

Abstract
Orotic acid (OA), an intermediate in pyrimidine metabolism, has been used for a variety of purposes, such as dietary supplements. Although it is well documented that OA induces fatty liver in a species-specific manner, the precise molecular mechanisms remain unclear. The present study investigated the role of the adenosine monophosphate-activated protein kinase (AMPK)-sterol regulatory element-binding protein-1 (SREBP-1) pathway in the OA-induced fatty liver. Treatment with OA suppressed the phosphorylation of AMPK via proteasomal degradation of upstream kinase LKB1 and induced activation of SREBP-1 in both human hepatoma cell lines and primary rat hepatocytes. OA-induced SREBP-1 transcriptional activity was suppressed by cotreatment with aminoimidazole carboxamide ribonucleotide (AICAR) or metformin, or by overexpression of constitutively active AMPK (CA-AMPK) in the human hepatoma cell line. Importantly, in vivo data corroborated these results. Feeding 1% OA with diet decreased the phosphorylation of AMPK and increased the maturation of SREBP-1 and the expression of SREBP-responsive genes in the rat liver. OA-induced lipid accumulation was also completely inhibited by rapamycin. Mouse hepatocytes and mice were resistant to OA-induced lipogenesis because of little if any response in AMPK and downstream effectors. In conclusion, OA induces hepatic lipogenesis, mediated predominantly by the AMPK/SREBP-1 pathway in rat hepatocytes and human hepatoma cell lines.
Keywords: lipidsh, toxicology, pharmacology, adenosine monophosphate-activated protein kinase, sterol regulatory element-binding protein-1
Induction of fatty liver in rats by orotic acid (OA; 6-carboxyuracil) administration was first observed in 1955 (1). Since then, OA has been investigated by many and has been used as a chemical inducer of fatty liver (2, 3). Although somewhat inconclusive, the reported mechanisms of OA-induced fatty liver include impairment of fatty acid oxidation, stimulation of lipogenesis, and reduction in lipid transport from the liver (4, 5). OA-induced fatty liver is species specific, and thus far, only the rat has been found to be susceptible. Durschlag and Robinson demonstrated that pharmacokinetic factors determined species specificity in OA-induced hepatotoxicity (6). However, the precise molecular mechanism by which OA induces fat accumulation, specifically in the rat liver, has remained unclear. Identification of molecular targets of OA effects and susceptibility are of great importance in terms of scientific as well as practical aspects. The major source of OA in a typical adult diet is milk and dairy products, which contribute about 0.005% of the total solids, a level at which rats do not show hepatic changes. However, considering the high content of OA in many dietary supplements, vitamin and mineral complexes, and muscle-building products widely sold on the market, we should be wary of possible health effects posed to humans.
AMP-activated protein kinase (AMPK), a heterotrimeric enzyme complex, is the key regulator of energy metabolism in cells. The energy-sensing motif of the enzyme monitors the energy status of cells and controls its activity by phosphorylation at T172 (7). In the liver, activation of AMPK phosphorylates and inactivates the rate-limiting enzymes of lipogenesis, such as acetyl-CoA carboxylase (ACC) (8). AMPK is classically regulated by various metabolic stresses that cause an increase in the AMP/ATP ratio or other cellular energy and metabolic states, such as glycogen, lipid, and NAD/NADH redox potential (9–11). However, recent studies have suggested that other unidentified pathways can regulate AMPK, regardless of cellular energy status. Metformin and rosiglitazone phosphorylate AMPK without any change in cellular energy state (12). Leptin has been found to stimulate AMPK through both AMP-dependent and AMP-independent mechanisms (13). AMPK is phosphorylated by upstream kinases, among which serine/threonine kinase 11 (LKB1) has been identified as a major AMPK kinase in the liver (14). LKB1 is considered to be constitutively active; however, in some cases, altered expression or activity of LKB1 is associated with the phosphorylation of AMPK (15). It is well documented that AMPK phosphorylation inhibits sterol regulatory element-binding protein-1 (SREBP-1), the key transcription factor responsible for fatty acid synthesis, through mammalian target of rapamycin (mTOR) and liver X receptor-α (LXRα) (16). Conversely, inhibition of AMPK has been suggested as a central event causing the development of chemical-induced fatty liver. Repressed AMPK activates anabolic pathways, such as fatty acid synthesis, and inhibits catabolic pathways, such as fatty acid oxidation. In studies performed with hepatocytes and in the livers of ethanol-fed mice, You et al. demonstrated that inhibition of AMPK leads to the activation of lipogenesis mediated by SREBP-1 (17). Similar results were obtained in studies with cigarette smoking and the saturated fatty acid palmitate (18, 19). AMPK positively regulated fatty acid oxidation by activating peroxisome proliferator-activated receptor-α (PPARα) and PPARγ coactivator (PGC)-1 (20). Thus, inhibition of AMPK by chemicals or physiological state may contribute to the intracellular accumulation of lipids.
The first step in OA metabolism is the reaction with phosphoribosyl pyrophosphate (PRPP) to form orotidine-5′-monophosphate. Depletion of PRPP by an OA dose is followed by a reduction in purine biosynthesis, including ATP, and defects in lipid transport from the liver (21). Recent studies have suggested an alternative mechanism of OA toxicity in which altered transcription of genes involved in fatty acid metabolism contribute to fatty liver; however, the exact mechanism of the gene expression changes remains to be elucidated (22). To understand the molecular mechanism of OA-induced fatty liver, we tested the effects of OA on the AMPK/SREBP-1 pathway in human hepatoma cell lines and rat or mouse hepatocytes as well as in rats and mice in vivo. Here, we report that OA activates SREBP-1 by AMPK inhibition in rat hepatocytes and human hepatoma cell lines in vitro and in rats in vivo.
MATERIALS AND METHODS
Materials
Anti-AMPK, anti-phosphor-AMPK (Thr172), anti-ACC, anti-phosphor-ACC (Ser79), anti-p70S6Kinase, anti-phosphor-p70S6Kinase (Thr389), anti-GAPDH, and anti-mTOR were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Anti-SREBP-1, anti-LKB1, anti-c-Myc, siLKB1 and anti-Flag were prepared from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Nile red, orotic acid monohydrate, aminoimidazole carboxamide ribonucleotide (AICAR), and metformin were obtained from Sigma Chemical Co. (St. Louis, MO). We purchased lactacystin and radicicol from Calbiochem (San Diego, CA). Rapamycin were obtained from Selleck Chemicals, LLC (Houston, TX).
Cell culture
SK-Hep1and HepG2 human hepatoma, H4IIEC3 rat hepatoma, and HeLa cells were cultured in Dulbecco's Minimal Essential Medium (DMEM; Gibco-BRL, Rockville, MD), supplemented with 10% fetal bovine serum, 3.7 mg/ml sodium bicarbonate, 100 units/ml penicillin, and streptomycin . Cells were incubated in a 37°C incubator in an atmosphere of 5% in air. SK-Hep1, an immortal human cell line derived from the ascetic fluid of a patient with adenocarcinoma of the liver, is used frequently as a representative hepatoma line. Rat and mouse hepatocytes were isolated from SD rats and C57/BL6 mice by collagenase perfusion as described (23, 24).
Real-time RT-PCR
Total RNA from livers and cells with or without OA were isolated using EASY BLUE total RNA extraction kit (INTRON Biotechnology, Korea), then reverse transcribed using SuperScriptTM III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Expression of mRNA was quantified by using SYBR Green PCR Master Mix and performed with ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA) with cDNA synthesis for FAS, LXRα, SREBP-1a, SREBP-1c, ACC, SCD1, L-PK, and GAPDH. The primers used for these genes are listed in Table 1. The data were analyzed using ABI Prism Software version 1.5 (Applied Biosystems) and were normalized with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels.
TABLE 1.
Sequences of real-time RT-PCR primers
| Gene | Forward Sequences | Reverse Sequences | Source |
| hSREBP-1a | 5′-CTGCTGACCGACATCGAAGAC-3′ | 5′-GATGCTCAGTGGCACTGACTCTTC-3′ | Ref. 46 |
| hSREBP-1c | 5′-GGAGGGGTAGGGCCAACG-3′ | 5′-AGGGGTGGAGCTCAACTG-3′ | Ref. 47 |
| hFASN | 5′-CGCGTGGCCGGCTACTCCTAC-3′ | 5′-CGGCTGCCACACGCTCCTCT-3′ | NM_004104 |
| hSCD1 | 5′-TGGTGATGTTCCAGAGGAGGTA-3′ | 5′-AATGTGGTGAAGTTGATGTGCC-3′ | NM_005063 |
| hACC1 | 5′-TTTGACCTCACTGCCATTCC-3′ | 5′-GCGACTTCCATACCGCATTAC-3′ | NM_198834 |
| hLPK | 5′-ATCATGCTGTCAGGGGAGAC-3′ | 5′-AGTGGGATCACGGCTTAGTG-3′ | NM_181871 |
| hGAPDH | 5′-GAGTCAACGGATTTGGTCGT-3′ | 5′-TTGATTTTGGAGGGATCTCG-3′ | NM_002046 |
| rSREBP-1 | 5′-CATGGATTGCACATTTGAAGAC-3′ | 5′-GCAGGAGAAGAGAAGCTCTCAG-3′ | Ref. 48 |
| rFAS | 5′-GGCCACCTCAGTCCTGTTAT-3′ | 5′-AGGGTCCAGCTAGAGGGTACA-3′ | NM_017332 |
| rSCD1 | 5′-TCCCCTCCTCCAAGGTCTAC-3′ | 5′-GCTCCACAAGCGATGAGC-3′ | NM_139192 |
| rLXRα | 5′-CAGGAAGAGATGTCCTTGTGG-3′ | 5′-TCTTCCACAACTCCGTTGC-3′ | NM_031627 |
| rACC1 | 5′-GGACAACACCTGTGTGGTAGAA-3′ | 5′0CGTGGGGATGTTCCCTCT-3′ | NM_022193 |
Western blot analysis
Chemicals were dissolved in DMSO or distilled water, and cells were incubated in serum-free or 1% serum media with or without chemicals. The final concentration of vehicle in media was maintained at 0.1% (v/v), and appropriate vehicle controls were used in all cases. Immunoblotting for SREBP-1, phosphor-AMPK (Thr172), AMPK, phosphor-ACC (Ser79), ACC, LKB1, phosphor-S6K1 (Thr389), S6K1, mTOR, phosphor-Raptor (Ser792), Raptor, and GAPDH were performed by established procedures. Bands were visualized using a chemiluminescence detection system (ChemiDocTM XRS system, Bio-Rad Laboratories, Inc., Hercules, CA) according to the manufacturer's instructions. Protein contents were determined using the method for DC protein assay (Bio-Rad Laboratories) using BSA as a standard.
Transient transfection and reporter gene assay
The luciferase reporter constructs, SRE-TK-Luc, were kind gifts from Dr. M. O. Lee (Seoul National University, Korea). The expression constructs constitutively active AMPK (CA-AMPK) and dominant-negative AMPK (DN-AMPK; T172A), which were provided by Dr. S. G. Kim (Seoul National University, Korea), were originally from Dr. J. Ha (Kyung Hee University, Korea) and Dr. Y. Kanno (Toho University, Japan), respectively. The plasmids of pCMV-RL were purchased from Promega Co. (Madison, WI). The expression plasmids of pEGFP-LKB1 were obtained from Dr. J. Ha. Cells were transfected with appropriate plasmid constructs (SRE-TK-Luc, CA-AMPK, DN-AMPK, and pEGFP-LKB1) by using the LipofectAMINE 2000 (Invitrogen) as recommended by the manufacturer, and they were incubated in 1% serum media with or without OA. pCMV-RL plasmids were cotransfected as an internal control. Luciferase activities were measured by using the Dual-Luciferase assay detection kit according to manufacturer's instructions (Promega). Luciferase activity was detected using a Centro LB960 luminometer (Berthold Technologies, Oak Ridge, TN). Transfection of siRNAs specific to LKB1 into SK-Hep1 cells was performed with LipofectAMINE 2000 (Invitrogen) according to the manufacturer's instructions. The cells were further incubated for 24 h prior to treatment with OA for 24 h.
Determination of adenine nucleotides
Frozen liver tissues of rat or mice and SK-Hep1, HepG2, primary rat, or mouse hepatocytes were washed with PBS and homogenized with perchloric acid (PCA). The samples were subjected to HPLC analysis for AMP and ATP as described previously (25).
Nile-red analysis
For Nile-red assay, SK-Hep1 cells were seeded in fluorescence plates. On the following day, the cells were incubated with or without OA. Cells were washed with PBS and stained with 100 ng/ml Nile-red fluorescent dye (Sigma-Aldrich, St. Louis, MO) in PBS. The stained cells were washed twice with PBS and then analyzed by fluorescence detection system (Molecular Devices, Sunnyvale, CA).
Animal experiments
Male Sprague-Dawley rats and C57BL/6 mice ages 4-5 weeks were purchased from Orient Bio, Korea, and housed in an air-conditioned room (24°C) with a 12 h light/dark cycle. After one week adaption period, animals were divided to three groups. Animals were given normal purified rodent diet (AIN-93G) or OA diet prepared by supplementation of 0.5% and 1% OA to the normal diet for seven days. They were euthanized after being fasted for 18 h. Livers were removed quickly and frozen at −70°C until subsequent protein analysis. In the separate study of mTOR effects on OA-fed rats, rats were divided into three groups. The rapamycin groups were given daily by gavage 2 mg/kg rapamycin dissolved in vehicle (DMSO/polyethylene glycol 400) with 1% OA mixed in diet as described above for seven days. The experiments using animals were carried out in accordance with animal experiment guidelines with approval of the Animal Care and Use Committee of Seoul National University.
Histological analysis and immunohistochemistry
The tissues were fixed with 4% paraformaldehyde in PBS and then transferred in paraffin. The tissues were embedded in paraffin blocks, cut into 5 µm sections with Probe-On-Plus Slides (Thermo Fisher Scientific, Inc., Waltham, MA), and stained with hematoxylin and eosin (H and E). Immunohistochemistry assay was performed using rabbit anti-SREBP-1 polyclonal antibody as previously described (26). Sections were counterstained with Meyer's hematoxylin. Negative controls with normal rabbit serum were processed in parallel, and no positive staining was observed.
Oil Red O staining
For Oil Red O staining, frozen liver tissues were cut into 7 µm with Probe-On-Plus Slides (Thermo Fisher Scientific) and affixed to microscope slides. Sections were reacted with Oil Red O solution buffer for 7 min at 60°C, and then incubated with 85% propylene glycol for 3 min. After rinsing with water, sections were stained with Harris hematoxylin for counterstaining.
Liver triglyceride content analysis
Liver TG content was determined by a modified Folch method described previously (27). Briefly, 50 mg liver tissue was homogenized in chloroform/methanol (2/1; v/v). After extraction at room temperature overnight, the organic phase was used for the determination of hepatic TG content using the serum triglyceride determination kit (Sigma-Aldrich).
Electrophoretic mobility shift assay
The labeled probe for the SRE of the FAS genes (5′-CATCCGGCATCACCCCACCGACGGCG-3′) were incubated with nuclear extract obtained from OA-treated cells. Electrophoretic mobility shift assay (EMSA) was carried out using Liteshift Chemiluminescent EMSA kit (Thermo Fisher Scientific, Inc., Rockford, IL) as recommended by the manufacturer. Reaction complexes were visualized using a chemiluminescence detection system (ChemiDocTM XRS system, Bio-Rad Laboratories).
Statistical analysis
All values are presented as the means ± SE of at least three independent experiments. Significant differences were obtained using Student's t-test. A difference was considered significant at P< 0.05.
RESULTS
OA activates SREBP-1 in rat hepatocytes and human hepatoma cell lines
SREBP-1 is a membrane-bound transcription factor that regulates lipid homeostasis by controlling the expression of genes for lipid metabolism. Increased hepatic lipogenesis by activation of SREBP-1 may contribute to the development of chemical-induced fatty liver (28). We first examined the effects of OA on SREBP-1 activation in various hepatic cells by Western blot analysis. Treatment with OA resulted in a substantial and dose-dependent increase in the amount of mature SREBP-1 protein in both human hepatoma SK-Hep1 and HepG2 cell lines. Similar results were obtained in primary rat hepatocytes in which the expression of mature SREBP-1 was increased, although a decrease in the precursor form was observed (Fig. 1A). A dose-dependent increase in the mRNA for Srebp-1c was observed in the SK-Hep1, HepG2, and primary rat hepatocytes (Fig. 1B).
Fig.1.

OA induces the activation of SREBP-1 and suppression of AMPK in human and rat liver cell lines and primary rat hepatocytes. Cells were treated with OA for 24 h in 1% serum media (A, B) and in serum-free condition, following incubation in serum-free media for 16 h (C, D). Cells were treated with 100 μM OA in the absence or presence of 500 μM metformin (Met) or AICAR for 24 h (D). Expression of proteins and mRNA was analyzed by Western blotting and by real-time PCR, respectively. Each bar represents the mean ± SE of three independent experiments. Results are expressed as fold changes over vehicle control. One representative blot of at least three independent experiments with similar results is shown. *P < 0.05, **P < 0.01, ***P < 0.001 compared with vehicle control.
OA inhibits AMPK phosphorylation in rat hepatocytes and human hepatoma cell lines
AMPK is a key energy sensor in the cells that ultimately control the lipid metabolism machinery. Although AMPK is classically regulated by the cellular energy status, it has been reported that chemicals or chemical mixtures can also regulate the activity of AMPK. To understand the potential mechanisms by which OA induces SREBP-1 activation, we examined the effects of OA on the phosphorylation of AMPK, because phosphorylation on the Thr172 residue is essential for its activation (29). Incubation of human and rat hepatoma cell lines with OA for 24 h resulted in the dose-dependent reduction in phosphorylation of AMPK despite increased expression of AMPK, resulting in the decrease in the pAMPK/AMPK ratio. Similar results were obtained when primary rat hepatocytes were treated with OA (Fig. 1C). Activation of AMPK leads to the phosphorylation and inhibition of ACC. We found that ACC phosphorylation was suppressed by OA treatment. The decreased phosphorylation of AMPK and ACC by OA was largely prevented by addition of metformin or AICAR (Fig. 1D).
Whereas rats are susceptible to OA-induced fatty liver, mice are known to be resistant (6). To understand the molecular mechanisms responsible for the species difference in OA effects, we examined the effects of OA on AMPK and SREBP-1 proteins in primary cultured mouse hepatocytes. Neither the maturation of SREBP-1 protein nor the phosphorylation of AMPK was affected by OA in these cells (supplementary Fig. I).
Activation of AMPK attenuates OA-induced expression of lipogenic genes
Phosphorylation of AMPK suppresses expression of SREBP-1 in vitro and in vivo (30, 31), whereas inhibition of AMPK augments expression of SREBP-1 and thus induces fatty liver. To identify the role of AMPK in the activation of SREBP-1 by OA, we transfected SK-Hep1 cells with a eukaryotic expression vector expressing constitutively active AMPK protein tagged with Myc epitope, and then the level of mature SREBP-1 was determined following OA treatment. Results are summarized in Fig. 2A. Basal and OA-induced expression of mature SREBP-1 protein was completely antagonized by CA-AMPK. Transfection with DN-AMPK tagged with Flag significantly increased the basal mature SREBP-1 expression, whereas no effect was shown in OA-treated cells (Fig. 2A). We next examined whether OA increased the expression of SREBP-1 target genes that are involved in fatty acid synthesis. When the levels of Fas, Srebp-1a, Srebp-1c, Acc, Scd-1, and L-Pk gene transcripts were measured by quantitative real-time PCR using gene-specific primers, we found that their mRNA expression was increased dose dependently in OA-treated cells. Combination treatment with AICAR prevented the OA-associated increase in the mRNA levels (Fig. 2B). Consequently, AICAR completely abrogated the effects of OA on intracellular lipid accumulation measured using Nile-red staining (Fig. 2C).
Fig.2.

Activation of AMPK attenuates OA-induced expression of lipogenic genes. SK-Hep1 cells were transfected with mock, CA-AMPK, or DN-AMPK for 18 h, and treated with OA for 24 h. Western blot analysis using a specific antibody was used to examine the expression of the proteins in the whole cell lysates. Band densities were determined using an image analysis system, normalized to that of GAPDH, and expressed as fold changes over vehicle control. One representative blot of at least three independent experiments with similar results is shown (A). Cells were treated with OA (0, 1, 10, 100 μM) in the absence or presence of 500 μM AICAR for 24 h. Total RNA was extracted, and the mRNA for each gene was analyzed by real-time PCR using gene-specific primers. Results are expressed as fold changes over vehicle control (B). Cells were treated with OA, and Nile-red fluorescence was observed after 48 h using a Molecular Device Gemini EM fluorometer (excitation 485 nm/emission 580 nm). The relative fluorescence intensity ratio for each sample was calculated by using a value of 1 for the vehicle control (C). Each bar represents the mean ± SE of three independent experiments. *P < 0.01, **P < 0.01, ***P < 0.001 compared with vehicle control.
We next performed reporter gene analysis using a reporter gene containing SRE sequences and a gel-shift assay in SK-Hep1 and HepG2 cells. Treatment of cells with OA resulted in a dose-dependent increase in luciferase activity (Fig. 3A). OA-induced luciferase activity was considerably diminished by cotreatment with metformin or AICAR or by cotransfection of CA-AMPK. Conversely, introduction of DN-AMPK to cells markedly augmented the OA effect on SRE luciferase activity (Fig. 3A). We also observed an increase in the nuclear SREBP-1-dependent gel-shift activity. Supershift analysis confirmed the DNA binding of SREBP-1 (Fig. 3B). Together, these data suggested that OA induces the maturation and transcriptional activity of SREBP-1 through AMPK inhibition in human and rat hepatocytes.
Fig.3.

Activation of AMPK attenuates OA-induced SREBP-1 transactivation. SRE luciferase transactivation was determined from the lysates of SK-Hep1 and HepG2 cells transfected with the pSRE-luc construct alone or with mock, CA-AMPK, or DN-AMPK and treated with OA in the absence or presence of 500 μM metformin or AICAR. Results are expressed as fold changes over vehicle control. Each bar represents the mean ± SE of three independent experiments. *P < 0.05, **P < 0.01 compared with mock-transfected vehicle control; #P < 0.05, ##P < 0.01 compared with DN-AMPK-transfected vehicle control (A). Gel shift analysis of SREBP-1 binding to the SRE was performed by the reaction of nuclear extracts with biotin-labeled SRE probe. For the competition assay, the nuclear extract was incubated with SREBP-1 antibody mixed with a labeled SRE probe. SS indicates a supershift of the SREBP1-DNA complex (B).
OA induces proteasomal degradation of LKB1
Control of protein stability via the proteasomal degradation pathway plays an important role in regulating many cellular processes. We asked whether the inhibition of AMPK phosphorylation by OA might be associated with the proteasomal degradation of AMPK kinase. We examined the effects of OA on the expression levels of various AMPK kinases. Treatment of SK-Hep1 cells with OA decreased the expression of LKB1, one of the major AMPK kinases expressed in the liver (Fig. 4A). Other AMPK kinases, such as CaMKKα or CaMKKβ, were unaffected by OA (data not shown). To determine whether LKB1 could mediate the OA effects on AMPK phosphorylation, we compared the results of LKB1-deficient cells with that of LKB1-expressing cells in their response to OA. To that end, we compared AMPK phosphorylation and SREBP-1 activation in HeLa cells deficient or reconstituted with LKB1 as well as in SK-Hep1 cells wild-type or silenced with LKB1 siRNA. Western blot analysis of AMPK is shown in Fig. 4B. AMPK phosphorylation was not affected by OA in either LKB1-deficient HeLa cells or LKB-silenced SK-Hep1 cells, whereas overexpression of LKB1 to HeLa cells restored the effect. Similar results were observed in SK-Hep1 cells. OA induced expression and activation of SREBP-1 and its target genes in SK-Hep1 cells. However, knockdown of LKB1 abolished the effects of OA (Fig. 4C).
Fig.4.

OA induces proteasomal degradation of LKB1. SK-Hep1, siLKB1-transfected SK-Hep1, HeLa, and HeLa cells transfected with mock or pEGFP-LKB1 expression vector were treated with OA for 24 h (A, B, C). HeLa cells transfected with pEGFP-LKB1 expression vector were treated with OA in the absence or presence of 3 h pretreatment with 5 μM lactacystin, a proteasomal degradation inhibitor. The radicicol and lactacystin treatment served as a control for proteasomal degradation and inhibition (D). Western blot analysis using a specific antibody was used to examine the expression of the proteins in the whole cell lysates. Band densities were determined using an image analysis system, normalized to that of GAPDH, and expressed as a percentage of the vehicle-treated control. Total RNA was extracted, and the mRNA for each gene was analyzed by real-time PCR using gene-specific primers. Results are expressed as fold changes over vehicle control. Each bar represents the mean ± SE of three independent experiments. **P < 0.01, ***P < 0.001 compared with vehicle control.
The stability of LKB1 is regulated by the interaction with hsp90, which prevents its degradation by the proteasome. Our results and those of other groups suggest that the stability of LKB1 is decreased by the hsp90 inhibitor radicicol and that cotreatment with the proteasomal inhibitor lactacystin prevents its degradation (Fig. 4D, left panel) (32). We next treated OA to HeLa cells expressing wild-type LKB1 in the absence or presence of lactacystin, and then examined intracellular levels of LKB1 by immunoblotting. These experiments revealed that the decrease in expression of LKB1 by OA was completely recovered by combination treatment with lactacystin (Fig. 4D). These data suggested that OA inhibits AMPK phosphorylation at least partly by proteasomal degradation of the upstream kinase LKB1.
OA activates SREBP-1 in rats in vivo
To test whether the data from our in vitro experiments could be reproduced in vivo, rats and mice were fed OA in the diet for seven days. Consistent with a previous study (33), rats fed OA showed increased hepatic TG levels and hepatic steatosis. Hematoxylin-eosin (HE) and Oil Red O staining of liver sections obtained from OA-treated rats clearly showed that rats were susceptible to OA-induced fatty liver (Fig. 5A). OA treatment significantly and dose-dependently increased the hepatic TG level by 2- to 4-fold compared with the control (Fig. 5B). Next we determined the expression and phosphorylation of the protein levels in the liver tissue. In line with the in vitro data shown above, the expression of AMPK and ACC were increased and the phosphorylation of the proteins were decreased, resulting in the significant decrease in the pAMPK/AMPK ratio and the pACC/ACC ratio (Fig. 5D). Similar results were obtained when we analyzed the expression and phosphorylation of Raptor in which we observed a net decrease in the pRaptor/Raptor ratio. The expression and phosphorylation of S6K1 were increased by OA treatment, leading to an increase in the pS6K1/S6K1 ratio. The level of mature SREBP-1 was increased in rat liver (Fig. 5D). Increases in protein expression, such as AMPK, ACC, and S6K, by OA in vitro and in vivo can be ascribed to the activation of mTOR, which remains to be explained in later experiments. However, little if any change was observed in mice in terms of fatty liver and phosphorylation of AMPK. Moreover, expression of mature SREBP-1 was even decreased in mouse liver (supplementary Fig. II). The AMP/ATP ratio was not changed in vitro or in vivo (supplementary Table I). To confirm that nuclear SREBP-1 is functional in vivo, we examined the mRNA levels of Lxr-α, SREBP-1, Acc, Scd-1, and Fas using real-time PCR. Increased expression of the genes was observed in the OA-fed group (Fig. 5C).
Fig.5.

Induction of fatty liver in the rats fed diet supplemented with OA. Male SD rats were fed diets containing 0, 0.5, or 1% OA for seven days. TG accumulation was assessed by HE and Oil Red O staining as well as by the determination of hepatic TG contents (A, B). LXR-α, SREBP-1, Acc, Scd-1, and Fas transcript levels were analyzed using real-time PCR (C). Western blot analysis using a specific antibody was used to examine the expression of the proteins in the rat liver homogenates. Band densities were determined using an image analysis system, normalized to that of GAPDH, and expressed as a percentage of the vehicle-treated control (D). Each bar represents the mean ± SE of the results obtained from five rats. *P < 0.05, **P < 0.01, ***P < 0.001 compared with vehicle control.
Rapamycin inhibits OA-induced fatty liver
S6K1, a member of the serine/threonine protein kinase family, is a downstream effector of the PI3K/Akt/mTOR signal transduction pathway, and its kinase activity regulates LXRα activation and subsequent lipogenic gene expression by SREBP-1 (34, 35). Inhibition of mTORC1 with rapamycin or knockdown of Raptor, an active component of mTORC1, blocks Akt-induced SREBP-1 activation and lipogenesis, indicating a close relationship between AMPK, mTORC1, and SREBP-1 activation (16). We asked whether activation of the mTOR/S6K1 pathway was linked to SREBP-1 activity, which then contributed to fat accumulation in the OA-fed rats. As expected, OA increased the phosphorylation of S6K1 and decreased the phosphorylation of Raptor (Fig. 5C). To determine the effect of rapamycin, an mTOR inhibitor, in the OA-induced fatty liver, rats were fed 1% OA and administered rapamycin for seven days. OA-induced lipid accumulation was completely inhibited by the treatment with rapamycin (Fig. 6A). HE and Oil Red O staining of liver sections clearly confirmed the results (Fig. 6B). Immunohistochemistry analyses showed significantly repressed SREBP-1 in the rapamycin-cotreatment liver (Fig. 6B). All the downstream effectors of SREBP-1, such as Lxr-α, Acc, Scd-1 and Fas, were consistently suppressed by rapamycin (Fig. 6C). These results support OA-induced fatty liver via mTOR-dependent SREBP-1 activation.
Fig.6.

Rapamycin (RPM) inhibits OA-induced lipogenesis. Male SD rats were divided into three groups, and then treated as follows for seven days: group I (control; Ctrl), normal diet + vehicle; group II (OA), 1% OA diet + vehicle; group III (OA + RPM), 1% OA diet + RPM. (A) Hepatic TG concentrations were determined using the serum triglyceride determination kit. (B) Fixed liver sections were subjected to HE and Oil Red O staining as well as immunohistochemistry assays with SREBP-1 antibody. Representative microphotographs of liver specimens from all groups of rats are shown. (C) The levels of mRNA of the indicated genes involved in lipogenesis were determined by real-time PCR. Each bar represents the mean ± SE of the results obtained from six rats. *P < 0.05, **P < 0.01, ***P < 0.001.
DISCUSSION
OA is an intermediate in pyrimidine metabolism that is readily synthesized in mammals and is also found in large quantities in cow's milk (36). Owing to the beneficial effects on the absorption of essential nutrients as well as on heart function (37), OA has been used as a dietary supplement and in vitamin and mineral complexes. OA has also been used worldwide as a tonic and for anabolic purposes. However, administration of large amounts of OA has been reported to induce fatty liver in the rat (1). The reported mechanisms of OA-induced fatty liver include impairment of fatty acid oxidation, stimulation of lipogenesis, and reduction in lipid transport from the liver (4, 5). Although liver fatty change by OA is evident and severe, the precise molecular mechanisms remain poorly understood. Additionally, the effects of OA are species specific; thus, the possible human health effects of OA have not been clearly identified (38).
Here, we describe the novel finding that the AMPK/mTOR/SREBP-1 pathway is involved in the OA-induced fatty liver. Our study demonstrated that OA inhibited the phosphorylation of AMPK and increased the activation of SREBP-1 in rat hepatocytes and human hepatoma cell lines. Activation of SREBP-1 and SREBP-regulated promoter by OA was mediated through AMPK inhibition, as verified by cotreatment with AMPK activators metformin and AICAR or by cotransfection of the constitutively active form of AMPK mutant in a SRE-luciferase assay. The decrease in the AMPK phosphorylation by OA can be ascribed, at least in part, to the proteasomal degradation of LKB1. Primary cultured mouse hepatocytes were resistant to OA treatment, as shown by the unchanged expression of pAMPK and mature SREBP-1. However, the OA responses in rat hepatocytes and human hepatoma cell lines were almost identical, indicating the possibility that humans might also be susceptible. Further experiments using the primary cultured human hepatocytes are needed to more clearly predict human health effects. Consistent with these in vitro findings, rats fed OA for seven days exhibited severe fatty liver with significantly reduced phosphorylation of hepatic AMPK and increased activation of SREBP-1. In contrast, cotreatment with rapamycin inhibited OA responses in the liver in terms of nuclear SREBP-1 activation and consequent hepatic TG accumulation. Figs. 1D and 5D show the increased expression of protein levels (including AMPK, ACC, Raptor, S6K1, and SREBP-1) which is supposed to be ascribed to mTOR activation by OA. mTOR regulates protein synthesis and cell growth by phosphorylating S6K1 and eukaryotic initiation factor 4E-binding protein 1 (4EBP1). Phosphorylation of S6K1 facilitates the recruitment and translation of a specific mRNA subset that contains a 5′ polypyrimidine tract (39). Phosphorylation of 4EBP1 releases eIF4E, which initiates translation (40).
The present study highlights the involvement of the AMPK/mTOR/SREBP-1 axis in OA-induced fatty liver. AMPK negatively regulates mTOR complex 1 (mTORC1), a central controller of cell growth, protein synthesis, mitochondrial biogenesis, and lipogenesis. Phosphorylation of AMPK by upstream kinase induces fatty acid oxidation and inhibits lipogenesis in the liver through suppression of SREBP-1 (30). Inhibition of mTORC1 with rapamycin or knockdown of Raptor, an active component of mTORC1, blocks Akt-induced SREBP-1 activation and lipogenesis, indicating a close relationship between AMPK, mTORC1, and SREBP-1 activation (16). The AMPK/mTOR/SREBP-1 axis has recently been shown to be a key molecular target in chemical- and diet-induced fatty liver. You et al. demonstrated that inhibition of AMPK plays a key role in ethanol-induced fatty liver by activating hepatic lipogenesis controlled by SREBP-1 and by suppressing hepatic free fatty acid oxidation (17). Vinciguerra et al. reported that high circulating free fatty acids, such as oleic acid, activates mTOR that is responsible for downregulation of PTEN, activation of Akt, and steatosis (41). Palmitate activates the mTOR/S6K pathway and induces insulin resistance in primary cultured rat hepatocytes (42). Impairment of lipid metabolism through the modulation of AMPK activity was also reported when hepatocytes were treated with side-stream cigarette smoke (18). They showed that second-hand cigarette smoke stimulates lipid accumulation in the liver and that this effect is mediated by AMPK and SREBP-1. Furthermore, they showed that inactivation of AMPK stimulates activation of SREBP-1 and leads to an increase in lipid accumulation. In a separate experiment, Syed et al. found that exposure of cigarette smoke condensate to human bronchial epithelial cells resulted in increased phosphorylation of mTOR, which is required for the insulin-stimulated induction of SREBP-1 expression (43). Note that the in vitro effects of cigarette smoking were recently verified in a cross-sectional study of 8,442 subjects. The prevalence rate of nonalcoholic fatty liver disease was significantly higher among cigarette smokers versus nonsmokers. Moreover, the prevalence ratio increases with the amount of cigarettes smoked per day (44). Although the precise effects of OA in humans are unknown, we must not exclude the possibility that OA consumption by diet and dietary supplements can induce fatty liver in humans. A quantitative risk assessment is needed to address the issue.
One of the novel findings in the present study is the identification of a molecular target that responds differently between the rat and mouse. Previously, the likely cause of the interspecies differences in OA hepatotoxicity has been thought to be metabolic differences. Mice accumulated less of the OA dose in the liver and excreted much more in the urine than did rats, thereby rendering them resistant to OA toxicity (6). Feeding an arginine-deficient diet to rats leads to a marked fatty liver by an elevated plasma concentration of OA. Additionally, mice, hamsters, and rabbits were resistant to fatty liver largely because of their high urinary excretion rate of OA (45). Apart from this pharmacokinetic issue, no other information has been available concerning species-specific mechanisms of OA hepatotoxicity. Our results showed that the most striking difference between the rat and mouse hepatocytes in primary culture is AMPK phosphorylation and SREBP-1 activation in response to OA exposure. Whereas decreased phosphorylation of AMPK and modulation of a series of downstream events leading to fatty acid synthesis and lipogenesis were observed in rat hepatocytes and human hepatoma cell lines, mouse hepatocytes were resistant to OA with regard to the AMPK/SREBP-1-dependent lipogenic pathway. Similar results were observed in animal studies using SD rats and C57BL/6 mice. These two species showed completely different responses to OA in terms of AMPK/SREBP-1 signaling and development of steatosis, indicating that not only pharmacokinetic but also pharmacodynamic factors participate in determining interspecies differences. Considering the very similar responses of human hepatoma cell lines and rat hepatocytes to the AMPK/SREBP-1 pathway, humans could be a species that may be sensitive to OA-induced fatty liver. Further experiments using primary cultured human hepatocytes and comprehensive human pharmacokinetic studies will be needed to define human health effects. The effects of OA could be even greater in obese and insulin-resistant diabetic patients if we consider the poor sensitivity of these patients to insulin-induced downstream effectors, including AMPK and SREBP-1. Regular ethanol consumption and cigarette smoke may also synergistically affect OA-induced fatty liver.
In summary, we demonstrated that OA induces the SREBP-1-induced lipogenesis pathway through mTOR-p70S6K1 activation and that reduced AMPK phosphorylation contributed to the activation of S6K1 phosphorylation. Moreover, we discovered that phosphorylation of AMPK is partially decreased by OA-induced LKB1 proteasomal degradation (Fig. 7). Our study provides some critical information needed for an understanding of the molecular mechanisms of OA-induced fatty liver and of species differences in susceptibility. OA induces lipogenesis, mediated predominantly by the AMPK/mTOR/SREBP-1 pathway in rat hepatocytes and human hepatoma cell lines.
Fig.7.
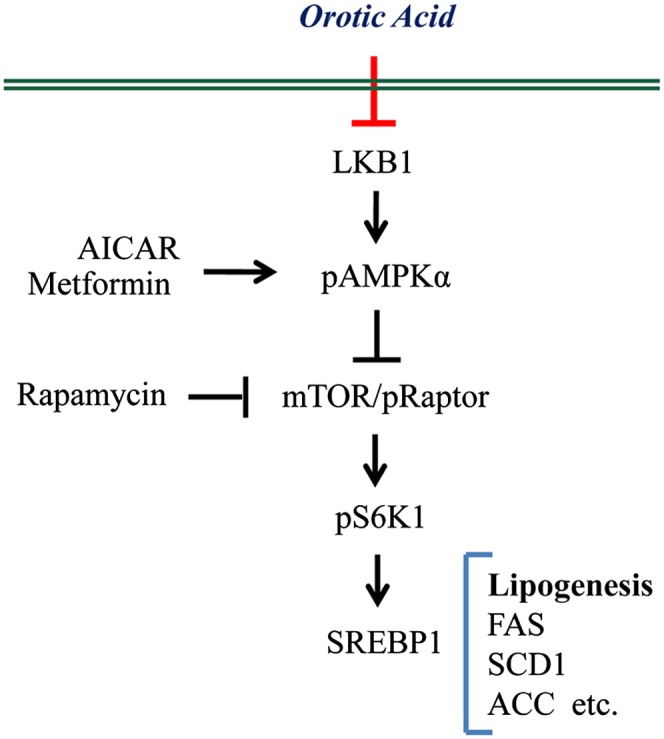
Proposed mechanism of OA-induced lipogenesis by the AMPK/SREBP-1 pathway.
Supplementary Material
Footnotes
- ACC
- acetyl-CoA carboxylase
- AICAR
- aminoimidazole carboxamide ribonucleotide
- AMPK
- adenosine monophosphate-activated protein kinase
- CA-AMPK
- constitutively active AMPK
- DN-AMPK
- dominant-negative AMPK
- HE
- hematoxylin-eosin
- LKB1(STK11)
- serine/threonine kinase 11
- mTOR
- mammalian target of rapamycin
- OA
- orotic acid
- PRPP
- phosphoribosyl pyrophosphate
- SCD1
- stearoyl-CoA desaturase-1
- SREBP
- sterol regulatory element-binding protein
- TG
- triglyceride
This work was supported by the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare & Family Affairs (Grant A100096); and by the National Research Foundation, Ministry of Education, Science and Technology (Grant 20100001706).
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of two figures and one tables.
REFERENCES
- 1.Standerfer S. B., Handler P. 1955. Fatty liver induced by orotic acid feeding. Proc. Soc. Exp. Biol. Med. 90: 270–271. [DOI] [PubMed] [Google Scholar]
- 2.Ferreira A. V., Parreira G. G., Porto L. C., Mario E. G., Delpuerto H. L., Martins A. S., Botion L. M. 2008. Fenofibrate prevents orotic acid-induced hepatic steatosis in rats. Life Sci. 82: 876–883. [DOI] [PubMed] [Google Scholar]
- 3.Buang Y., Wang Y. M., Cha J. Y., Nagao K., Yanagita T. 2005. Dietary phosphatidylcholine alleviates fatty liver induced by orotic acid. Nutrition. 21: 867–873. [DOI] [PubMed] [Google Scholar]
- 4.Miyazawa S., Furuta S., Hashimoto T. 1982. Reduction of beta-oxidation capacity of rat liver mitochondria by feeding orotic acid. Biochim. Biophys. Acta. 711: 494–502. [PubMed] [Google Scholar]
- 5.Hebbachi A. M., Seelaender M. C., Baker B. W., Gibbons G. F. 1997. Decreased secretion of very-low-density lipoprotein triacylglycerol and apolipoprotein B is associated with decreased intracellular triacylglycerol lipolysis in hepatocytes derived from rats fed orotic acid or n-3 fatty acids. Biochem. J. 325: 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durschlag R. P., Robinson J. L. 1980. Species specificity in the metabolic consequences of orotic acid consumption. J. Nutr. 110: 822–828. [DOI] [PubMed] [Google Scholar]
- 7.Hawley S. A., Davison M., Woods A., Davies S. P., Beri R. K., Carling D., Hardie D. G. 1996. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 271: 27879–27887. [DOI] [PubMed] [Google Scholar]
- 8.Hardie D. G., Corton J., Ching Y. P., Davies S. P., Hawley S. 1997. Regulation of lipid metabolism by the AMP-activated protein kinase. Biochem. Soc. Trans. 25: 1229–1231. [DOI] [PubMed] [Google Scholar]
- 9.Kawanaka K., Nolte L. A., Han D. H., Hansen P. A., Holloszy J. O. 2000. Mechanisms underlying impaired GLUT-4 translocation in glycogen-supercompensated muscles of exercised rats. Am. J. Physiol. Endocrinol. Metab. 279: E1311–E1318. [DOI] [PubMed] [Google Scholar]
- 10.Taylor E. B., Ellingson W. J., Lamb J. D., Chesser D. G., Winder W. W. 2005. Long-chain acyl-CoA esters inhibit phosphorylation of AMP-activated protein kinase at threonine-172 by LKB1/STRAD/MO25. Am. J. Physiol. Endocrinol. Metab. 288: E1055–E1061. [DOI] [PubMed] [Google Scholar]
- 11.Rafaeloff-Phail R., Ding L., Conner L., Yeh W. K., McClure D., Guo H., Emerson K., Brooks H. 2004. Biochemical regulation of mammalian AMP-activated protein kinase activity by NAD and NADH. J. Biol. Chem. 279: 52934–52939. [DOI] [PubMed] [Google Scholar]
- 12.Fryer L. G. D., Parbu-Patel A., Carling D. 2002. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 277: 25226–25232. [DOI] [PubMed] [Google Scholar]
- 13.Minokoshi Y., Kim Y. B., Peroni O. D., Fryer L. G., Müller C., Carling D., Kahn B. B. 2002. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 415: 339–343. [DOI] [PubMed] [Google Scholar]
- 14.Hawley S. A., Boudeau J., Reid J. L., Mustard K. J., Udd L., Mäkelä T. P., Alessi D. R., Hardie D. G. 2003. Complexes between the LKB1 tumor suppressor, STRADalpha/beta and MO25alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2: 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Branvold D. J., Allred D. R., Beckstead D. J., Kim H. J., Fillmore N., Condon B. M., Brown J. D., Sudweeks S. N., Thomson D. M., Winder W. W. 2008. Thyroid hormone effects on LKB1, MO25, phospho-AMPK, phospho-CREB, and PGC-1α in rat muscle. J. Appl. Physiol. 105: 1218–1227. [DOI] [PubMed] [Google Scholar]
- 16.Porstmann T., Santos C. R., Griffiths B., Cully M., Wu M., Leevers S., Griffiths J. R., Chung Y. L., Schulze A. 2008. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8: 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.You M., Matsumoto M., Pacold C. M., Cho W. K., Crabb D. W. 2004. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 127: 1798–1808. [DOI] [PubMed] [Google Scholar]
- 18.Yuan H., Shy J. Y. J., Martins-Green M. 2009. Second-hand smoke stimulates lipid accumulation in the liver by modulating AMPK and SREBP-1. J. Hepatol. 51: 535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y., Song P., Xu J., Zhang M., Zou M. H. 2007. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J. Biol. Chem. 282: 9777–9788. [DOI] [PubMed] [Google Scholar]
- 20.Lee W. J., Kim M., Park H. S., Kim H. S., Jeon M. J., Oh K. S., Koh E. H., Won J. C., Kim M. S., Oh G. T., et al. 2006. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARα and PGC-1. Biochem. Biophys. Res. Commun. 340: 291–295. [DOI] [PubMed] [Google Scholar]
- 21.Kelley W. N., Greene M. L., Fox I. H., Rosenbloom F. M., Levy R. I., Seegmiller J. E. 1970. Effects of orotic acid on purine and lipoprotein metabolism in man. Metabolism. 19: 1025–1035. [DOI] [PubMed] [Google Scholar]
- 22.Griffin J. L., Bonney S. A., Mann C., Hebbachi A. M., Gibbons G. F., Nicholson J. K., Shoulders C. C., Scott J. 2004. An integrated reverse functional genomic and metabolic approach to understanding orotic acid-induced fatty liver. Physiol. Genomics. 17: 140–149. [DOI] [PubMed] [Google Scholar]
- 23.French C. T., Hanneman W. H., Chubb L. S., Billings R. E., Andersen M. E. 2004. Induction of CYP1A1 in primary rat hepatocytes by 3,3′,4,4’,5-pentachlorobiphenyl: evidence for a switch circuit element. Toxicol. Sci. 78: 276–286. [DOI] [PubMed] [Google Scholar]
- 24.Honkakoski P., Negishi M. 1998. Protein serine/threonine phosphatase inhibitors suppress phenobarbital-induced Cyp2b10 gene transcription in mouse primary hepatocytes. Biochem. J. 330: 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wynants J., Van Belle H. Single-run high-performance liquid chromatography of nucleotides, nucleosides, and major purine bases and its application to different tissue extracts. 1985. Anal. Biochem. 144: 258–266. [DOI] [PubMed] [Google Scholar]
- 26.Wang L. H., Choi Y. L., Hua X. Y., Shin Y. K., Song Y. J., Youn S. J., Yun H. Y., Park S. M., Kim W. J., Kim H. J., et al. 2006. Increased expression of sonic hedgehog and altered methylation of its promoter region in gastric cancer and its related lesions. Mod. Pathol. 19: 675–683. [DOI] [PubMed] [Google Scholar]
- 27.Bruce C. R., Anderson M. J., Carey A. L., Newman D. G., Bonen A., Kriketos A. D., Cooney G. J., Hawley J. A. 2003. Muscle oxidative capacity is a better predictor of insulin sensitivity than lipid status. J. Clin. Endocrinol. Metab. 88: 5444–5451. [DOI] [PubMed] [Google Scholar]
- 28.You M., Fischer M., Deg M. A., Crabb D. W. 2002. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J. Biol. Chem. 277: 29342–29347. [DOI] [PubMed] [Google Scholar]
- 29.Stein S. C., Woods A., Jones N. A., Davison M. D., Carling D. 2000. The regulation of AMP-activated protein kinase by phosphorylation. Biochem. J. 345: 437–443. [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., et al. 2001. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108: 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shklyaev S., Aslanidi G., Tennant M., Prima V., Kohlbrenner E., Kroutov V., Campbell-Thompson M., Crawford J., Shek E. W., Scarpace P. J., et al. 2003. Sustained peripheral expression of transgene adiponectin offsets the development of diet-induced obesity in rats. Proc. Natl. Acad. Sci. USA. 100: 14217–14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boudeau J., Deak M., Lawlor M. A., Morrice N. A., Alessi D. R. 2003. Heat-shock protein 90 and Cdc37 interact with LKB1 and regulate its stability. Biochem. J. 370(Pt. 3): 849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Creasey W. A., Hankin L., Handschumacher R. E. 1961. Fatty livers induced by orotic acid. J. Biol. Chem. 236: 2064–2070. [PubMed] [Google Scholar]
- 34.Brown E. J., Beal P. A., Keith C. T., Chen J., Shin T. B., Schreiber S. L. 1995. Control of p70S6 kinase by kinase activity of FRAP in vivo. Nature. 377: 441–446. [DOI] [PubMed] [Google Scholar]
- 35.Hwahng S. H., Ki S. H., Bae E. J., Kim H. E., Kim S. G. 2009. Role of adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway in repression of liver X receptor-alpha-dependent lipogenic gene induction and hepatic steatosis by a novel class of dithiolethiones. Hepatology. 49: 1913–1925. [DOI] [PubMed] [Google Scholar]
- 36.Hurlbert R. B., Potter V. R. 1954. Nucleotide metabolism. J. Biol. Chem. 209: 1–21. [PubMed] [Google Scholar]
- 37.Richards S. M., Conyers R. A. J., Fisher J. L., Rosenfeldt F. L. 1997. Cardioprotection by orotic acid: metabolism and mechanism of action. J. Mol. Cell. Cardiol. 29: 3239–3250. [DOI] [PubMed] [Google Scholar]
- 38.Robinson J. L. 1980. Bovine milk orotic acid: variability and significance for human nutrition. J. Dairy Sci. 63: 865–871. [Google Scholar]
- 39.Dufner A., Thomas G. 1999. Ribosomal S6 kinase signaling and the control of translation. Exp. Cell Res. 253: 100–109. [DOI] [PubMed] [Google Scholar]
- 40.Gingras A. C., Raught B., Sonenberg N. 2004. mTOR signaling to translation. Curr. Top. Microbiol. Immunol. 279: 169–197. [DOI] [PubMed] [Google Scholar]
- 41.Vinciguerra M., Veyrat-Durebex C., Moukil M. A., Rubbia-Brandt L., Rohner-Jeanrenaud F., Foti M. 2008. PTEN down-regulation by unsaturated fatty acids triggers hepatic steatosis via an NF-kappaBp65/mTOR-dependent mechanism. Gastroenterology. 134: 268–280. [DOI] [PubMed] [Google Scholar]
- 42.Mordier S., Iynedjian P. B. 2007. Activation of mammalian target of rapamycin complex 1 and insulin resistance induced by palmitate in hepatocytes. Biochem. Biophys. Res. Commun. 362: 206–211. [DOI] [PubMed] [Google Scholar]
- 43.Syed D. N., Afaq F., Kweon M. H., Hadi N., Bhatia N., Spiegelman V. S., Mukhtar H. 2007. Green tea polyphenol EGCG suppresses cigarette smoke condensate-induced NF-kappaB activation in normal human bronchial epithelial cells. Oncogene. 26: 673–682. [DOI] [PubMed] [Google Scholar]
- 44.Xu C. F., Yu C. H., Xu L., Miao M., Li Y. M. 2010. Is cigarette smoking an independent risk factor or a cofactor for nonalcoholic fatty liver disease? Hepatology. 52: 803–804. [DOI] [PubMed] [Google Scholar]
- 45.Milner J. A., Hassan A. S. 1981. Species specificity of arginine deficiency-induced hepatic steatosis. J. Nutr. 111: 1067–1073. [DOI] [PubMed] [Google Scholar]
- 46.Harrison W. J., Bull J. J., Seltmann H., Zouboulis C. C., Philpott M. P. 2007. Expression of lipogenic factors galectin-12, resistin, SREBP-1, and SCD in human sebaceous glands and cultured sebocytes. J. Invest. Dermatol. 127: 1309–1317. [DOI] [PubMed] [Google Scholar]
- 47.Sekiya M., Hiraishi A., Touyama M., Sakamoto K. 2008. Oxidative stress induced lipid accumulation via SREBP1c activation in HepG2 cells. Biochem. Biophys. Res. Commun. 375: 602–607. [DOI] [PubMed] [Google Scholar]
- 48.Howell G., 3rd, Deng X., Yellaturu C., Park E. A., Wilcox H. G., Raghow R., Elam M. B. 2009. N-3 polyunsaturated fatty acids suppress insulin-induced SREBP-1c transcription via reduced trans-activating capacity of LXRα. Biochim. Biophys. Acta. 1791: 1190–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.