

Abstract
Many important and complex laboratory procedures require an input of high quality, intact RNA. A degraded sample or the presence of impurities can lead to disastrous results in downstream experimental applications. It is therefore, of utmost importance to use solid techniques with numerous safeguards and quality control checks to ensure a superior sample. Herein, we detail a protocol to isolate total RNA from whole zebrafish embryos using a commercially available chemical denaturant and subsequent cleanup to remove traces of DNA and impurities using a commercial RNA isolation kit. As RNA is relatively unstable and easily prone to cleavage by RNAses, most protocols assay gene expression using a cDNA product that is directly synthesized from an RNA template. We detail a procedure to convert RNA into the more stable cDNA product using a commercially available kit. Throughout these procedures there are numerous quality control checks to ensure that the sample is not degraded or contaminated. The end product of these protocols is cDNA that is suitable for microarray analysis, RT-PCR or long-term storage.
Protocol
Part 1: Extraction of Total RNA using TRIzol Reagent
When working with RNA it is very important to work in an environment that is free of RNAses. Simple precautions such as having reserved pipettes for use only with RNA procedures and spraying the work area with a decontaminant reagent (e.g., RNAse Away) before beginning the procedure are very helpful.
To attain a sufficient amount of RNA pool 50 zebrafish embryos in a 1.5 ml microfuge tube and remove as much water as possible with a pipette.
Immediately add 250 μl of TRIzol reagent1 to the microfuge tube containing the embryos. TRIzol reagent contains phenol and should only be used under a fume hood.
Lyse and homogenize the embryos with a pellet pestle (approximately 20 strokes) until the tissue is sufficiently disrupted.
When the cells are sufficiently homogenized, add 750 μl TRIzol reagent to equal a total volume of 1 ml.
To permit complete dissociation of nucleoprotein complexes, incubate homogenized samples for 5 minutes at room temperature. At this stage, the sample can be flash frozen in liquid nitrogen and stored at -80°C or the protocol can be continued.
To continue add 0.2 ml of chloroform and rock the tube for 15 seconds to mix.
Incubate the sample for 2 minutes at room temperature and then centrifuge at 12,000 x g for 15 minutes at 4°C.
The mixture will separate into a lower red phenol-chloroform phase, an interphase, and a colorless upper aqueous phase. The upper aqueous phase contains the RNA. The top layer should comprise of about 60% of the initial volume of TRIzol (approximately 0.6 ml). Transfer the top aqueous layer into a new microfuge tube using a 1 ml pipette being careful not to transfer any of the interphase layer.
To precipitate the RNA add 0.5 ml isopropanol.
Allow the sample to sit at room temperature for 10 minutes and then centrifuge the sample at 12,000 x g for 10 minutes at 4°C. RNA will form a gel-like pellet on the bottom of the tube.
Without disturbing the pellet, remove the supernatant with a pipette and wash the pellet in 1 ml 75% ethanol. Mix the sample by gentle inversion and centrifuge at 7,500 x g for 5 minutes at 4°C.
After centrifugation, remove the ethanol with a pipette and allow sample to dry while inverted for 10 minutes.
Resuspend the pellet by adding 100 μl of RNAse-free water and incubate the sample at 55°C for 10 minutes. It is recommended to finger vortex frequently during incubation to aid in RNA rehydration. Optionally, you can check sample quality and quantity using a NanoDrop ND-1000 spectrophotometer or directly continue to the next step.
Part 2: RNA Cleanup using the Qiagen RNEasy Mini Kit
The Qiagen RNEasy Mini Kit2 is useful in removing contaminants and impurities from an RNA sample isolated from other methods. For this procedure, β-mercaptoethanol, ethanol, and nuclease-free water are needed in addition to the components included in the kit. An optional DNAse treatment is recommended during this procedure.
Upon first use of the kit, Buffer RPE will need to be prepared by adding 4 volumes of 100% ethanol to 1 volume of Buffer RPE.
On the day of the procedure, Buffer RLT will need to be prepared fresh. Calculate the total amount of Buffer RLT that will be needed. A volume of 350 μl is needed per sample. To prepare Buffer RLT, add 10 μl of β-mercaptoethanol for every 1 ml of buffer. This should be done under the fume hood.
To each sample add 350 μl of Buffer RLT and 250 μl of 100% ethanol. Mix well by pipetting up and down several times.
Transfer the sample, which should be approximately 700 μl, into an RNEasy column that is placed in a 2 ml collection tube and centrifuge at 8,000 x g for 1 minute at room temperature.
After centrifuging, discard the flow through and add 700 μl of Buffer RW1 to an RNEasy spin column.
Centrifuge the sample at 8,000 x g for 1 minute at room temperature and then discard the flow through.
- DNAse treatment with Qiagen RNAse-Free DNAse Kit: DNAse treatment is optional but is highly recommended for the highest quality samples.
- When you first receive the DNAse treatment kit, you will need to prepare the DNAse I stock solution by adding 550 μl nuclease-free water to the DNAse. Mix gently by inverting and do not vortex. Aliquot into single use vials containing 10 μl of the stock solution. You can store the stock solution at -20°C for up to 9 months. The thawed aliquots can be stored at 4°C for 6 weeks if needed, but do not refreeze the aliquots after thawing.
- To prepare the DNAse I incubation mix add 70 μl Buffer RDD to 10 μl DNAse I stock solution per sample. Mix by gentle inversion and centrifuge tube briefly to collect residual liquid form the sides of the tube. Add 80 μl DNAse I incubation mix directly to the RNEasy spin column. Incubate the DNAse treatment for 30 minutes at room temperature.
Following DNAse treatment add 350 μl Buffer RW1 to the spin column and centrifuge at 8,000 x g for 1 minute at room temperature.
After centrifugation discard the flow through and add 500 μl Buffer RPE to the spin column. Incubate sample for 5 minutes at room temperature.
Centrifuge sample at 8,000 x g for 1 minute at room temperature and discard the flow through.
After centrifugation add 500 μl 75% ethanol to the spin column.
Repeat centrifugation at 8,000 x g, but for 2 minutes at room temperature.
After centrifugation discard the flow through and allow the column to dry for 5 minutes.
Immediately centrifuge at 8,000 x g for 5 minutes at room temperature to remove any left over traces of ethanol.
After centrifugation is complete transfer the spin column into a new 1.5 ml collection tube and add 10 μl nuclease-free water. Incubate for 1 minute and then centrifuge at 10,000 x g for 1 minute at room temperature. The RNA will be in the flow through.
Add another 10 μl of nuclease-free water to the spin column and incubate at room temperature for 1 minute. Centrifuge sample at 10,000 x g for 1 minute at room temperature.
After centrifugation remove and discard the spin column. RNA will be in the flow through.
Check the quantity and quality of the RNA using a NanoDrop ND-1000 spectrophotometer following the manufacturer’s instructions.
In addition, run a denaturing gel or use an Agilent 2100 bioanalyzer to check the integrity of the RNA.
Samples should be stored at -80°C until cDNA synthesis can be performed.
Part 3: cDNA Synthesis using the Invitrogen SuperScript First-Strand System
RNA is easily prone to cleavage by RNAses leading to degradation. As a result many gene expression protocols have been developed to use a more stable cDNA product that has been directly synthesized from the RNA. Here we detail synthesis of first-strand cDNA using the Invitrogen SuperScript First-Strand Synthesis System3. Each cDNA synthesis reaction can accommodate up to 5 μg of RNA.
Before beginning the procedure mix and briefly centrifuge each component.
Prepare RNA/primer mixture in a sterile 0.5 ml microfuge tube as outlined in Table 1.
Incubate the sample at 65°C for 5 minutes.
Immediately transfer sample to ice slurry for 10 minutes.
While the sample is cooling prepare a master mix with the components outlined in Table 2. The volumes are listed per 1 reaction. Adjust volumes accordingly for the total number of reactions.
After cooling, add 9 μl of the master mix prepared in step 5 to each RNA/primer mixture. Mix gently and collect by brief centrifugation. The total volume of each sample should be 19 μl.
Incubate sample at 42°C for 2 minutes in a thermocycler.
Add 1 μl (50 units) of SuperScript II RT to each tube, mix, and incubate at 42°C for 60 minutes in a thermocycler.
Terminate the reaction at 70°C for 15 minutes and then chill sample to 4°C. A thermocycler can be programmed for steps 8 and 9.
After the sample has reached 4°C, transfer the reaction to an ice bath.
On ice add 1 μl of RNAse H to each tube and incubate for 20 minutes at 37°C. The total volume is now 21 μl. Addition of RNAse H to your sample will degrade the RNA template and leave only the single strand cDNA product.
Part 4: cDNA Isolation and Precipitation
A 1.5 ml phase lock gel tube will be used in the isolation of the cDNA product. Prepare a 1.5 ml phase lock gel tube by centrifugation at 12,000 x g for 2 minutes. The gel should all be at the bottom of the tube.
Add 81.5 μl phenol (Tris-saturated, buffered to pH 8.0), 81.5 μl of 24:1 chloroform isoamyl alcohol, and the sample (21 μl) into the phase lock gel tube. Mix several times by gentle inversion.
Centrifuge the sample at 12,000 x g for 5 minutes at room temperature.
The cDNA should be in the upper aqueous phase. Transfer the upper phase into a clean 1.5 ml microfuge tube. The total volume should be around 20 μl.
Add 1X volume of 3 M NaOAc, pH 5.2 and mix gently. For example, if the volume of the upper aqueous phase was equal to 20 μl, you will need to add 2 μl of 3 M NaOAc, pH 5.2.
Add 7 μl of 5 mg/ml glycogen and mix gently.
Add 1X volume of isopropanol and mix gently. Generally this is around 30 μl of isopropanol.
Allow sample to incubate at room temperature for 10 minutes and then centrifuge at 12,000 x g for 20 minutes at room temperature.
A small cDNA pellet should form on the bottom of the tube. Carefully remove the supernatant with a pipette.
Add 500 μl of 70% ethanol and mix by inversion.
Centrifuge the sample at 12,000 x g for 5 minutes.
After centrifugation remove the supernatant with a pipette and repeat the 70% ethanol wash and centrifugation (steps 10 and 11).
After centrifugation remove the supernatant with a pipette. If there is liquid remaining on the sides of the tube, it can be gathered by briefly spinning the tube. Remove any excess liquid and allow the pellet to dry at room temperature for 5 minutes.
Add 20 μl of nuclease-free water to rehydrate the pellet. Place the sample at 55°C for 5 minutes for rehydration of the pellet.
The quantity and quality of the cDNA product is then checked using a NanoDrop ND-1000 spectrophotometer following the manufacturer’s instructions. The cDNA can be stored at -20°C.
Representative Results
After RNA cleanup, approximately 15 μg of high quality total RNA can be expected from 50 zebrafish embryos. To assess total RNA quality, a spectrophotometer, gel electrophoresis, and an Agilent 2100 Bioanalyzer can be used. A NanoDrop ND-1000 spectrophotometer can be used to assess the absorbance at 260 nm in comparison to the absorbance at 280 nm (Figure 1). The A260/A280 ratio can reveal the presence of contaminants and give evidence of possible degradation. An A260/A280 ratio of 1.9-2.0 is considered acceptable. In addition, it is highly recommended that an RNA denaturation gel be run to assess RNA integrity. RNA strands will separate on basis of their size, with smaller strands migrating further down the gel. A non-degraded sample will appear as a smear with two strong bands. The smear is a result of a population of different sized mRNA strands and the bands correspond to 28S and 18S rRNA. The 28S band should be about twice as intense as the 18S band (Figure 2). Alternatively, total RNA integrity can be assessed using an Agilent 2100 bioanalyzer. Two distinct peaks representing the 28S and 18S rRNA is expected (Figure 3). The 28S peak should be larger than the 18S peak with the area under the peaks approximately 2:1, respectively. Two distinct peaks will not be observed in degraded samples. RNA samples exhibiting degradation should not be carried through subsequent experimental procedures.
Spectrophotometric analysis can be used to assess the quality of the cDNA product (Figure 4). The A260/A280 should be around 1.8. cDNA reactions with an input of 5 μg of total RNA routinely yield 1-2 μg of cDNA product in our laboratory.
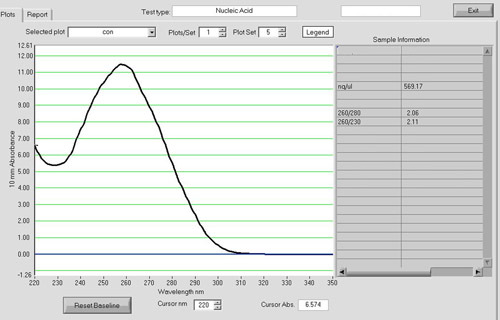 Figure 1. A NanoDrop spectrum of RNA sample. After RNA cleanup, RNA quantity and quality can be assessed using a NanoDrop ND-1000 spectrophotometer. Approximately 15 μg of high quality total RNA is routinely yielded from 50 zebrafish embryos with an A260/A280 ratio around 1.9-2.0.
Figure 1. A NanoDrop spectrum of RNA sample. After RNA cleanup, RNA quantity and quality can be assessed using a NanoDrop ND-1000 spectrophotometer. Approximately 15 μg of high quality total RNA is routinely yielded from 50 zebrafish embryos with an A260/A280 ratio around 1.9-2.0.
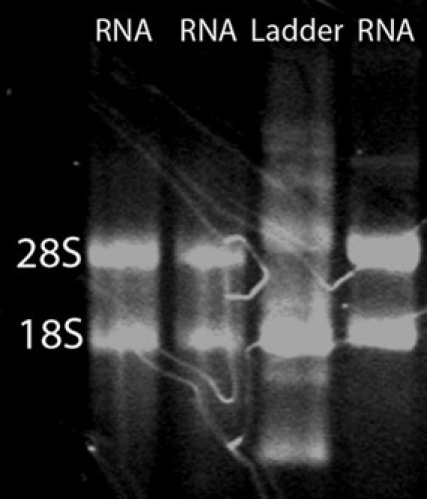 Figure 2.
An RNA denaturation gel. To assess the integrity of the total RNA isolation, an RNA denaturation gel can be ran. A smear with two bright bands corresponding to 28S and 18S rRNA is expected. The 28S band should be approximately twice as intense as the 18S band.
Figure 2.
An RNA denaturation gel. To assess the integrity of the total RNA isolation, an RNA denaturation gel can be ran. A smear with two bright bands corresponding to 28S and 18S rRNA is expected. The 28S band should be approximately twice as intense as the 18S band.
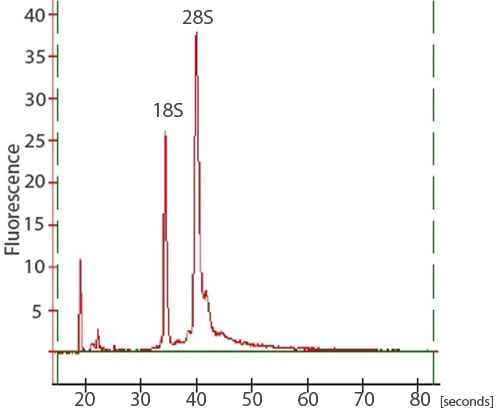 Figure 3. A trace of an RNA sample from a bioanalyzer. To assess the integrity of the total RNA isolation, samples may also be analyzed on an Agilent 2100 bioanalyzer. Two sharp peaks, corresponding to the 28S and 18S rRNA, should be visible. The 28S peak should be larger than the 18S peak with the area under the peaks approximately 2:1, respectively.
Figure 3. A trace of an RNA sample from a bioanalyzer. To assess the integrity of the total RNA isolation, samples may also be analyzed on an Agilent 2100 bioanalyzer. Two sharp peaks, corresponding to the 28S and 18S rRNA, should be visible. The 28S peak should be larger than the 18S peak with the area under the peaks approximately 2:1, respectively.
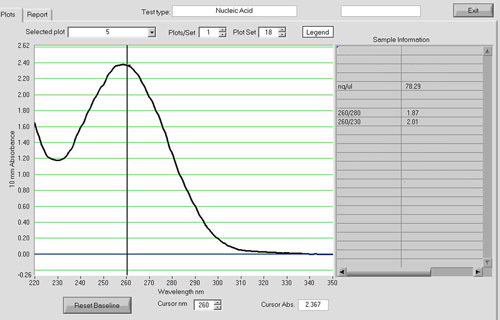 Figure 4. A NanoDrop spectrum of a cDNA sample. Spectrophotometric analysis with a NanoDrop ND-1000 can be used to assess the quantity and quality of the cDNA product. The A260/A280 ratio should be around 1.8. cDNA reactions with an input of 5 ug of total RNA routinely yield 1-2 ug of cDNA in our laboratory.
Figure 4. A NanoDrop spectrum of a cDNA sample. Spectrophotometric analysis with a NanoDrop ND-1000 can be used to assess the quantity and quality of the cDNA product. The A260/A280 ratio should be around 1.8. cDNA reactions with an input of 5 ug of total RNA routinely yield 1-2 ug of cDNA in our laboratory.
Table 1. Components to add for RNA/primer mixtures at step 2 of cDNA synthesis.
| Component | Volume |
| Total RNA (up to 5 μg) | 10 μl |
| 10 mM dNTP mix | 1 μl |
| Oligo(dT)12-18 (0.5 μg/ μl) | 1 μl |
| DEPC-treated water | n μl |
| Total volume | 10 μl |
Table 2. Reaction mixture for step 5 of cDNA synthesis. Volumes listed are per reaction. Increase volumes of each component for the total number of reactions.
| Component | Volume |
| 10X RT Buffer | 2 μl |
| 25 mM MgCl2 | 4 μl |
| 0.1 mM DTT | 2 μl |
| RNAseOUT | 1 μl |
Discussion
The fragility of the RNA molecule is the most critical consideration that should be remembered throughout this protocol. Sterile equipment that is RNAse-free should always be used in an RNAse-free zone of the laboratory. It is helpful to reserve a part of the laboratory to be used for RNA procedures only. This area should be frequently sprayed with an RNAse removing product, such as RNAse Away. The RNA samples should always be handled with gloves and kept on ice to prevent degradation.
This protocol took advantage of commercially available reagents and kits. Alternatively, there are numerous additional reagents and RNA isolation kits available on the market for total RNA isolations. The reagents and kits included in this protocol have worked well and routinely yielded high quality RNA in our laboratory.
As an alternative, double stranded cDNA synthesis can be performed, instead of the first strand synthesis demonstrated in this protocol. Double stranded cDNA is sometimes desired as it provides increased stability. In addition, some protocols require double stranded cDNA products as an input. However, if the samples are going to be kept in house for downstream applications, first strand synthesis is sufficient and is much more economical.
References
- TRIzol Reagent Manual. 2007. [Google Scholar]
- RNEasy Mini Handbook. 4th ed 2006. [Google Scholar]
- SuperScript First-Strand Synthesis System for RT-PCR. 2003. Version E. [Google Scholar]