med Chem-Anticancer Agents. 2002:2255–2266. doi: 10.2174/1568011023354218. [DOI] [PubMed] [Google Scholar]
- 32.Saif MW, Sellers S, Diasio RB, Douillard JY. A phase I Dose-Escalation Study of Edotecarin (J-107088) Combined with Infusional 5-Fluorouracil and Leucovorin in Patients with Advanced/Metastatic Solid Tumors. Anti-Cancer Drugs. 2010;21:716–723. doi: 10.1097/CAD.0b013e32833cb658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Staker BL, Feese MD, Cushman M, Pommier Y, Zembower D, Stewart L, Burgin AB. Structures of Three Classes of Anticancer Agents bound to the Human Topoisomerase I-DNA Covalent Complex. J Med Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 34.Zembower DE, Zhang HP, Lineswala JP, Kuffel MJ, Aytes SA, Ames MM. Indolocarbazole Poisons of Human Topoisomerase I: Regioisomeric Analogues of ED-110. Bioorg Med Chem Lett. 1999;9:145–150. doi: 10.1016/s0960-894x(98)00710-0. [DOI] [PubMed] [Google Scholar]
- 35.Strumberg D, Pommier Y, Paull K, Jayaraman M, Nagafuji P, Cushman M. Synthesis of Cytotoxic Indenoisoquinoline Topoisomerase I Poisons. J Med Chem. 1999;42:446–457. doi: 10.1021/jm9803323. [DOI] [PubMed] [Google Scholar]
- 36.Winestock CH, Plaut GWE. Synthesis and Properties of Certain Substituted Lumazines. J Org Chem. 1961;26:4456–4462. [Google Scholar]
- 37.Finch P, Merchant Z. Structures of D-Arabinose and D-Glucose Oximes. Journal of the Chemical Society-Perkin Transactions. 1975;1:1682–1686. [Google Scholar]
- 38.Snyder JR. An NMR Investigation of the Aldopentose Oximes. Carbohydr Res. 1990;198:1–13. [Google Scholar]
- 39.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. Synthesis of Benz[d]indeno[1,2-b]pyran-5,11-diones: Versatile Intermediates for the Design and Synthesis of Topoisomerase I Inhibitors. Bioorg Med Chem Lett. 2006;16:1846–1849. doi: 10.1016/j.bmcl.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 40.Morrell A, Antony S, Kohlhagen G, Pommier Y, Cushman M. A Systematic Study of Nitrated Indenoisoquinolines Reveals a Potent Topoisomerase I Inhibitor. J Med Chem. 2006;49:7740–7753. doi: 10.1021/jm060974n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ley SV, Norman J, Griffith WP, Marsden SP. Tetrapropylammonium Perruthenate, Pr4n+Ruo4-, Tpap - a Catalytic Oxidant for Organic-Synthesis. Synthesis. 1994:639–666. [Google Scholar]
- 42.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 43.Paull KD, Hamel E, Malspeis L. Prediction of Biochemical Mechanism of Action from the In Vitro Antitumor Screen of the National Cancer Institute. In: Foye WO, editor. Cancer Chemotherapeutic Agents. American Chemical Society; Washington, DC: 1995. pp. 9–45. [Google Scholar]
- 44.Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, Pommier Y. Differential Induction of Topoisomerase I-DNA Cleavage Complexes by the Indenoisoquinoline MJ-III-65 (NSC 706744) and Camptothecin: Base Sequence Analysis and Activity against Camptothecin-Resistant Topoisomerase I. Cancer Res. 2003;63:7428–7435. [PubMed] [Google Scholar]
- 45.Cinelli MA, Morrell AE, Dexheimer TS, Agama K, Agrawal S, Pommier Y, Cushman M. The Structure-Activity Relationships of A-Ring-substituted Aromathecin Topoisomerase I Inhibitors Strongly Support a Camptothecin-like Binding Mode. Bioorg Med Chem. 2010;18:5535–5552. doi: 10.1016/j.bmc.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao X, Antony S, Pommier Y, Cushman M. On the Binding of Indeno[1,2-c]isoquinolines in the DNA-Topoisomerase I Cleavage Complex. J Med Chem. 2005;48:3231–3238. doi: 10.1021/jm050017y. [DOI] [PubMed] [Google Scholar]
- 47.Halgren TA. Merck Molecular Force Field. 1. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J Comput Chem. 1996;17:490–519. [Google Scholar]
- 48.Ioanoviciu A, Antony S, Pommier Y, Staker BL, Stewart L, Cushman M. Synthesis and Mechanism of Action Studies of a Series of Norindenoisoquinoline Topoisomerase I Poisons Reveal an Inhibitor with a Flipped Orientation in the Ternary DNA-Enzyme-Inhibitor Complex as Determined by X-ray Crystallographic Analysis. J Med Chem. 2005;48:4803–4814. doi: 10.1021/jm050076b. [DOI] [PubMed] [Google Scholar]
- 49.Song YL, Cushman M. The Binding Orientation of a Norindenoisoquinoline in the Topoisomerase I-DNA Cleavage Complex Is Primarily Foverned by pi-pi Stacking Interactions. J Phys Chem B. 2008;112:9484–9489. doi: 10.1021/jp8005603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cinelli MA, Cordero B, Dexheimer TS, Pommer E, Cushman M. Synthesis and Biological Evaluation of 14-(Aminoalkyl-aminomethyl)aromathecins as Topoisomerase I Inhibitors: Inventigating the Supothesis of Shared Structure-Activity Relationships. Bioorg Med Chem. 2009;17:7145–7155. doi: 10.1016/j.bmc.2009.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cinelli MA, Morrell A, Dexheimer TS, Scher ES, Pommier Y, Cushman M. Design, Synthesis, and Biological Evaluation of 14-Substituted Aromathecins as Topoisomerase I Inhibitors. J Med Chem. 2008;51:4609–4619. doi: 10.1021/jm800259e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bailly C, Qu XG, Anizon F, Prudhomme M, Riou JF, Chaires JB. Enhanced Binding to DNA and Topoisomerase I Inhibition by an Analog of the Antitumor Antibiotic Rebeccamycin Containing an Amino Sugar Residue. Mol Pharm. 1999;55:377–385. doi: 10.1124/mol.55.2.377. [DOI] [PubMed] [Google Scholar]
- 53.Delcros JG, Tomasi S, Carrington S, Martin B, Renault J, Blagbrough IS, Uriac P. Effect of Spermine Conjugation on the Cytotoxicity and Cellular Transport of Acridine. J Med Chem. 2002;45:5098–5111. doi: 10.1021/jm020843w. [DOI] [PubMed] [Google Scholar]
- 54.Dallavalle S, Giannini G, Alloatti D, Casati A, Marastoni E, Musso L, Merlini L, Morini G, Penco S, Pisano C, Tinelli S, De Cesare M, Beretta GL, Zunino F. Synthesis and Cytotoxic Activity of Polyamine Analogues of Camptothecin. J Med Chem. 2006;49:5177–5186. doi: 10.1021/jm060285b. [DOI] [PubMed] [Google Scholar]
- 55.Kaluzhny DN, Tatarskiy VV, Dezhenkova LG, Plikhtyak IL, Miniker TD, Shchyolkina AK, Strel’tsov SA, Chilov GG, Novikov FN, Kubasova IY, Smirnova ZS, Mel’nik SY, Livshits MA, Borisova OF, Shtil AA. Novel Antitumor L-Arabinose Derivative of Indolocarbazole with High Affinity to DNA. Chem Med Chem. 2009;4:1641–1648. doi: 10.1002/cmdc.200900227. [DOI] [PubMed] [Google Scholar]
- 56.Erickson JA, Jalaie M, Robertson DH, Lewis RA, Vieth M. Lessons in Molecular Recognition: The Effects of Ligand and Protein Flexibility on Molecular Docking Accuracy. J Med Chem. 2004;47:45–55. doi: 10.1021/jm030209y. [DOI] [PubMed] [Google Scholar]
- 57.Facompre M, Carrasco C, Colson P, Houssier C, Chisholm JD, Van Vranken DL, Bailly C. DNA Binding and Topoisomerase I Poisoning Activities of Novel Disaccharide Indolocarbazoles. Mol Pharm. 2002;62:1215–1227. doi: 10.1124/mol.62.5.1215. [DOI] [PubMed] [Google Scholar]
- 58.Bailly C, Qu XG, Graves DE, Prudhomme M, Chaires JB. Calories from Carbohydrates: Energetic Contribution of the Carbohydrate Moiety of Rebeccamycin to DNA Binding and the Effect of Its Orientation on Topoisomerase I Inhibition. Chem Biol. 1999;6:277–286. doi: 10.1016/S1074-5521(99)80073-8. [DOI] [PubMed] [Google Scholar]
- 59.Anizon F, Belin L, Moreau P, Sancelme M, Voldoire A, Prudhomme M, Ollier M, Severe D, Riou JF, Bailly C, Fabbro D, Meyer T. Syntheses and Biological Activities (Topoisomerase Inhibition and Antitumor and Antimicrobial Properties) of Rebeccamycin Analogues Bearing Modified Sugar Moieties and Substituted on the Imide Nitrogen with a Methyl Group. J Med Chem. 1997;40:3456–3465. doi: 10.1021/jm9702084. [DOI] [PubMed] [Google Scholar]
- 60.Carrasco C, Facompre M, Chisholm JD, Van Vranken DL, Wilson WD, Bailly C. DNA Sequence Recognition by the Indolocarbazole Antitumor Antibiotic AT2433-B1 and Its Dastereoisomer. Nucleic Acids Res. 2002;30:1774–1781. doi: 10.1093/nar/30.8.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ohkubo M, Nishimura T, Kawamoto H, Nakano M, Honma T, Yoshinari T, Arakawa H, Suda H, Morishima H, Nishimura S. Synthesis and Biological Activities of NB-506 Analogues Modified at the Glucose Group. Bioorg Med Chem Lett. 2000;10:419–422. doi: 10.1016/s0960-894x(00)00004-4. [DOI] [PubMed] [Google Scholar]
- 62.Guano F, Pourquier P, Tinelli S, Binaschi M, Bigioni M, Animati F, Manzini S, Zunino F, Kohlhagen G, Pommier Y, Capranico G. Topoisomerase Poisoning Activity of Novel Disaccharide Anthracyclines. Mol Pharm. 1999;56:77–84. doi: 10.1124/mol.56.1.77. [DOI] [PubMed] [Google Scholar]
- 63.Wall ME, Wani MC, Nicholas AW, Manikumar G, Tele C, Moore L, Truesdale A, Leitner P, Besterman JM. Plant Antitumor Agents. 30. Synthesis and Structure-Activity of Novel Camptothecin Analogs. J Med Chem. 1993;36:2689–2700. doi: 10.1021/jm00070a013. [DOI] [PubMed] [Google Scholar]
- 64.Jaxel C, Kohn KW, Wani MC, Pommier Y. Structure-Activity Study of the Actions of Camptothecin Derivatives on Mammalian Topoisomerase I: Evidence for a Specific Receptor Site and a Relation to Antitumor Activity. Cancer Res. 1989;49:1465–1469. [PubMed] [Google Scholar]
- 65.Brand J, Huhn T, Groth U, Jochims JC. Nitrosation of Sugar Oximes: Preparation of 2-Glycosyl-1-hydroxydiazene-2-oxides. Chem–Eur J. 2006;12:499–509. doi: 10.1002/chem.200500325. [DOI] [PubMed] [Google Scholar]
- 66.Den Otter HP. Some Derivatives of Glucamine and Galactamine. Rec Trav Chem Pays-Bas. 1937;56:1196–1202. [Google Scholar]
- 67.Winestock CH, Aogaichi T, Plaut GWE. The Substrate Specificity of Riboflavin Synthetase. J Biol Chem. 1963;238:2866–2874. [PubMed] [Google Scholar]
- 68.Pourquier P, Ueng LM, Fertala J, Wang D, Park HJ, Essigmann JM, Bjornsti MA, Pommier Y. Induction of Reversible Complexes between Eukaryotic DNA Topoisomerase I and DNA-containing Oxidative Base Damages. 7,8-Dihydro-8-Oxoguanine and 5-Hydroxycytosine. J Biol Chem. 1999;274:8516–8523. doi: 10.1074/jbc.274.13.8516. [DOI] [PubMed] [Google Scholar]
- 69.Dexheimer TS, Pommier Y. DNA Cleavage Assay for the Identification of Topoisomerase I Inhibitors. Nat Protocol. 2008;3:1736–1750. doi: 10.1038/nprot.2008.174. [DOI] [PMC free article] [PubMed] [Google Scholar]