

Abstract
Autosomal Dominant Hypercholesterolemia (ADH), characterized by isolated elevation of plasmatic LDL cholesterol and premature cardiovascular complications, is associated with mutations in 3 major genes: LDLR (LDL receptor), APOB (apolipoprotein B) and PCSK9 (proprotein convertase subtilisin-kexin type 9). Through the French ADH Research Network, we collected molecular data from 1358 French probands from eleven different regions in France. Mutations in the LDLR gene were identified in 1003 subjects representing 391 unique events with 46.0% missense, 14.6% frameshift, 13.6% splice, and 11.3% nonsense mutations, 9.7% major rearrangements, 3.8% small in frame deletions/insertions, and 1.0% UTR mutations. Interestingly, 175 are novel mutational events and represent 45% of the unique events we identified, highlighting a specificity of the LDLR mutation spectrum in France. Furthermore, mutations in the APOB gene were identified in 89 probands and in the PCSK9 gene in 10 probands. Comparison of available clinical and biochemical data showed a gradient of severity for ADH-causing mutations: FH=PCSK9>FDB>‘Others’ genes. The respective contribution of each known gene to ADH in this French cohort is: LDLR 73.9%, APOB 6.6%, PCSK9 0.7%. Finally, in 19.0% of the probands, no mutation was found, thus underscoring the existence of ADH mutations located in still unknown genes. © 2010 Wiley-Liss, Inc.
Keywords: Autosomal Dominant Hypercholesterolemia, Mutation screening, French population, Genotype/phenotype correlation
INTRODUCTION
Hypercholesterolemia is a major risk factor for atherosclerosis and its premature cardiovascular complications. Hypercholesterolemia can be multifactorial or less frequently monogenic, leading to Autosomal Dominant Hypercholesterolemia (ADH; MIM# 143890) characterized by an elevation of plasmatic LDL cholesterol levels and xanthoma, xanthelasma, arcus corneae or premature coronary heart disease. The diagnosis of ADH is difficult, due to the overlap of cholesterol values between monogenic and multifactorial forms. DNA testing provides an unequivocal diagnosis and allows the identification of affected relatives at an early age when they can be offered lifestyle advice and appropriate lipid-lowering therapies (Humphries et al. 2008).
The first ADH causative gene identified was LDLR encoding the LDL receptor (Goldstein et al. 1973). This disease was named FH for Familial Hypercholesterolemia (MIM# 606945) and its heterozygous prevalence was estimated at 1/500. To date, over 1000 mutations in LDLR have been implicated in ADH (Villéger et al. 2002; Leigh et al. 2008). Subsequently, a second gene was involved after the discovery of hypercholesterolemic patients with normal LDL receptor activity (Innerarity et al. 1987). They carried a missense mutation (p.Arg3527Gln previously named p.Arg3500Gln) in APOB, encoding apolipoprotein B, the main ligand for the LDL receptor (Soria et al. 1989). This new molecular disorder was called FDB for Familial Defective apolipoprotein B-100 (MIM# 144010) and its frequency has been estimated at 1/250 in Switzerland and 1/1250 in Northern Europe and the US (Rabès et al. 2000). Subsequently, we identified a third ADH-causative gene: proprotein convertase subtilisin-kexin type 9 (PCSK9; MIM# 607787) (Abifadel et al. 2003). PCSK9 has been shown to degrade LDL receptor independently of its catalytic activity (McNutt et al. 2007). Very recently, we mapped a fourth major locus for ADH at 16q22.1 that we named HCHOLA4 (Marques-Pinheiro et al. 2010). Finally, the proportion of ADH patients for whom the disease is not explained by a mutation in, either, LDLR, APOB, or PCSK9 was estimated at 15.25 % (Varret et al. 2008). The aim of this study was to assess the molecular epidemiology of ADH in a representative French population.
MATERIALS AND METHODS
Proband and family recruitment
ADH probands and families were recruited by the French National Research Network on Hypercholesterolemia that includes numerous clinicians from different cities in France. Since 2005, they selected probands meeting the following inclusion criteria: total and LDL-cholesterol levels above the 95 th percentile when compared with a sex-and age-matched French population (STANISLAS cohort, B. Herbeth, G. Siest & S Visvikis-Siest, personal communication; Siest et al. 1998), autosomal dominant transmission of hypercholesterolemia in the family. Venous blood samples were sent to 3 genetic laboratories certified for ADH molecular diagnosis (A.S., A.C. & JP.R.) where DNA was extracted. The number of probands included (1358) and the diversity of their geographical origin (11 different French regions), constitute a representative sample of the French population. The study was performed in accordance with French bioethics regulations and all subjects gave informed consent.
Candidate gene analysis
The APOB-p.Arg3527Gln mutation was detected as previously described (Rabès et al. 1997) or by sequencing (NM_000384.2). The promoters, the 18 exons of LDLR (NM_000527.3), and the 12 exons of PCSK9 (NM_174936.3), as well as close flanking intronic sequences, were amplified. Primer sequences and annealing temperatures are available on request. Electrophoregrams were analyzed using Gensearch®, a DNA sequence analysis software developed by PhenoSystems SA, Belgium (http://www.phenosystems.com). Detection of deletions/duplications of one or more exons of LDLR was performed with SALSA MLPA kit (P062) and data were analyzed with Coffalyser software (MRC-Holland). In all subjects, genes were studied sequentially: at first, the APOB-p.Arg3527Gln mutation was looked for and the LDLR gene was sequenced. If no mutation was found, the search for a deletion/duplication of LDLR was performed. Finally, if no deletion/duplication was discovered, the PCSK9 gene was sequenced.
Nomenclature
All existing and new mutations were described following the recommendations of the Human Genome Variation Society at http://www.hgvs.org/mutnomen. Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence. Furthermore, amino acid variants now follow the standard nomenclature with the initiating methionine given as number one, rather than the historical numbering from the first residue of the mature peptide. Hence, 21 or 27 has been added to all original amino acid numbering for LDL receptor or apo B, respectively. Variants in the 5′ untranslated region are now numbered from the nucleotide immediately preceding the A of the initiating methionine.
In silico prediction of effect of molecular event on LDL receptor
The causal effect of each new molecular event was estimated with in silico prediction of protein function using the following tools: NetGene2 (http://www.cbs.dtu.dk/services/NetGene2), NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html), Polyphen (http://genetics.bwh.harvard.edu/pph), SIFT (http://sift.jcvi.org), Pmut (http://mmb2.pcb.ub.es:8080/PMut) and SNP3D (http://www.snps3d.org). The reference sequences used for LDLR were P01130.1 (SwissProt) orNP_000518.1 (NCBI RefSeq).
Statistical analysis
When possible, we collected clinical and/or biochemical data under fasting conditions and without any cholesterol lowering drug. Plasma levels of total-, LDL-, HDL-cholesterol, triglycerides, and clinical signs of the disease were not available for all probands, thus sample size is different among each lipid parameter as presented in Supp. Figure S1. Lipid levels were expressed as multiples of median (MoM) for age and gender of a reference French population: the STANISLAS cohort. Comparison of quantitative values (lipid levels and age) was performed by the Mann-Whitney test with Graph Pad Prism 5.03 software. Results are presented with the median and range from minimum to maximum MoM values. Comparison of qualitative values was performed with the Chi-Square Test (or Fisher Test for N<5) online with the StatPages at http://www.statpages.org.
RESULTS AND DISCUSSION
Through the ADH French Research Network, we collected molecular data from 1358 French ADH probands and found 1111 molecular events: 1012 (91.1%) LDLR mutations in 1003 (73.9%) probands, 9 with two LDLR variants each; 89 (8.0%) APOB-p.Arg3527Gln mutation in 89 (6.6%) probands including 2 probands also heterozygous for a LDLR mutation; 10 (0.9%) PCSK9 mutations in 10 (0.7%) probands. For the 258 remaining probands (19.0%), no mutation was identified in the three major ADH genes.
Variations in LDLR
Variations in LDLR were identified in 1003 probands representing 391 unique events distributed as follows: 46.0% missense; 11.3% nonsense; 14.6% frameshift; 3.8% small in frame deletions, insertions, or indels; 13.6% splice; 1.0% in 5′UTR; and 9.7% large deletions or duplications (complete list available on request). In accordance with the known heterogeneity of the French population, this distribution was similar to that reported worldwide (Leigh et al. 2008) (Supp. Table S1). However, splice mutations were significantly more abundant in this French cohort (p=0.002), probably indicating a technological bias. Indeed, systematic sequencing of intronic sequences is a more recent practice (Amsellem et al. 2002).
Within the 283 variations newly reported in this French population, 175 were novel mutational events (Tables 1, 2, and 3) and represent 45% (175/391) of the unique events we identified and 22% (222/1003) of probands with a variation in LDLR (1 with two new LDLR variants). Furthermore, LDLR mutational events newly reported in France represent 72% (283/391) of the unique events reported here and 41% (416/1003) of LDLR variation carriers. This highlights a higher level of allelic heterogeneity for LDLR and indicates a specificity of the spectrum of LDLR mutations in France when compared to other countries. Another method for genetic diagnosis of ADH is based on a DNA-array platform that is able to detect 242 different point mutations in LDLR and 3 in APOB (Lipochip version 8, http://www.progenika.com). The Lipochip used to screen clinically diagnosed FH patients in Spain was able to detect mutations in 78% of all carriers (Alonso R et al. 2009). If the Lipochip (version 8) had been used to screen this French cohort, it only would have detected 40% of the mutation carriers, thus indicating the need for specific national screening strategies.
Table 1.
New mutational events leading to abnormal protein size
| Location | cDNA (HGVS) | Protein (HGVS) | Predicted protein | Number of probands | Familial segregation (*) |
|---|---|---|---|---|---|
| nonsense | |||||
| exon 2 | c.102C>A | p.Cys34X | 33 AA, truncated or no protein | 1 | na |
| exon 4 | c.535G>T | p.Glu179X | 178 AA, truncated or no protein | 1 | na |
| exon 4 | c.539G>A | p.Trp180X | 179 AA, truncated or no protein | 3 | na |
| exon 10 | c.1532T>G | p.Leu511X | 510 AA, truncated or no protein | 1 | na |
| exon 11 | c.1598G>A | p.Trp533X | 532 AA, truncated or no protein | 1 | na |
| exon 11 | c.1685G>A | p.Trp562X | 561 AA, truncated or no protein | 1 | yes (3 - 1) |
| exon 13 | c.1860G>A | p.Trp620X | 619 AA, truncated or no protein | 1 | yes (2 - 1) |
| exon 14 | c.1997G>A | p.Trp666X | 665 AA, truncated or no protein | 1 | na |
| exon 17 | c.2446A>T | p.Lys816X | 815 AA, truncated or no protein | 1 | na |
| frameshifts | |||||
| exon 3 | c.244del | p.Cys82AlafsX124 | 81 AA with 124 novel AA, truncated or no protein | 1 | na |
| exon 4 | c.350_372dup | p.Gln125ThrfsX89 | 124 AA with 89 novel AA, truncated or no protein | 1 | na |
| exon 4 | c.357del | p.Lys120SerfsX86 | 119 AA with 86 novel AA, truncated or no protein | 1 | na |
| exon 4 | c.374_375insCTGA | p.Gln125HisfsX2 | 124 AA with 2 novel AA, truncated or no protein | 1 | yes (2 - 2) |
| exon 4 | c.450dup | p.Ala151ArgfsX29 | 150 AA with 29 novel AA, truncated or no protein | 1 | na |
| exon 4 | c.482_488del | p.Ile161SerfsX43 | 160 AA with 43 novel AA, truncated or no protein | 1 | na |
| exon 4 | c.609del | p.Cys204AlafsX2 | 203 AA with 2 novel AA, truncated or no protein | 1 | na |
| exon 4 | c.664_681delinsCCGACTG | p.Cys222ProfsX14 | 221 AAwith 14 novel AA, truncated or no protein | 1 | na |
| exon 4 | c.666_687del | p.Cys222X | 221 AA, truncated or no protein | 1 | na |
| exon 4 | c.673_682delinsTGCAA | p.Lys225CysfsX13 | 224 AAwith 13 novel AA, truncated or no protein | 2 | na |
| exon 4 | c.681_682insTGAG | p.Glu228X | 227 AA, truncated or no protein | 1 | na |
| exon 4 | c.682del | p.Glu228ArgfsX37 | 227 AA with 37 novel AA, truncated or no protein | 2 | yes (3 - 1) |
| exon 5 | c.752dup | p.Ser252GlnfsX5 | 251 AA with 5 novel AA, truncated or no protein | 1 | yes (2 - 1) |
| exon 5 | c.781del | p.Cys261AlafsX4 | 260 AA with 4 novel AA, truncated or no protein | 1 | na |
| exon 6 | c.865del | p.Cys289AlafsX81 | 288 AA with 81 novel AA, truncated or no protein | 1 | na |
| exon 6 | c.875dup | p.Asp293GlyfsX8 | 292 AA with 8 novel AA, truncated or no protein | 2 | na |
| exon 7 | c.951del | p.Glu317AspfsX53 | 316 AA with 53 novel AA, truncated or no protein | 1 | na |
| exon 7 | c.1008del | p.Tyr336X | 335 AA, truncated or no protein | 3 | na |
| exon 7 | c. 1031 del | p.Phe344SerfsX26 | 343 AA with 26 novel AA, truncated or no protein | 3 | na |
| exon 7 | c.1042del | p.Ala348ProfsX22 | 347 AA with 22 novel AA, truncated or no protein | 1 | na |
| exon 8 | c.1099_1104delinsGT | p.Leu367ValfsX2 | 366 AA with 2 novel AA, truncated or no protein | 1 | yes (2 - 0) |
| exon 9 | c.1343del | p.Gln448ArgfsX3 | 447 AA with 3 novel AA, truncated or no protein | 1 | na |
| exon 10 | c.1496_1497del | p.Ser499CysfsX36 | 498 AA with 36 novel AA, truncated or no protein | 1 | na |
| exon 10 | c.1549_1555del | p.Ser517GlnfsX29 | 516 AA with 29 novel AA, truncated or no protein | 1 | na |
| exon 11 | c.1610del | p.Gly537GlufsX11 | 536 AA with 11 novel AA, truncated or no protein | 1 | na |
| exon 11 | c.1632del | p.Gly546AlafsX2 | 545 AA with 2 novel AA, truncated or no protein | 2 | yes (3 - 0) |
| exon 12 | c.1718del | p.Gly573AlafsX92 | 572 AA with 92 novel AA, truncated or no protein | 1 | na |
| exon 12 | c.1737del | p.Ser580ProfsX85 | 579 AA with 85 novel AA, truncated or no protein | 4 | na |
| exon 12 | c.1749_1753del | p.Ser584LeufsX17 | 583 AA with 17 novel AA, truncated or no protein | 1 | na |
| exon 13 | c.1886del | p.Phe629SerfsX36 | 628 AA with 36 novel AA, truncated or no protein | 1 | na |
| exon 13 | c.1934dup | p.Asn645LysfsX24 | 644 AA with 24 novel AA, truncated or no protein | 1 | na |
| exon 13 | c.1948_1952dup | p.Asp651GlufsX16 | 650 AA with 16 novel AA, truncated or no protein | 1 | yes (3 - 0) |
| exon 13 | c.1961_1965dup | p.His656SerfsX11 | 655 AA with 11 novel AA, truncated or no protein | 1 | na |
| exon 14 | c.2013_2014del | p.Leu672GlufsX44 | 671 AA with 44 novel AA, truncated or no protein | 1 | na |
| exon 14 | c.2030_2042del | p.Cys677SerfsX28 | 676 AA with 28 novel AA, truncated or no protein | 1 | na |
| exon 15 | c.2187_2197del | p.Lys730HisfsX48 | 729 AA with 48 novel AA, truncated or no protein | 1 | na |
| exon 15 | c.2230del | p.Arg744AspfsX21 | 743 AA with 21 novel AA, truncated or no protein | 1 | yes (2 - 0) |
| exon 16 | c.2318del | p.Gly773AlafsX15 | 772 AA with 15 novel AA, truncated or no protein | 1 | na |
| exon 17 | c.2403_2406del | p.Leu802AlafsX126 | 801 AA with 126 novel AA, truncated or no protein | 1 | na |
| exon 17 | c.2416del | p.Val806SerfsX123 | 805 AA with 123 novel AA, truncated or no protein | 2 | na |
| exon 17 | c.2509del | p.His837ThrfsX92 | 836 AA with 92 novel AA, truncated or no protein | 1 | na |
| major rearangements | MLPA results | Predicted protein if recombinaison between Alu not affecting splice sites | |||
| Prom | c.1-?_1060+?del | del from prom. to exon 7 | no in phase ATG within exon 8, no protein | 1 | yes (2 - 2) |
| Prom | c.1-?_3428+?del | del from prom. to exon 18 | no protein | 1 | yes (2 - 2) |
| exon 1 | c.1-?_67+?del | del of exon 1 | no in phase ATG within exon 2, no protein | 2** | na |
| exon 1 | c.1-?_3428+?del | del of exons 1 to 18 | no protein | 1 | na |
| exon 2 | c.68-?_817+?dup dup of exons 2 to | 5 | p.Gly24Val273 dup, elongated protein (249 AA) | 2** | na |
| exon 2 | c.68-?_1586+?del | del of exons 2 to 10 | p.Val23AlafsX19, truncated protein | 1 | na |
| exon 2 | c.68-?_1705+?del | del of exons 2 to 11 | p.Val23Asp;Gly24_Asp569del, shortened protein (545 AA) | 2** | na |
| exon 2 | c.68-?_2140+?del | del of exons 2 to 14 | p.Val23Glu;Gly24_Glu714del, shortened protein (690 AA) | 1 | na |
| exon 2 | c.68-?_2547+?del | del of exons 2 to 17 | p.Val23GlufsX9, truncated protein | 1 | na |
| exon 3 | c.191-?_313+?del | del of exon 3 | p.Leu64Ser;Ser65_Pro105del, shortened protein (40 AA) | 1 | na |
| exon 3 | c.191-?_694+?del | del of exons 3 and 4 | p.Leu64Ser;Ser65_Ala232del, shortened protein (167 AA) | 1 | na |
| exon 4 | c.314-?_940+?dup | dup of exons 4 to 6 | p.Gly314Ala;Pro106_Cys313 dup, elongated protein (207 AA) | 1 | yes (3 - 1) |
| exon 5 | c. 695-?_1586+?del | del of exons 5 to 10 | p.Ala233ValfsX18, truncated protein | 1 | na |
| exon 9 | c.1187-?_3428+?del | del of exons 9 to 18 | no protein | 2** | na |
| exon 11 | c.1586-?_1705+?del | del of exon 11 | p.Phe530SerfsX10, truncated protein | 2** | yes (3 - 3), na |
| exon 12 | c.1706-?_1845+?dup | dup of exon 12 | p.Asp616Ile fsX96, truncated protein | 1 | na |
| exon 12 | c.1706-?_2389+?del | del of exons 12 to 16 | p.Asp569Val;Leu570_Val797del, shortened protein (227 AA) | 3** | na |
| exon 13 | c.1846-?_2140+?dup | dup of exons 13 and 14 | p.Glu714GlyfsX29, truncated protein | 4* | na |
| 1 and 8 | c.1-?_190+?del 1061?_1845+?del | del of exons 1-2 and 8 to 12 | no in phase ATG within exon 3, no protein | 2** | na |
na: not available.
nb of affected carriers - nb of unaffected non carriers.
all unrelated carriers may present different events since the exact breakpoints of these major rearrangements are unknown.
Table 2.
New intronic and in frame deletion or insertion variations
| Location | cDNA (HGVS) | Protein (HGVS) | Number of probands | Familial segregation (*) | Splice modification prediction | ||
|---|---|---|---|---|---|---|---|
| NetGene2 | NNSPLICE | Conclusion | |||||
| intronic events | |||||||
| intron 4 | c.693_694+20del | 1 | na | new DS at +60 | new DS at +60 | deleterious | |
| intron 4 | c.694+1G>T | 1 | na | loss of DS | loss of DS | deleterious | |
| intron 6 | c.940+14del | 1 | na | loss of DS | loss of DS | deleterious | |
| intron 6 | c.940+1G>A | 1 | na | loss of DS | loss of DS | deleterious | |
| intron 6 | c.940+1G>C | 1 | na | loss of DS | loss of DS | deleterious | |
| intron 6 | c.940+2T>A | 1 | na | loss of DS | loss of DS | deleterious | |
| Intron 6 | c.941-12G>A# | 1 | yes (3 - 0) | no change | no change | benign | |
| intron 7 | c.1060+24C>A | 1 | na | new DS at +11 | no change | ? | |
| intron 7 | c.1060+26 T>G | 1 | na | no change | no change | benign | |
| intron 8 | c.1187-1G>A | 1 | na | loss of AS | na | ? | |
| intron 9 | c.1358+3_1358+8del | 1 | yes (7 - 8) | loss of DS | loss of DS | deleterious | |
| intron 9 | c.1359-4T>C | 1 | na | no change | no change | benign | |
| intron 9 | c.1359-25T>A | 1 | na | no change | no change | benign | |
| intron 10 | c.1587-2A>T | 2 | yes (2 - 2) | loss of AS | loss of AS | deleterious | |
| intron 11 | c.1705+2_+3insC | 1 | na | loss of DS | loss of DS | deleterious | |
| intron 11 | c.1706-2A>T | 1 | na | new AS at 1715 | na | ? | |
| intron 11 | c.1706-24T>C | 1 | na | no change | no change | benign | |
| intron 15 | c.2311+1G>T | 2 | yes (4 - 2) | loss of DS | loss of DS | deleterious | |
| intron 16 | c.2389+14G>A | 1 | na | no change | no change | benign | |
| intron 17 | c.2547+5G>C | 1 | na | no change | new DS at +114 | ? | |
| in frame deletions or insertions | |||||||
| exon 4 | c.316_336del | p.Pro106_Asp112del | 2 | yes (3 - 1) | no change | no change | benign |
| exon 4 | c.516_524dup | p.Cys173_Asp175dup | 1 | yes (2 - 0) | no change | no change | benign |
| exon 4 | c.648_656del | p.Asp217_Gly219del | 1 | na | no change | no change | benign |
| exon 4 | c.667_693del | p.Lys223_Cys231del | 1 | na | no change | new DS at +59 | ? |
| exon 4 | c.673_681dup | p.Lys225_Asp227dup | 1 | na | no change | no change | benign |
| exon 4 | c.682_683insAAATCTGAC | p.Asp227_Glu228InsLysSerAsp | 1 | na | no change | no change | benign |
| exon 7 | c.964_966del | p.Asn322del | 1 | na | no change | no change | benign |
| exon 11 | c.1629_1652del | p.Lys543_Asp551delinsAsn | 1 | na | no change | no change | benign |
| exon 12 | c.1730_1738del | p.Trp577_Asp579del | 1 | na | no change | no change | benign |
| exon 12 | c.1829_1831del | p.Ser610del | 2 | yes (5 - 1) | no change | no change | benign |
| exonic events | |||||||
| exon 9 | c.1194C>T | p.Ile398Ile | 1 | na | no change | no change | benign |
| exon 12 | c.1813C>T | p.Leu605Leu | 1 | yes (4 - 1) | new DS at 1813 | new DS at 1813 | deleterious |
| exon 14 | c.2140G>C | p.Glu714Gln | 1 | na | loss of DS | loss of DS | deleterious |
Splice modification predicted with NetGene2 (http://www.cbs.dtu.dk/services/NetGene2) and NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html) softwares.
na: not available.
nb of affected carriers - nb of unaffected non carriers. DS: donor site. AS: Acceptor site.
Variation effect tested by RT-PCR.
The reference sequences used forLDLR were P01130.1 (SwissProt) or NP_000518.1 (NCBI RefSeq).
Table 3.
New missense variations
| Prediction of damaging effect at the protein level | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Loca-tion | cDNA (HGVS) | Protein (HGVS) | Number of probands | FSA(*) | Polyphen | SIFT | Pmut | SNPs3D, deleterious (SVM score) | Conclusion | ||
| structural effect | protein domain | damaging | |||||||||
| exon 2 | c.100T>G | p.Cys34Gly | 1 | yes (3-0) | S-S bond disrupted | Extracell. | probably | Not Tolerated | Neutral | yes (-3.80) | deleterious |
| exon 3 | c.233G>A | p.Arg78His | 1 | na | Extracell. | benign | Tolerated | Neutral | no (0.74) | benign | |
| exon 3 | c.244T>G | p.Cys82Gly | 3 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Pathological | yes (-3.75) | deleterious |
| exon 3 | c.255G>T | p.Gln85His | 1 | na | Extracell. | benign | Tolerated | Neutral | no (1.36) | benign | |
| exon 3 | c.265T>G | p.Cys89Gly | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Pathological | yes (-3.75) | deleterious |
| exon 3 | c.270T>A | p.Asp90Glu | 4 | yes (4-3, 3-1), na | LB site disrupted | Extracell. | probably | Not Tolerated | Neutral | yes (-2.04) | deleterious |
| exon 3 | c.291C>G | p.Asn97Lys | 1 | na | Close to LB site | Extracell. | possibly | Not Tolerated | Neutral | yes (-1.11) | deleterious |
| exon 3 | c.310T>C | p.Cys104Arg | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Pathological | yes (-3.26) | deleterious |
| exon 4 | c.362G>A | p.Cys121Tyr | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Pathological | yes (-3.18) | deleterious |
| exon 4 | c.382T>C | p.Cys128Arg | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Pathological | yes (-4.21) | deleterious |
| exon 4 | c.383G>T | p.Cys128Phe | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Pathological | yes (-3.86) | deleterious |
| exon 4 | c.416A>T | p.Asp139Val | 1 | yes (2-2) | LB site disrupted | Extracell. | probably | Not Tolerated | Neutral | yes (-3.82) | deleterious |
| exon 4 | c.416A>G | p.Asp139Gly | 1 | na | LB site disrupted | Extracell. | probably | Not Tolerated | Neutral | yes (-2.10) | deleterious |
| exon 4 | c.427T>G | p.Cys143Gly | 1 | yes (4-2) | S-S bond disrupted | Extracell. | probably | Not Tolerated | Neutral | yes (-2.70) | deleterious |
| S-S bond | |||||||||||
| exon 4 | c.464G>A | p.Cys155Tyr | 1 | na | disrupted, HdpC and Overpacking at BS | Extracell. | probably | Not Tolerated | Pathological | yes (-2.62) | deleterious |
| exon 4 | c.501C>G | p.Cys167Trp | 3 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Pathological | yes (-3.99) | deleterious |
| exon 4 | c.589T>G | p.Cys197Gly | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Neutral | yes (-3.47) | deleterious |
| exon 4 | c.598T>A | p.Phe200lle | 1 | na | Extracell. | benign | Tolerated | Neutral | no (1.38) | benign | |
| exon 4 | c.611G>A | p.Cys204Tyr | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Pathological | yes (-3.06) | deleterious |
| exon 4 | c.641G>C | p.Trp214Ser | 1 | na | Close to LB site | Extracell. | probably | Tolerated | Pathological | yes (-2.05) | deleterious |
| exon 4 | c.669G>C | p.Lys223Asn | 2 | na | Extracell. | benign | Tolerated | Neutral | no (1.11) | benign | |
| exon 4 | c.680A>T | p.Asp227Val | 1 | yes (4-1) | LB site disrupted | Extracell. | probably | Not Tolerated | Neutral | yes (-3.18) | deleterious |
| exon 4 | c.693C>G | p.Cys231Trp | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Pathological | yes (-3.71) | deleterious |
| exon 5 | c.793A>T | p.Ser265Cys | 1 | na | Extracell. | probably | Not Tolerated | Neutral | yes (-2.03) | deleterious | |
| exon 6 | c.869T>G | p.Ile290Ser | 1 | na | Close to LB site | Extracell. | probably | Not Tolerated | Neutral | yes (-1.95) | deleterious |
| exon 6 | c.887G>A | p.Cys296Tyr | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Pathological | yes (-1.81) | deleterious |
| exon 6 | c.914G>C | p.Trp305Ser | 1 | na | Extracell. | probably | Not Tolerated | Neutral | possibly (-0.10) | deleterious | |
| exon 7 | c.965A>T | p.Asn322lle | 1 | na | Extracell. | probably | Not Tolerated | Neutral | yes (-1.41) | deleterious | |
| exon 7 | c.1007A>G | p.Tyr336Cys | 1 | na | Close to LB site | Extracell. | probably | Tolerated | Neutral | possibly not (0.13) | benign |
| exon 7 | c.1019 1020delins TG | p.Cys340Leu | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Neutral | yes (-3.27) | deleterious |
| exon 7 | c.1055G>T | p.Cys352Phe | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Pathological | yes (-3.27) | deleterious |
| exon 8 | c.1061A>C | p.Asp354Ala | 1 | na | LB site disrupted, HdpC and CC at BS | Extracell. | probably | Not Tolerated | Neutral | yes (-2.25) | deleterious |
| exon 8 | c.1067A>C | p.Asp356Ala | 1 | la | Close to LB site | Extracell. | probably | Not Tolerated | Pathological | yes (-1.58) | deleterious |
| exon 8 | c.1103G>C | p.Cys368Ser | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Neutral | yes (-3.27) | deleterious |
| exon 8 | c.1153C>G | p.Leu385Val | 1 | na | Extracell. | benign | Tolerated | Neutral | possibly (-0.23) | benign | |
| exon 9 | c.1223A>C | p.Glu408Ala | 2 | na | YWTD-EGF | possibly | Tolerated | Neutral | possibly (-0.47) | ? | |
| exon 9 | c.1288G>C | p.Val430Leu | 1 | na | YWTD-EGF | possibly | Tolerated | Neutral | yes (-1.19) | ? | |
| exon10 | c.1424C>T | p.Ala475Val | 2 | na | YWTD-EGF | benign | Tolerated | Neutral | no (0.60) | benign | |
| exon10 | c.1460A>G | p.Asn487Ser | 1 | na | YWTD-EGF | probably | Tolerated | Neutral | yes (-0.90) | ? | |
| exon 10 | c.1487G>T | p.Gly496Val | 1 | na | Overpacking at BS | YWTD-EGF | probably | Tolerated | Neutral | possibly (-0.37) | ? |
| exon 10 | c.1519A>G | p.Lys507Glu | 1 | na | YWTD-EGF | benign | Tolerated | Neutral | possibly not (0.12) | benign | |
| exon 10 | c.1546G>A | p.Gly516Ser | 1 | na | YWTD-EGF | benign | Tolerated | Neutral | yes (-1.93) | benign | |
| exon 10 | c.1567G>T | p.Val523Leu | 1 | na | YWTD-EGF | possibly | Not Tolerated | Neutral | yes (-2.47) | deleterious | |
| exon 10 | c.1577C>G | p.Pro526Arg | 1 | na | Overpacking and CC at BS | YWTD-EGF | probably | Not Tolerated | Neutral | yes (-2.63) | deleterious |
| exon 11 | c.1597T>C | p.Trp533Arg | 1 | na | CC and HdpC at BS | YWTD-EGF | probably | Not Tolerated | Pathological | yes (-4.04) | deleterious |
| exon 11 | c.1606T>G | p.Trp536Gly | 1 | na | YWTD-EGF | probably | Tolerated | Pathological | yes (-3.12) | deleterious | |
| exon11 | c.1625T>G | p.Ile542Ser | 1 | na | HdpC and Cavity creation at BS | YWTD-EGF | probably | Not Tolerated | Pathological | yes (-4.06) | deleterious |
| exon 11 | c.1633G>A | p.Gly545Arg | 2 | na | Overpacking at BS | YWTD-EGF | probably | Not Tolerated | Pathological | yes (-1.73) | deleterious |
| exon 11 | c.1644T>G | p.Asn548Lys | 2 | yes (3-1) | YWTD-EGF | probably | Not Tolerated | Neutral | yes (-2.06) | deleterious | |
| exon11 | C.1687OT | p.Pro563Ser | 1 | na | YWTD-EGF | probably | Not Tolerated | Neutral | yes (-3.18) | deleterious | |
| exon11 | c.1703T>C | p.Leu568Pro | 1 | na | YWTD-EGF | probably | Not Tolerated | Neutral | yes (-3.01) | deleterious | |
| exon 11 | c.1705G>T | p.Asp569Tyr | 1 | na | YWTD-EGF | probably | Not Tolerated | Pathological | yes (-4.15) | deleterious | |
| exon 12 | c.1727A>C | p.Tyr576Ser | 1 | na | New cavity at BS | YWTD-EGF | probably | Not Tolerated | Neutral | yes (-3.17) | deleterious |
| exon 12 | c.1736A>G | p.Asp579Gly | 1 | na | YWTD-EGF | probably | Not Tolerated | Pathological | yes (-3.53) | deleterious | |
| exon 12 | c.1793T>C | p.Ile598Thr | 1 | yes (2-1) | YWTD-EGF | possibly | Not Tolerated | Neutral | yes (-1.68) | deleterious | |
| exon 12 | c.1844A>T | p.Glu615Val | 3 | yes (5-8) | YWTD-EGF | probably | Not Tolerated | Neutral | yes (-3.48) | deleterious | |
| exon 13 | c.1853T>G | p.Val618Gly | 1 | na | YWTD-EGF | probably | Not Tolerated | Neutral | yes (-2.90) | deleterious | |
| exon13 | c.1856T>C | p.Phe619Ser | 2 | na | YWTD-EGF | probably | Not Tolerated | Neutral | yes (-2.90) | deleterious | |
| exon 13 | c.1907G>A | p.Gly636Asp | 1 | na | YWTD-EGF | probably | Not Tolerated | Neutral | yes (-3.01) | deleterious | |
| exon13 | c.1928C>T | p.Ala643Val | 1 | na | YWTD-EGF | benign | Tolerated | Pathological | no (1.55) | benign | |
| exon 13 | C.1945OT | p.Pro649Ser | 1 | yes (2-0) | YWTD-EGF | probably | Tolerated | Neutral | yes (-2.16) | ? | |
| exon 13 | c.1955T>C | p.Met652Thr | 1 | na | YWTD-EGF | probably | Not Tolerated | Neutral | yes (-1.46) | deleterious | |
| exon 13 | c.1958T>G | p.Val653Gly | 1 | na | YWTD-EGF | probably | Tolerated | Neutral | possibly (-0.45) | ? | |
| exon 13 | c.1973T>C | p.Leu658Pro | 1 | na | YWTD-EGF | possibly | Tolerated | Neutral | possibly not (0.32) | benign | |
| exon 13 | c.1975A>C | p.Thr659Pro | 4 | yes (2-0) | HdpC at BS | YWTD-EGF | benign | Tolerated | Neutral | yes (-2.04) | benign |
| exon14 | c.2094C>G | p.Cys698Trp | 1 | na | S-S bond disrupted | Extracell. | probably | Not Tolerated | Pathological | yes (-2.53) | deleterious |
| exon14 | c.2120A>T | p.Asp707Val | 1 | na | YWTD-EGF | probably | Not Tolerated | Pathological | yes (-3.00) | deleterious | |
| exon14 | c.2132G>C | p.Cys711Ser | 2 | na | S-S bond disrupted | YWTD-EGF | probably | Not Tolerated | Pathological | yes (-2.32) | deleterious |
| exon 14 | c.2140G>C | p.Glu714Gln | 1 | na | CC at ES | Extracell. | benign | Tolerated | Neutral | no (0.85) | benign |
| exon17 | c.2482T>C | p.Tyr828His | 1 | na | Cytoplasm. | probably | Not Tolerated | Neutral | possibly (-0.09) | deleterious | |
Prediction of damaging effect at the protein level performed with Polyphen (http://genetics.bwh.harvard.edu/pph), SIFT (http://sift.jcvi.org), Pmut (http://mmb2.pcb.ub.es/pmut) and SNP3D (http://www.snps3d.org) softwares.
na: not available.
nb of affected carriers - nb of unaffected non carriers. S-S: Disulfide. LB: Ligand Binding. BS: Buried Site. ES: Exposed Site. HdpC: Hydrophobicity Change. CC: Charge Change. The reference sequences used for LDLR were P01130.1 (SwissProt) orNP_000518.1 (NCBI RefSeq). Underlined: prediction different from the three others Bold: prediction of damaging effect was similar with either Polyphen, SIFT, Pmut or SNPs3D.
New mutational events leading to abnormal protein size
All nonsense mutations (9) and frameshift variations (41) were deemed as FH-causing mutations, since their theoretical consequence is the synthesis of a truncated protein (Table 1). Prediction of the damaging effect remained difficult for the 19 major rearrangements detected by MLPA since the exact breakpoints were not investigated (Table 1). The main mechanism reported to explain large deletions or duplications is homologous recombination between Alu sequences that are numerous in LDLR (Lehrman et al. 1987). Only introns 9 and 13 do not contain Alu sequences and no major deletion or duplication involving one of these two introns has been reported to date. In the 1990s, deletion breakpoints were sequenced in 14 of the 39 deletions reported in LDLR, and 12 involved an Alu repeat at both endpoints (Hobbs et al. 1992, Nissen et al. 2006). FH Potenza is a 5 kb deletion that joins a coding sequence in exon 13 to an Alu repetitive element in intron 15 (Lehrman et al. 1986). FH Helsinki is a 9.5 kb deletion that does not involve Alu sequences at either end of the deletion (Aalto-Setälä et al. 1989). Except for these two examples, data indicate that large deletions or duplications are mainly due to homologous recombination between two Alu sequences located in deep intronic sequences, far from splice sites. Therefore, in accordance with this observation and with respect to the translation frame of LDLR exons, protein variants could be predicted (Table 1).
New intronic variations and small in frame deletions, insertions, or indels
The majority of FH-causing variations within LDLR have been investigated at the DNA level, but only a small number of these were corroborated by cellular functional studies. From these few studies and from in silico analyses, it is now possible to predict the damaging effect at the protein level. The putative causal effect of each new event was also estimated through Familial Segregation Analysis (FSA) when available.
From the 20 new intronic variations, 10 (50%) were predicted to be deleterious by NetGene2 and NNSPLICE predictor tools, and this could be supported by FSA in three pedigrees. Six (30%) were predicted to be benign with both tools. Surprisingly, the only one for which FSA could be performed revealed the presence of the c.941-12G>A variation in the three affected members analyzed (Table 2). Furthermore, RT-PCR analysis of monocyte mRNA showed an abnormal splicing of intron 6 (data not shown). Four (20%) intronic variations were predicted to be deleterious by only one of the two tools (Table 2).
The 10 in frame del/ins were predicted to be benign, except c.667_693del27bp that was predicted to create a new donor splice site 59 bp downstream with NNSPLICE (Table 2). FSA could be performed for three families, thus indicating that even if predicted to be benign, the familial inheritance of these variations suggested causality. Interestingly, the silent variation p.Leu605Leu was predicted to create a new donor site at position 1813 with a predicted score at 0.58 when the physiological one remains at 0.50 (NNSPLICE). This new donor splice site could lead to: the substitution of p.Leu605 by a threonine, the deletion of 11 amino acids, a frameshift, and a premature termination 49 codons downstream. Furthermore, FSA showed that p.Leu605Leu was carried by the 4 affected family members and not by the unaffected, thus supporting causality. The use of RT-PCR analysis of LDLR mRNA from isolated blood cells is necessary to support this point as has been shown for another silent mutation, p.Arg406Arg (Bourbon et al. 2007).
New missense variations
Seventy new missense variations were detected here (Table 3). For 28 substitutions, prediction of a damaging effect was similar with either Polyphen, SIFT, Pmut or SNPs3D. For 36 variations, only one prediction was different from the three others and was not always given by the same software (underlined in Table 3). Finally, 6 missense variations (“?” last column, Table 3) were predicted neutral twice and pathogenic twice. Altogether, these analyses showed that 51 (73%) of the new missense variations were very probably deleterious, whereas 13 (19%) were very probably benign. Interestingly, the missense variation c.2140G>C (p.Glu714Gln) was predicted to be benign in Table 3, but to create the loss of the intron 14 donor splice site in Table 2.
New promotor variations
Four new DNA variations were found in the promoter sequence: c.-140C>T, c.-155_-150 delACCCCAinsTT, c.-219dupA and c.-267A>G. The first two fall within sterol regulatory elements, SRE1 (-130 to -144) and SRE2 (-145 to -161), respectively (Südhof et al. 1987; Liu J et al. 2000). The third one falls within a cis-acting element FP1 (-219 to -238) (Mehta et al. 1996). The last one falls close to the 3'end of FP2 (-268 to -280).
In conclusion, 78% (136/175) of the new molecular events identified in LDLR were very probably FH-causing mutations and were present in 79% (176/222) probands, whereas 16% (28/175) were very probably benign and were present in 16% (35/222) of probands, suggesting that the ADH-causing mutation remains to be identified in this last group. Altogether, these observations confirm the care needed in the interpretation of novel sequence variants and the relevance of functional analysis. Moreover, these results underscore the care needed in the overall interpretation of in silico predictions, FSA and in vitro functional studies.
Variations in APOB, PCSK9 and other genes
The APOB-p.Arg3527Gln mutation was identified in 89 probands and 10 mutations in PCSK9 were found (Abifadel 2003, Allard 2005, Abifadel 2010 personal data). The respective contribution of each gene to ADH was 73.9% LDLR, 6.6% APOB, 0.7% PCSK9 and 19.0% “Others”. The identification of this “Others” group of ADH patients clearly demonstrates that there is at least one other disease gene involved in ADH. Furthermore, because of the numerous proteins involved in cholesterol homeostasis, this new group of patients is very probably a heterogeneous class of molecular defects. This is supported by the identification of the LDLRAP1 gene (also known as ARH) which encodes a protein required for clathrin-mediated internalization of the LDL receptor by liver cells (Garcia et al. 2001), but also by our recent report of the localization of a new ADH gene at 16q22.1 (Marques et al. 2010). The percentage of this new group of ADH reported here (19%) is in the range of previously reported large cohort studies that estimated it between 12% and 48% (Varret et al. 2008).
Clinical and biological features of subjects from the four molecular groups
The four molecular groups were named FH, FDB, PCSK9 and «Others» for carrying a mutation in LDLR, APOB, PCSK9 and other genes, respectively. The four groups were composed of 190F/192M, 43F/12M, 4F/6M and 30F/21M, respectively, showing a significant difference in the sex ratio between FH/FDB, FDB/«Others», and PCSK9/«Others». Significant differences in the ages of patients were also observed across the four groups. «Others” [median of 46 years old, range: 9-78 (N=51)] were significantly older than FH [median of 37 years old, range: 2-64 (N=382)] (p=0.002), FDB [median of 37 years old, range: 5-61 (N=55)] (p=0.047), and PCSK9 [median of 38 years old, range: 3-49 (N=10)] (p=0.042). To overcome these differences for age and sex of patients among the four groups, we adjusted lipid values for age and gender of a reference French population and expressed them as multiples of median (MoM).
Total and LDL cholesterol levels were significantly higher for FH and PCSK9 when compared to FDB and «Others», and for FDB when compared to «Others” (Supp. Figure S1, panels A and B). As expected, no significant differences were observed for HDL cholesterol levels between FH, FDB and PCSK9 (Supp. Figure S1, panel C). Interestingly, HDL cholesterol levels were significantly higher for «Others” when compared to FH and PCSK9. Triglycerides levels were significantly higher for FH than FDB, as previously reported (Miserez and Keller 1995; Ejarque et al. 2008), and for «Others” when compared to FDB or PCSK9 (Supp. Figure S1, panel D).
Frequency of tendon xanthomas was significantly different only between FH and «Others», with 57% (70/123) and 14% (5/35), respectively (p<0.0001). Frequency of evidence of CHD was also significantly different between FH and «Others», with 68% (44/65) and 41% (11/27), respectively (p<0.016).
Based on the results presented here, a gradient of severity could be drawn for ADH: FH = PCSK9 > FDB > «Others». «Others” seemed to be the less severe group with total and LDL-cholesterol levels significantly lower and presence of xanthomas or evidence of CHD rarer. Furthermore, the age of probands was higher, thus suggesting that it may be diagnosed later in life. Another feature of «Others” was an HDL-cholesterol level significantly higher that should be protective against CHD. This observation could explain the lower frequency of CHD in this group when compared to FH.
CONCLUSION
In conclusion, mutations in LDLR remain the main cause of ADH, and their already large spectrum is here widened with the report of 175 new sequence variations. We also demonstrated the specificity of the spectrum of LDLR gene mutations in the French population when compared to other countries, thus underscoring the requirement of specific national molecular screening strategies. More than ¾ of these variations likely cause familial hypercholesterolemia as inferred from the predicted effect on structure and 16% are probably benign, with the remainder requiring careful interpretation and further functional analyses to avoid a false positive diagnosis. Although it had been stated that most human ADH mutations in LDLR and other genes had been documented, the relatively high number of new mutations reported here suggests that a substantial proportion of mutations across all human communities remains unidentified.
This is the largest French ADH cohort ever reported and it allowed statistical analysis of clinical and biochemical data. The comparison of the four molecular groups showed, for the first time, that a significant gradient of severity could be established for ADH: FH = PCSK9 > FDB > «Others». Finally, we report a more precise estimation of the percentage of ADH nonLDLR/nonAPOB/nonPCSK9 patients that is close to 19%.
Acknowledgments
We thank all probands and family members for their cooperation. This work was supported by grants from Pfizer, Fondation de France, GIS-Maladies Rares, PHRC (AOM06024) and ANR (ANR-05-PCOD-017, ANR-06-MRAR-038, ANR-08-GENO-002-01). M.M. and A.M. are supported by grants from Ministère de l'Education Nationale et de la Technologie (France). M.A is supported by grants from Région Ile de France and Conseil de la Recherche de l'Université Saint-Joseph (Beirut, Lebanon).
SUPPORTING INFORMATION
Supp. Table S1.
Compared distribution of each type of mutation in the LDLR gene between the French cohort and worldwide reports*
| % in the French cohort (All probands) | % in the French cohort (Unique events) | % in worldwide reported unique events* | p value ** | |
|---|---|---|---|---|
| Missense | 47.7 | 46.0 | 47 | 0.793 |
| Nonsense | 17.8 | 11.3 | 9 | 0.143 |
| Frameshift | 11.8 | 14.6 | 19 | 0.053 |
| In frame deletions, insertions or indels | 2.7 | 3.8 | 4 | 0.743 |
| Splice | 11.2 | 13.6 | 8 | 0.002 |
| 5′ UTR | 0.4 | 1.0 | 2 | 0.130 |
| Major rearangements | 8.5 | 9.7 | 11 | 0.491 |
| N | 1012 | 391 | 1066 | |
Leigh et al.2008.
Chi2 test.
Supp. Figure S1.
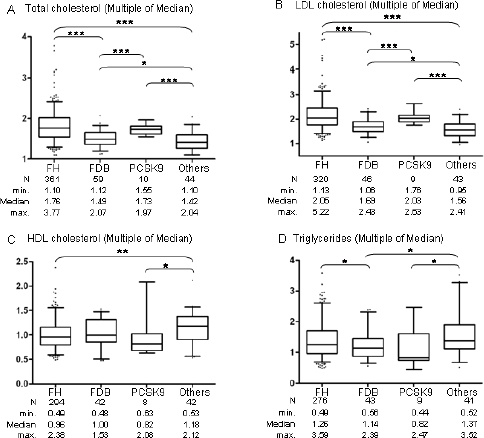
Comparison of lipid levels among the four molecular groups of patients. Panel A: Multiple of Median (MoM) for Total cholesterol levels. Panel B: Multiple of Median (MoM) for LDL cholesterol levels. Panel C: Multiple of Median (MoM) for HDL cholesterol levels. Panel D: Multiple of Median (MoM) for triglycerides levels. Results are presented with the median and range from minimum to maximum MoM values for each group. Median Mann-Whitney Test: * p < 0.05, ** p < 0.01, *** p < 0.001.
References
- Aalto-Setälä K, Helve E, Kovanen PT, Kontula K. Finnish type of low density lipoprotein receptor gene mutation (FH-Helsinki) deletes exons encoding the carboxy-terminal part of the receptor and creates an internalization-defective phenotype. J Clin Invest. 1989;84:499–505. doi: 10.1172/JCI114192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derré A, Villéger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- Allard D, Amsellem S, Abifadel M, Trillard M, Devillers M, Luc G, Krempf M, Reznick Y, Girardet J-P, Fredenrich A, Junien C, Varret M, Boileau C, Benlian P Rabès J-P. Novel mutations of the PCSK9 gene cause variable phenotype of Autosomale Dominant Hypercholesterolemia. Hum Mut. 2005;26:497–507. doi: 10.1002/humu.9383. [DOI] [PubMed] [Google Scholar]
- Alonso R, Defesche JC, Tejedor D, Castillo S, Stef M, Mata N, Gomez-Enteria P, Martinez-Faedo C, Forga L, Mata P. Genetic diagnosis of familial hypercholesterolemia using a DNA-array based platform. Clin Biochem. 2009;42:899–903. doi: 10.1016/j.clinbiochem.2009.01.017. [DOI] [PubMed] [Google Scholar]
- Amsellem S, Briffaut D, Carrié A, Rabès JP, Girardet JP, Fredenrich A, Moulin P, Krempf M, Reznik Y, Vialettes B, de Gennes JL, Brukert E, Benlian P. Intronic mutations outside of Alu-repeat-rich domains of the LDL receptor gene are a cause of familial hypercholesterolemia. Hum Genet. 2002;111:501–10. doi: 10.1007/s00439-002-0813-4. [DOI] [PubMed] [Google Scholar]
- Bourbon M, Sun XM, Soutar AK. A rare polymorphism in the low density lipoprotein (LDL) gene that affects mRNA splicing. Atherosclerosis. 2007;195:e17–20. doi: 10.1016/j.atherosclerosis.2007.01.034. [DOI] [PubMed] [Google Scholar]
- Ejarque I, Real JT, Martinez-Hervas S, Chaves FJ, Blesa S, Garcia-Garcia AB, Millan E, Ascaso JF, Carmena R. Evaluation of clinical diagnosis criteria of familial ligand defective apoB 100 and lipoprotein phenotype comparison between LDL receptor gene mutations affecting ligand-binding domain and the R3500Q mutation of the apoB gene in patients from a South European population. Transl Res. 2008;151:162–7. doi: 10.1016/j.trsl.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M, Calandra S, Bertolini S, Cossu F, Grishin N, Barnes R, Cohen JC, Hobbs HH. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Sciences. 2001;292:1394–8. doi: 10.1126/science.1060458. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Schrott HG, Hazzard WR, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest. 1973;52:1544–68. doi: 10.1172/JCI107332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1:445–66. doi: 10.1002/humu.1380010602. [DOI] [PubMed] [Google Scholar]
- Humphries SE, Norbury G, Leigh S, Hadfield SG, Nair D. What is the clinical utility of DNA testing in patients with familial hypercholesterolaemia? Curr Opin Lipidol. 2008;19:362–368. doi: 10.1097/MOL.0b013e32830636e5. [DOI] [PubMed] [Google Scholar]
- Innerarity TL, Weisgraber KH, Arnold KS, Mahley RW, Krauss RM, Vega GL, Grundy SM. Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc Natl Acad Sci USA. 1987;84:6919–23. doi: 10.1073/pnas.84.19.6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrman MA, Russell DW, Goldstein JL, Brown MS. Exon-Alu recombination deletes 5 kilobases from the low density lipoprotein receptor gene, producing null phenotype in familial hypercholesterolemia. PNAS. 1986;83:3679–3683. doi: 10.1073/pnas.83.11.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrman MA, Russell DW, Goldstein JL, Brown MS. Alu-Alu recombination deletes splice acceptor sites and produces secreted low density lipoprotein receptor in a subject with familial hypercholesterolemia. J Bio Chem. 1987;262:3354–3361. [PubMed] [Google Scholar]
- Leigh SEA, Foster AH, Whittall RA, Hubbart CS ans Humphries SE. Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database. Annals of Hum Genet. 2008;72:485–498. doi: 10.1111/j.1469-1809.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Ahlborn TE, Briggs MR, Kraemer FB. Identification of a novel sterol-independent regulatory element in the human low density lipoprotein receptor promoter. J Biol Chem. 2000;275:5214–21. doi: 10.1074/jbc.275.7.5214. [DOI] [PubMed] [Google Scholar]
- Marques-Pinheiro A, Marduel M, Rabès JP, Devillers M, Villéger L, Allard D, Weissenbach J, Guerin M, Zair Y, Erlich D, Junien C, Munnich A, Krempf M, Abifadel M, Jaïs JP, The French Research Network on ADH. Boileau C, Varret M. A fourth locus for autosomal dominant hypercholesterolemia maps at 16q22.1. Eur J Hum Genet. 2010 doi: 10.1038/ejhg.2010.94. Jun 23. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNutt MC, Lagace TA, Horton JD. Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem. 2007;282:20799–20803. doi: 10.1074/jbc.C700095200. [DOI] [PubMed] [Google Scholar]
- Mehta KD, Chang R, Underwood J, Wise J, Kumar A. Identification of a novel cis-acting element participating in maximal induction of the human low density lipoprotein receptor gene transcription in response to low cellular cholesterol levels. J Biol Chem. 1996;271:33616–3362. doi: 10.1074/jbc.271.52.33616. [DOI] [PubMed] [Google Scholar]
- Miserez AR, Keller U. Differences in the phenotypic characteristics of subjects with familial defective apolipoprotein B-100 and familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1995;15:1719–29. doi: 10.1161/01.atv.15.10.1719. [DOI] [PubMed] [Google Scholar]
- Nissen PH, Damgaard D, Stenderup A, Nielsen GG, Larsen ML, Faergeman O. Genomic characterization of five deletions in the LDL receptor gene in Danish Familial Hypercholesterolemic subjects. BMC Med Gene. 2006;7:55. doi: 10.1186/1471-2350-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabès JP, Varret M, Saint-Jore B, Erlich D, Jondeau G, Krempf M, Giraudet P, Junien C, Boileau C. Familial Ligand-Defective Apolipoprotein B-100: Simultaneous Detection of the Arg3500 -> Gln and Arg3531 -> Cys mutations in a French Population. Human Mutation. 1997;10:160–163. doi: 10.1002/(SICI)1098-1004(1997)10:2<160::AID-HUMU8>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Rabès JP, Varret M, Devillers M, Aegerter P, Villéger L, Krempf M, Junien C, Boileau C. R3531C mutation in the Apolipoprotein B gene is not sufficient to cause hypercholesterolemia. Arterio. Thromb. Vasc. Biol. 2000;20:e76–e82. doi: 10.1161/01.atv.20.10.e76. [DOI] [PubMed] [Google Scholar]
- Siest G, Visvikis S, Herbeth B, Gueguen R, Vincent-Viry M, Sass C, Beaud B, Lecomte E, Steinmetz J, Locuty J, Chevrier P. Objectives, design and recruitment of a familial and longitudinal cohort for studying gene-environment interactions in the field of cardiovascular risk: the Stanislas cohort. Clin Chem Lab Med. 1998;36:35–42. doi: 10.1515/CCLM.1998.007. [DOI] [PubMed] [Google Scholar]
- Soria LF, Ludwig EH, Clarke HR, Vega GL, Grundy SM, McCarthy BJ. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proc Natl Acad Sci USA. 1989;86:587–91. doi: 10.1073/pnas.86.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Van der Westhuyzen DR, Goldstein JL, Brown MS, Russell DW. Three direct repeats and a TATA-like sequence are required for regulated expression of the human low density lipoprotein receptor gene. J Biol Chem. 1987;262:10773–9. [PubMed] [Google Scholar]
- Varret M, Abifadel M, Rabès JP, Boileau C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clin Genet. 2008;73:1–13. doi: 10.1111/j.1399-0004.2007.00915.x. [DOI] [PubMed] [Google Scholar]
- Villéger L, Abifadel M, Allard D, Rabès JP, Thiart R, Kotze MJ, Béroud C, Junien C, Boileau C, Varret M. The UMD-LDLR Database: Additions to the software and 490 new entries to the database. Hum Mutat. 2002;20:81–87. doi: 10.1002/humu.10102. [DOI] [PubMed] [Google Scholar]