

Abstract
Severe inflammation characterizes rapidly progressive glomerulonephritides, and expression of the kinin B1 receptor (B1R) associates with inflammation. Delayed B1R blockade reduces renal inflammation in a model of unilateral ureteral obstruction, but whether B1R modulates the pathophysiology of glomerulonephritides is unknown. Here, we observed an association of B1R protein expression and inflammation, in both glomeruli and the renal interstitium, in biopsies of patients with glomerulonephritides, Henoch-Schönlein purpura nephropathy, and ANCA-associated vasculitis. In the nephrotoxic serum–induced glomerulonephritis model, we observed upregulation of the B1R receptor; treatment with a B1R antagonist beginning 2 weeks after the onset of disease reduced both glomerular and tubular lesions and improved renal function. B1R blockade reduced renal chemokine expression and macrophage accumulation. Collectively, our data demonstrate that blockade of the kinin B1R has significant potential for the treatment of glomerulonephritis.
Rapidly progressive glomerulonephritides constitute a group of kidney diseases sharing common pathologic features. These nephropathies are characterized by severe glomerular inflammation, mainly composed of infiltrating macrophages and T cells. This leads to the formation of glomerular crescents composed of immune and proliferating epithelial cells from the Bowman's capsule. After the primary glomerular insult, inflammation and lesions extend to the tubulointerstitial compartment and induce tubular damage and progressive interstitial fibrosis, leading to the loss of renal function; therefore, renal inflammation is a key player in the initiation and the progression of these pathologies. It has been suggested that any strategy or agent able to limit or attenuate renal inflammation should significantly slow the progression of glomerulonephritides to ESRD.1
Endogenous kinins (bradykinin-related peptides) are produced through cleavage of kininogens by kallikreins. These peptides mediate their biologic effects by activation of two G protein–coupled receptors: a constitutively expressed B2 receptor (B2R) and an inducible B1 receptor (B1R).2 Bradykinin and kallidin are the natural agonists of the B2R, whereas their metabolites, generated by the enzyme kininase I, des-Arg9-BK and Lys-des-Arg9-BK, respectively, are specific for the B1R. The B1R is poorly expressed under physiologic conditions and induced, mainly under inflammatory conditions, in a variety of tissues.3 In addition, a number of studies support the in vivo generation of B1 agonist under pathologic conditions.3,4 Under experimental renal inflammation, a marked upregulation of the B1R expression along the rat nephron has been reported.5,6 In this inflammatory context, it has been shown that the B1R agonist is able to upregulate the expression of its own receptor via the activation of transcription factor NF-κB.7,8
The activated B1R stimulates the release of proinflammatory cytokines including TNF-α and IL-19 and is also involved in leukocyte accumulation and activation.10,11 Little is known about the role of the B1R in human (patho)physiology, but in animal models, an attenuated inflammatory response has been observed in B1R knockout mice in pleurisy and paw edema12 as well as in a model of intestinal ischemia and reperfusion injury.13 Specific inflamed organ-induced B1R expression and the availability of an orally active B1R antagonist (B1Ra) has made the B1R a potential therapeutic target for the treatment of pathologies related to chronic inflammation, such as atherosclerosis, airway inflammation, diabetic neuropathy, arthritis, and neuropathic pain.14,15 Indeed, since the work of Gougat et al.16 describing the selectivity of this orally active B1Ra, a number of original in vivo studies have reported its therapeutic potential in inflammatory bowel disease,17 skin inflammatory diseases,18 sensory polyneuropathy,19 and neuropathic pain.20
The role of the B1R in renal inflammation has been poorly investigated. We recently reported in the unilateral ureteral obstruction (UUO) model that preventive and, much more interesting, delayed treatment with an orally active nonpeptide B1Ra blocked monocyte/macrophage infiltration involving a direct inhibition of chemokine expression by resident renal cells. This was associated with reduced tubulointerstitial fibrosis.21 The promising results obtained using the UUO model led us to study the role and the therapeutic potential of the B1R in glomerulonephritides.
We show here for the first time induction of the B1R associated with overt inflammation in two of the most common forms of human glomerulonephritides (pediatric and adult): Henoch-Schönlein purpura nephropathy (HSPN) and ANCA-associated crescentic glomerulonephritis. We then investigated whether, after establishment of inflammation, delayed blockade of the B1R inhibited the progression of nephrotoxic serum (NTS)-induced glomerulonephritis, the mouse immune model of progressive glomerulonephritis.
Results
B1R mRNA Is Expressed in Human Renal Biopsies of Henoch-Schönlein Purpura
To investigate whether the B1R was induced in patients with rapidly progressive glomerulonephritis, we analyzed B1R mRNA expression in biopsies from pediatric patients with HSPN (n=5) compared with patients with minimal change nephrotic syndrome (MCNS; n=3). MCNS is different from glomerulonephritis because it is a syndrome characterized by discrete lesions of glomerular epithelial cells but without signs of inflammation and crescent formation. A significant induction of B1R-mRNA was found in renal biopsies of HSPN compared with MCNS (Figure 1).
Figure 1.

B1R mRNA is upregulated in HSPN. B1 mRNA expression levels in renal biopsies was quantified by real-time PCR. *P < 0.05 versus MCNS.
B1R Is Induced in Human Renal Biopsies of Henoch-Schönlein Purpura and ANCA-Associated Vasculitis
To support these results on the level of mRNA expression, we retrospectively analyzed biopsies from pediatric patients with HSPN (n=20), from adult patients with ANCA-associated vasculitis (AAV; n=13), and from patients with MCNS (n=9; Figure 2A). As control kidney tissue, we used the nonpathologic regions of kidneys from patients who underwent kidney tumorectomy (n=10). All of the biopsies were scored for inflammation, and B1R-positive cells were identified by immunohistochemical staining with the specific, in-house–developed human B1R antibody K21N.7 The B1R localization in these different nephropathies can be found together with clinical data and anatomopathologic scores in Figure 2A.
Figure 2.

Representative human B1R expression in normal and pathologic human kidney biopsies. (A) Clinical data, anatomopathologic scores, and B1R localization from patients selected for immunohistochemical analysis of human renal biopsies are summarized. Data are means ± SEM. M/F, male/female; P-creat, plasma creatinine; Cr Clear., creatinine clearance; Prot., proteinuria; Inflam., inflammation. (B) Immunohistochemical staining of B1R in renal biopsies. Control, nonpathologic regions of renal tumors. Magnification, ×200.
B1R staining was absent in nonpathologic regions of kidney sections of renal tumors (Figure 2B, Control) and in biopsies obtained from patients with MCNS (Figure 2B, MCNS). All MCNS biopsies scored negative for inflammation (Figure 2A). Very interestingly, in HSPN biopsies, we observed epithelial and interstitial B1R staining associated with clear inflammation (score 2+; Figure 2, HSPN ×200 and enlargement). In addition, in biopsies of AAV, with severe glomerular and interstitial inflammation (score 3+; Figure 2A), we observed B1R staining in the tubulointerstitial cellular compartment together with B1R expression in crescentic glomeruli (Figure 2B, AAV ×200 and enlargement).
Renal B1R Expression Is Induced in the Mouse Model of NTS-Induced Glomerulonephritis
The studies on human biopsies suggested specific association of B1R expression with glomerular and interstitial inflammation in human glomerulonephritis. To study the role of B1R induction in rapidly progressive glomerulonephritis, we used the murine model of NTS-induced glomerulonephritis.22 Consistent with what we observed in human biopsies, renal B1R mRNA was induced in this model 2 weeks after NTS administration and persisted until at least week 6, the latest time point in our study (Figure 3). Because our K21N antibody against B1R was raised against the last 21 amino acids of the carboxy-terminal domain of the human B1R, which is absent in the rodent B1R, and because no commercially available antibody works in our hands, we have been unsuccessful in detecting B1R protein in the mouse kidney sections.
Figure 3.
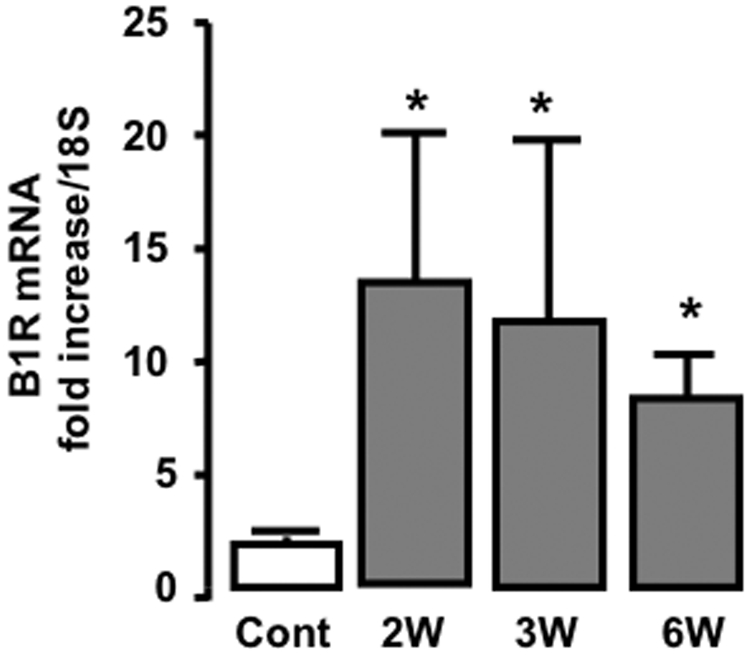
B1R mRNA expression is induced during NTS-induced nephritis. The B1R mRNA expression is assessed by B1R mRNA quantification using real-time PCR (n=6 mice per group). Cont, control mice; W, week. *P < 0.05 versus control.
Delayed B1Ra Treatment Improves Glomerular and Tubular Lesions and Blood Creatinine in NTS-Induced Glomerulonephritis
We next investigated whether delayed oral B1Ra treatment was effective in protecting the development of lesions during established immune glomerulonephritis. Vehicle-treated mice developed progressive renal injury from week 2 to week 6 as shown by increased crescent formation, cast deposits, and tubular atrophy. In contrast, delayed treatment with a B1Ra (SSR240612, 10 mg/kg every other day), initiated 2 weeks after NTS injection and continued until week 6, showed substantial improvement of the glomerulonephritis-induced lesions (Figure 4, A through E).
Figure 4.

Delayed B1Ra treatment reduces renal lesions and improves renal function. (A and B) Representative photographs of crescent and cast formation x400. (A through E) Crescent (A and C) and cast formation (B and D), as well as tubular atrophy (E), were assessed by periodic acid-Schiff staining after 2 and 6 weeks of NTS in vehicle-treated (○ or □) or B1Ra-treated (● or ■) mice. (F) Renal function is evaluated by measuring blood creatinine (n=6 mice per group). Cont, control mice; W, week. *P < 0.05 versus control; #P < 0.05 versus 6 weeks.
This model of glomerulonephritis induces progressive decline of renal function. We measured plasma creatinine in control mice and NTS vehicle– or B1Ra-treated mice at week 6. Compared with control mice, NTS mice exhibited increased plasma creatinine levels. Very interestingly, we observed that reduction of renal lesions by delayed B1Ra treatment was associated with significant improvement of renal function (Figure 4F).
B1R Blockade Reduces Fibrosis during NTS-Glomerulonephritis
We next evaluated the therapeutic effect of delayed B1R blockade on the progression of renal fibrosis. We immunostained sections for the extracellular matrix protein collagen III followed by histomorphometric analysis of the cortical collagen content. As shown in Figure 5, NTS administration significantly induced interstitial and glomerular fibrosis in vehicle-treated mice (Figure 5, B, E, and G) compared with control mice (Figure 5, A, D, and G). This effect was significantly inhibited by delayed B1Ra treatment (Figure 5, C, F, and G).
Figure 5.

B1R blockade inhibits the development of renal fibrosis. (A through F) The development of renal fibrosis is quantified by immunohistologic staining for collagen III in control mice (A and D) and vehicle-treated (B and E) or B1Ra-treated (C and F) mice 6 weeks after NTS administration. Representative pictures are shown. (G) Quantitative analysis of collagen III immunostaining (n=6 mice per group). Cont, control mice; W, week. *P < 0.05 versus control; #P < 0.05 versus 6 weeks. Magnifications: ×200 in A through C; ×400 in D through F.
Delayed B1R Blockade Reduces NTS-Induced Chemokine Expression and Renal Inflammation
We previously demonstrated that the B1R can modulate chemokine expression and inflammatory cell recruitment.21 We analyzed the effect of delayed B1R blockade in the glomerulonephritis model on CCL2 (Figure 6A), CCL7 (Figure 6B), and CCL5 (Figure 6C) mRNA expression, three chemokines known to be involved in the inflammatory process.22–24 We observed that delayed B1Ra treatment blunted NTS-induced chemokine upregulation (Figure 6).
Figure 6.

Delayed B1Ra treatment inhibits chemokine expression during NTS-glomerulonephritis. (A through C) CCL2 (A), CCL7 (B), and CCL5 (C) expression is quantified using real-time PCR 2 and 6 weeks after NTS injection in vehicle-treated (□) or B1Ra-treated (■) mice (n=6 mice per group). Cont, control mice; W, week. *P < 0.05 versus control; #P < 0.05 versus 6 weeks.
We next assessed the effect of B1R blockade on inflammatory cell recruitment. Because macrophages have been shown to be crucial for the progression of glomerulonephritis, we quantified macrophage accumulation by immunohistologic staining for F4/80. We observed that NTS administration induced a strong macrophage infiltrate in the interstitial space and in crescentic glomeruli (Figure 7, B and E) compared with control mice (Figure 7, A and D). The inhibitory effect of B1R blockade on renal chemokine expression was associated with a reduced macrophage infiltrate after 6 weeks of glomerulonephritis (Figure 7, C, F, and G). We also studied the effect of the treatment on the renal expression of CD4 (Figure 8A), CD8 (Figure 8B), and Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN) (Figure 8C), three markers for CD4+ and CD8+ T lymphocytes and dendritic cells (DCs), respectively. We found that B1Ra treatment significantly inhibited the expression of these markers during NTS- glomerulonephritis (Figure 8).
Figure 7.

B1R blockade inhibits macrophage infiltration. (A through F) Macrophage number is quantified by immunohistologic staining for F4/80 in control mice (A and D) and vehicle-treated (B and E) or B1Ra-treated (C and F) mice 6 weeks after NTS administration. Representative pictures are shown. (G) Quantitative analysis of F4/80 immunostaining (n=6 mice per group). Cont, control mice; W, week. *P < 0.05 versus control; #P < 0.05 versus 6 weeks. Magnifications: ×200 in A through C; ×400 in D through F.
Figure 8.

Delayed B1R blockade blunts NTS-induced upregulation of mRNA markers of T lymphocytes and DCs. (A through C) Expression of CD4 (A), CD8 (B), and DC-SIGN (C) mRNA was quantified using real-time PCR 2 and 6 weeks after NTS injection in vehicle-treated (□) or B1Ra-treated (■) mice (n=6 mice per group). Cont, control mice; W, week. *P < 0.05 versus control; #P < 0.05 versus 6 weeks.
Discussion
Collectively, our results show that the renal B1R is induced in human rapidly progressive glomerulonephritis and that delayed administration of a B1Ra can reduce the development of renal inflammation and lesions and improve renal function in the murine experimental model of NTS-induced glomerulonephritis. Glomerulonephritides constitute a group of diseases characterized by strong inflammation leading to crescent formation, tubular atrophy, fibrosis, and progressive renal failure. As recently reviewed, future therapy for these nephropathies should ideally target the renal inflammatory response1; however, nontoxic immunomodulatory molecules are still lacking.1 Because of its inducible character at the site of inflammation in different tissues, it has been suggested that blocking the proinflammatory B1R will be potentially without significant adverse effects. This has drawn attention to the B1R as a new therapeutic target for the treatment of pathologies related to chronic inflammation, such as airway inflammation, diabetic neuropathy, arthritis, and chronic and neuropathic pain.15 We and others previously described that B1R blockade can reduce the development of inflammation and subsequent fibrosis in the UUO model21,25; however, it remained unknown whether blockade of the B1R can inhibit immune nephropathies.
The association between B1R expression and inflammation is well described both in vitro and in vivo in animals; however, little is known about the expression of the B1R in humans. We investigated whether the B1R was expressed in human renal biopsies obtained from patients with rapidly progressive glomerulonephritis, AAV and Henoch-Schönlein purpura. Although undetectable in biopsies from control subjects and patients with MCNS, the human B1R is clearly induced in these nephropathies, as evidenced by the localization on tubular epithelial cells of diagnostic biopsies from patients with HSPN as well as in crescentic glomeruli from patients with vasculitis. The latter is consistent with a previous study showing the presence of the B1R agonist by using specific antipeptide antibodies, mainly in glomeruli but also in proximal and distal tubuli from patients with vasculitis.26 B1R staining in the interstitial inflammatory and fibrotic space is consistent with previous reports describing B1R expression in macrophages and fibroblasts in vitro.3,9,27 Although additional experiments are needed to define better the interstitial cells that express the B1R, the most important conclusion to be drawn from our data is that the B1R is not detectable in control kidney biopsies but clearly is expressed in pathologic biopsies from patients with glomerulonephritis.
We next evaluated the therapeutic potential of B1R delayed blockade in the murine model of accelerated NTS-glomerulonephritis. To mimic the human pathologic situation, whereby patients are treated in the presence of overt pathology, we chose to administer the B1Ra 2 weeks after the onset of the pathology. We observed reduction of renal lesions including crescent formation, cast deposits, tubular atrophy, and interstitial fibrosis. Moreover, after 4 weeks of treatment, we showed a significant improvement of renal function. As evidenced by a number of studies, interstitial inflammation constitutes an early and major event in response to NTS.22,28,29 We showed here that delayed B1Ra treatment reduced significantly macrophage recruitment and diminished the mRNA expression of markers for CD4+ and CD8+ T lymphocytes, three populations of immune cells involved in the inflammatory response in the NTS model.22,28,29 The role of the B1R on T lymphocyte recruitment has been poorly investigated, leading to controversial results. A recent study demonstrated that B1R knockout mice showed more severe disease in a model of experimental autoimmune encephalomyelitis and this was associated with enhanced Th17 cell infiltration.30 In contrast, in a model of murine lung inflammation, B1R blockade showed no effect on T lymphocyte number.31 Independent of these controversial results on the B1R, our results are in agreement with a study that conditional ablation of macrophages 2 weeks after the induction of NTS-glomerulonephritis improved renal lesions and interstitial fibrosis and that this effect was associated with a reduction of CD4+ cells.29
DCs are known to be present in the healthy tubulointerstitium, where they perform sentinel functions. During acute renal injury, DCs alert the immune system and regulate T cell activation. The role of these cells in experimental glomerulonephritis has been debated recently. Depletion of DCs has been shown to be deleterious in the acute phase of NTS-glomerulonephritis (i.e., 2 weeks after NTS).32 Indeed, the authors showed that these cells induce IL-10 producing CD4+ cell infiltration with anti-inflammatory effects32; however, using the NOH model, in which transgenic mice express the model antigens ovalbumin and hen egg lysosyme in glomerular podocytes, it was observed that during chronic crescentic glomerulonephritis, renal DCs are a link between the glomerular injury and the progression of kidney disease toward the tubulointerstitial compartment.33 This suggests that targeting DCs could improve chronic kidney disease progression. Our data support this hypothesis by showing that the protective effect of delayed B1R blockade is associated with a reduction of DC-SIGN mRNA in our progressive model of NTS-glomerulonephritis.
A number of chemokines are involved in renal inflammatory cell infiltration.34 The role of CCL2/MCP1,22,23 CCL5/RANTES,22 and CCL7/MCP324 is clearly established in this process. Our data show reduced renal expression of these three chemokines in response to B1Ra treatment in the NTS model. This is consistent with previous reports showing that in the NTS model, CCL2/MCP1 knockout mice or mice treated with an anti–CCL2-neutralizing antibody present less inflammation, fewer renal lesions, and less renal fibrosis compared with control mice.22,24 Moreover, mice treated with a CCL5 antagonist showed reduced inflammation and improved renal function, without significant modification of renal lesions.22
Although we are aware that human chronic kidney disease is much more complex than in experimental models and that clinical trials for such disease need studies of long duration, we propose that B1R blockade with an orally active B1Ra might be an efficient strategy to prevent or significantly slow the progression of inflammatory kidney diseases in humans and should be evaluated against and in combination with established therapies.
Concise Methods
Drug
B1Ra SSR240612 was a gift of Sanofi-Aventis R&D Montpellier-France (P. Carayon16). For in vivo experiments, this compound was dissolved in water containing 2% DMSO to obtain a 1-g/L solution. The SSR240612 solution was diluted with distilled water and administered by gavage. Final DMSO concentration was 0.01%.
Human Kidney Specimens
We retrospectively analyzed paraffin sections of renal biopsies from patients (pediatric and adults) who underwent diagnostic evaluation from 2001 through 2005. A recent approved pediatric clinical project (PHRC-AOL 0507302-2006) allowed us to obtain mRNA from biopsies stored in a commercially available RNase inhibitor solution (RNAlater; Ambion). Informed consent was given by the patients or their parents for use of part of the biopsy for scientific purpose. All procedures and use of tissue were performed according to national ethical guidelines and were in accordance with the Declaration of Helsinki.
Animals
The mice were housed in a pathogen-free environment. All experiments reported were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by a local animal care and use committee.
Glomerulonephritis
The protocol and the preparation of the NTS used have been previously described.22 Briefly CD-1 male mice of 6 weeks of age were preimmunized by subcutaneous injection of 200 μg of normal sheep IgG (Sigma) in Freunds complete adjuvant (Sigma). Five days later, immunization was performed by intravenous injection of 50 μl of NTS into the tail vein for 3 consecutive days.22 Mice were maintained on a standard mouse chow and tap water. Treatment with the B1Ra at 10 mg/kg (dosage selected on the basis of literature data)16,19–21 was initiated 2 weeks after nephritis induction and continued throughout the time of nephritis. The dosage of the B1Ra was given orally every other day. Control group received by oral route the 0.01% DMSO solution, which had no significant effect on body weight (animals receiving the B1Ra, body weight at 6 weeks 39.08 ± 4.80 g versus control animals receiving only the 0.01% DMSO solution 38.08 ± 1.40 g). Mice were killed at various time points, and the kidneys were removed and divided into different parts according to the different protocols used. Plasma was collected to measure blood creatinine (Creatinine CP; HORIBA-ABX) with the COBAS Mira Plus device.
Histologic Analysis and Immunohistochemistry
From paraffin-embedded biopsies, we performed routine histology. Four-micrometer sections were cut and used for routine staining (hematoxylin-eosin and periodic acid-Schiff staining) and immunohistochemistry. All human biopsies were scored (from 0+ to 3+) by an expert pathologist who was blinded to the clinical data to evaluate the presence of inflammatory infiltrates, glomerulosclerosis, and interstitial fibrosis. For immunohistochemistry, human and mouse renal tissue were first de-waxed in toluene and rehydrated through a series of graded ethanol washes before endogenous peroxidase blockage. Specific primary antibodies were incubated (1 hour at room temperature) on human or mouse sections for the detection of B1R (1:5000),7 collagen type III (1:500; Interchim), and anti-mouse F4/80 (1:250; RM2900; Caltag laboratories Inc., Burlingame, CA). For visualization, we used the Dako Envision system. Sections were finally counterstained with hematoxylin. Negative controls for the immunohistochemical procedures included substitution of the primary antibody with nonimmune sera.
Histomorphometric analyses were performed as described previously,35 using a commercially available image analysis software that allows rebuilding of a kidney section from adjacent individual captures (Explora Nova Mosaïc software, La Rochelle, France). The number of glomerular crescents was determined, by blinded analysis, of at least 100 glomeruli on periodic acid-Schiff–stained kidney sections, and only glomeruli that had two or more layers of cells in Bowman's space were considered crescentic as previously reported.36 Atrophic tubules were identified by their irregular, thickened basement membranes.
Isolation of RNA
Total RNA was isolated from mouse kidney sections or renal biopsies using QIAGEN RNeasy Mini kit, eluted in 20 μl of RNase-free water and treated by DNase (TURBO DNA-free kit; Ambion) according to the manufacturer's protocol. A total of 1.5 μl of this solution was used for quantification by a NanoDrop instrument (ND-1000 spectrophometer).
cDNA Synthesis
A total of 100 ng of total RNA was used for cDNA synthesis by High Capacity cDNA Archive Kit (Applied Biosystems) in a volume of 100 μl following the manufacturer's protocol. We then diluted cDNA by a factor of 1:2.
Quantification of Gene Expression by Real-Time Quantitative PCR
Real-time PCR was performed using the ABI PRISM 7900 HT. PCR amplification was performed in a total volume of 25 μl containing 25 ng of cDNA sample, 300 nM of forward and reverse primer and 12.5 μl of Power SYBR Green PCR Master Mix (Applied Biosystems). The reaction mixture was preheated at 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute.
Statistical Analysis
Data are expressed as means ± SD. ANOVA with post hoc Tukey α test was performed for comparison between the various groups. P < 0.05 was considered statistically significant.
Disclosures
None.
Acknowledgments
This work was funded in part by the “111 des Arts” project and by the “Egide-Van Gogh” collaborative project. J.K. was supported by the Fondation pour la Recherche Médicale Fellowship Grant. J.L.B. was supported by INSERM and the “Direction Régionale Clinique” (CHU de Toulouse, Toulouse, France) under the Interface program. D.J.S. was supported by research grant DK30932 from the National Institutes of Health. B1Ra SSR240612 was a kind gift of Sanofi-Aventis R&D Montpellier-France (P. Carayon).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
REFERENCES
- 1. Kitching AR, Holdsworth SR, Hickey MJ: Targeting leukocytes in immune glomerular diseases. Curr Med Chem 15: 448–458, 2008 [DOI] [PubMed] [Google Scholar]
- 2. Regoli D, Barabe J: Pharmacology of bradykinin and related kinins. Pharmacol Rev 32: 1–46, 1980 [PubMed] [Google Scholar]
- 3. Leeb-Lundberg LM, Marceau F, Muller-Esterl W, Pettibone DJ, Zuraw BL: International union of pharmacology: XLV. Classification of the kinin receptor family: From molecular mechanisms to pathophysiological consequences. Pharmacol Rev 57: 27–77, 2005 [DOI] [PubMed] [Google Scholar]
- 4. Marceau F: Kinin B1 receptors: A review. Immunopharmacology 30: 1–26, 1995 [DOI] [PubMed] [Google Scholar]
- 5. Marin-Castano ME, Schanstra JP, Praddaude F, Pesquero JB, Ader JL, Girolami JP, Bascands JL: Differential induction of functional B1-bradykinin receptors along the rat nephron in endotoxin induced inflammation. Kidney Int 54: 1888–1898, 1998 [DOI] [PubMed] [Google Scholar]
- 6. Schanstra JP, Marin-Castano ME, Praddaude F, Tack I, Ader JL, Girolami JP, Bascands JL: Bradykinin B(1) receptor-mediated changes in renal hemodynamics during endotoxin-induced inflammation. J Am Soc Nephrol 11: 1208–1215, 2000 [DOI] [PubMed] [Google Scholar]
- 7. Schanstra JP, Bataille E, Marin Castano ME, Barascud Y, Hirtz C, Pesquero JB, Pecher C, Gauthier F, Girolami JP, Bascands JL: The B1-agonist [des-Arg10]-kallidin activates transcription factor NF-kappaB and induces homologous upregulation of the bradykinin B1-receptor in cultured human lung fibroblasts. J Clin Invest 101: 2080–2091, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phagoo SB, Poole S, Leeb-Lundberg LM: Autoregulation of bradykinin receptors: Agonists in the presence of interleukin-1beta shift the repertoire of receptor subtypes from B2 to B1 in human lung fibroblasts. Mol Pharmacol 56: 325–333, 1999 [DOI] [PubMed] [Google Scholar]
- 9. Tiffany CW, Burch RM: Bradykinin stimulates tumor necrosis factor and interleukin-1 release from macrophages. FEBS Lett 247: 189–192, 1989 [DOI] [PubMed] [Google Scholar]
- 10. Ahluwalia A, Perretti M: Involvement of bradykinin B1 receptors in the polymorphonuclear leukocyte accumulation induced by IL-1 beta in vivo in the mouse. J Immunol 156: 269–274, 1996 [PubMed] [Google Scholar]
- 11. Bockmann S, Paegelow I: Kinins and kinin receptors: Importance for the activation of leukocytes. J Leukoc Biol 68: 587–592, 2000 [PubMed] [Google Scholar]
- 12. Pesquero JB, Araujo RC, Heppenstall PA, Stucky CL, Silva JA, Jr, Walther T, Oliveira SM, Pesquero JL, Paiva AC, Calixto JB, Lewin GR, Bader M: Hypoalgesia and altered inflammatory responses in mice lacking kinin B1 receptors. Proc Natl Acad Sci U S A 97: 8140–8145, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Souza DG, Lomez ES, Pinho V, Pesquero JB, Bader M, Pesquero JL, Teixeira MM: Role of bradykinin B2 and B1 receptors in the local, remote, and systemic inflammatory responses that follow intestinal ischemia and reperfusion injury. J Immunol 172: 2542–2548, 2004 [DOI] [PubMed] [Google Scholar]
- 14. Duchene J, Ahluwalia A: The kinin B(1) receptor and inflammation: New therapeutic target for cardiovascular disease. Curr Opin Pharmacol 9: 125–131, 2009 [DOI] [PubMed] [Google Scholar]
- 15. Campos MM, Leal PC, Yunes RA, Calixto JB: Non-peptide antagonists for kinin B1 receptors: New insights into their therapeutic potential for the management of inflammation and pain. Trends Pharmacol Sci 27: 646–651, 2006 [DOI] [PubMed] [Google Scholar]
- 16. Gougat J, Ferrari B, Sarran L, Planchenault C, Poncelet M, Maruani J, Alonso R, Cudennec A, Croci T, Guagnini F, Urban-Szabo K, Martinolle JP, Soubrie P, Finance O, Le Fur G: SSR240612 [(2R)-2-[((3R)-3-(1,3-benzodioxol-5-yl)-3-[[(6-methoxy-2-naphthyl)sulfonyl]amino]propanoyl)amino]-3-(4-[[2R,6S)-2,6-dimethylpiperidinyl]methyl]pheny l)-N-isopropyl-N-methylpropanamide hydrochloride], a new nonpeptide antagonist of the bradykinin B1 receptor: Biochemical and pharmacological characterization. J Pharmacol Exp Ther 309: 661–669, 2004 [DOI] [PubMed] [Google Scholar]
- 17. Hara DB, Leite DF, Fernandes ES, Passos GF, Guimaraes AO, Pesquero JB, Campos MM, Calixto JB: The relevance of kinin B1 receptor upregulation in a mouse model of colitis. Br J Pharmacol 154: 1276–1286, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pietrovski EF, Otuki MF, Regoli D, Bader M, Pesquero JB, Cabrini DA, Zampronio AR: The non-peptide kinin receptor antagonists FR 173657 and SSR 240612: Preclinical evidence for the treatment of skin inflammation. Regul Pept 152: 67–72, 2009 [DOI] [PubMed] [Google Scholar]
- 19. Dias JP, Ismael MA, Pilon M, de Champlain J, Ferrari B, Carayon P, Couture R: The kinin B1 receptor antagonist SSR240612 reverses tactile and cold allodynia in an experimental rat model of insulin resistance. Br J Pharmacol 152: 280–287, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Quintao NL, Passos GF, Medeiros R, Paszcuk AF, Motta FL, Pesquero JB, Campos MM, Calixto JB: Neuropathic pain-like behavior after brachial plexus avulsion in mice: The relevance of kinin B1 and B2 receptors. J Neurosci 28: 2856–2863, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Klein J, Gonzalez J, Duchene J, Esposito L, Pradere JP, Neau E, Delage C, Calise D, Ahluwalia A, Carayon P, Pesquero JB, Bader M, Schanstra JP, Bascands JL: Delayed blockade of the kinin B1 receptor reduces renal inflammation and fibrosis in obstructive nephropathy. FASEB J 23: 134–142, 2009 [DOI] [PubMed] [Google Scholar]
- 22. Lloyd CM, Minto AW, Dorf ME, Proudfoot A, Wells TN, Salant DJ, Gutierrez-Ramos JC: RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J Exp Med 185: 1371–1380, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Segerer S, Nelson PJ, Schlondorff D: Chemokines, chemokine receptors, and renal disease: From basic science to pathophysiologic and therapeutic studies. J Am Soc Nephrol 11: 152–176, 2000 [DOI] [PubMed] [Google Scholar]
- 24. Tesch GH, Schwarting A, Kinoshita K, Lan HY, Rollins BJ, Kelley VR: Monocyte chemoattractant protein-1 promotes macrophage-mediated tubular injury, but not glomerular injury, in nephrotoxic serum nephritis. J Clin Invest 103: 73–80, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang PH, Cenedeze MA, Campanholle G, Malheiros DM, Torres HA, Pesquero JB, Pacheco-Silva A, Camara NO: Deletion of bradykinin B1 receptor reduces renal fibrosis. Int Immunopharmacol 9: 653–657, 2009 [DOI] [PubMed] [Google Scholar]
- 26. Kahn R, Herwald H, Muller-Esterl W, Schmitt R, Sjogren AC, Truedsson L, Karpman D: Contact-system activation in children with vasculitis. Lancet 360: 535–541, 2002 [DOI] [PubMed] [Google Scholar]
- 27. Ricupero DA, Romero JR, Rishikof DC, Goldstein RH: Des-Arg(10)-kallidin engagement of the B1 receptor stimulates type I collagen synthesis via stabilization of connective tissue growth factor mRNA. J Biol Chem 275: 12475–12480, 2000 [DOI] [PubMed] [Google Scholar]
- 28. Cook HT, Singh SJ, Wembridge DE, Smith J, Tam FW, Pusey CD: Interleukin-4 ameliorates crescentic glomerulonephritis in Wistar Kyoto rats. Kidney Int 55: 1319–1326, 1999 [DOI] [PubMed] [Google Scholar]
- 29. Duffield JS, Tipping PG, Kipari T, Cailhier JF, Clay S, Lang R, Bonventre JV, Hughes J: Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol 167: 1207–1219, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schulze-Topphoff U, Prat A, Prozorovski T, Siffrin V, Paterka M, Herz J, Bendix I, Ifergan I, Schadock I, Mori MA, Van Horssen J, Schroter F, Smorodchenko A, Han MH, Bader M, Steinman L, Aktas O, Zipp F: Activation of kinin receptor B1 limits encephalitogenic T lymphocyte recruitment to the central nervous system. Nat Med 15: 788–793, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gama Landgraf R, Sirois P, Jancar S: Differential modulation of murine lung inflammation by bradykinin B1 and B2 selective receptor antagonists. Eur J Pharmacol 460: 75–83, 2003 [DOI] [PubMed] [Google Scholar]
- 32. Scholz J, Lukacs-Kornek V, Engel DR, Specht S, Kiss E, Eitner F, Floege J, Groene HJ, Kurts C: Renal dendritic cells stimulate IL-10 production and attenuate nephrotoxic nephritis. J Am Soc Nephrol 19: 527–537, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heymann F, Meyer-Schwesinger C, Hamilton-Williams EE, Hammerich L, Panzer U, Kaden S, Quaggin SE, Floege J, Grone HJ, Kurts C: Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J Clin Invest 119: 1286–1297, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kluth DC, Erwig LP, Rees AJ: Multiple facets of macrophages in renal injury. Kidney Int 66: 542–557, 2004 [DOI] [PubMed] [Google Scholar]
- 35. Schanstra JP, Neau E, Drogoz P, Arevalo Gomez MA, Lopez Novoa JM, Calise D, Pecher C, Bader M, Girolami JP, Bascands JL: In vivo bradykinin B2 receptor activation reduces renal fibrosis. J Clin Invest 110: 371–379, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heeringa P, van Goor H, Itoh-Lindstrom Y, Maeda N, Falk RJ, Assmann KJ, Kallenberg CG, Jennette JC: Lack of endothelial nitric oxide synthase aggravates murine accelerated anti-glomerular basement membrane glomerulonephritis. Am J Pathol 156: 879–888, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]