

Abstract
In this review, we summarize key observations supporting the idea of immunosenescence in the CNS. We provide a discussion of senescent changes that affect microglial cells and emphasize differences between laboratory rodents and humans. Microglial immunosenescence may explain why humans but not rodents develop neurofibrillary degeneration.
Keywords: Aging, immune system, neurofibrillary degeneration, immunosenescence
In the space of approximately fifteen years (1985–2000), neuroimmunological thinking went from viewing the CNS as a sheltered place, i.e. an immunologically privileged organ, to believing that the CNS has its own immune system which is dangerous and produces detrimental neuroinflammation that causes neurodegeneration, in other words, that there is hardly any privilege at all. Even for those with a progressive mindset this kind of conceptual shift must be nothing short of astounding. However, the radical shift in opinions may be explained simply by the fact that both of these extreme views became dogmatic before they were thoroughly scrutinized. We now know that the brain’s immune privilege is not complete, i.e. it exists only in the sense that immunological reactions occur a bit slower and with lower intensity than they do in the periphery, and we are beginning to realize that neuroinflammation is not an autodestructive process, but occurs as a reparative tissue response to neuronal injury and disease. It is clear furthermore, with some hindsight, that a key event contributing to this paradigm shift in neuroimmunology was the rediscovery of microglial cells during the 1980’s and 90’s, a development that was discussed in some detail just recently [1].
Notwithstanding these caveats, we believe it is not only reasonable but also accurate to view microglia as the brain’s immune system. Clearly, something special had to occur, evolutionarily speaking, to endow a vulnerable organ with limited regenerative capacity, such as the CNS, with immunological surveillance while at the same time protecting it from the detrimental side effects of potentially fulminant immune reactions. Nature’s answer to this predicament is microglia, cells that function as both neuroprotective glia and immunologically competent cells at the same time. One might say that nature has struck a compromise to all-out immune surveillance via blood-borne leukocytes by colonizing the CNS early in development with a special population of cells that in a functional sense represent a hybrid between leukocytes and neuroprotective glia, and naturally, it is a job well done. However, it is normal for all living things to age, and just like immunological defenses decline with aging in the periphery the brain’s immune system also is subject to aging. We are only beginning to grasp this latter notion and while the consequences of CNS immunosenescence are far from clear, in this paper we will summarize what is known about microglial aging, reiterating and expanding upon the microglial dysfunction hypothesis, a theory first posited some time ago in the context of Alzheimer’s disease (AD) and which claims that the neurofibrillary degeneration of AD is largely the result of declining microglial cell function and viability [2]. Thus, while immunosenescence in the periphery accounts for increased susceptibility of the elderly to infections and cancer, the sequelae of CNS immunosenescence may be related primarily to waning microglial neuroprotection resulting in aging-related neurodegenerative changes. There is only one assumption that has to be made in order for the microglial dysfunction theory to stand, and that is the belief that microglia are entirely beneficial and supportive in terms of maintaining CNS homeostasis and ensuring neuronal stability and viability. Observations that support a neuroprotective role of microglia have previously been reviewed and the interested reader is referred to these earlier papers and references therein [3–7].
Features of microglial senescence in rodents
There are a number of aspects to microglial senescence, and these include cell morphology, replication, biochemistry, and function. The first detailed study undertaken in this regard was by Vaughan and Peters, who described the evolution of ultrastructural differences in microglia from rats that were aged between 3 and 30 months [8]. The authors observed that microglia, much more so than astrocytes and oligodendrocytes, show a remarkable accumulation of membrane –bound inclusion material that bears resemblance to lipofuscin. They also noted changes in cell shape and in cell number, that is, with advancing age microglial cells were transformed from multipolar (ramified) to more elongated and/or spherical forms, and their total numbers increased by 65% over the 27-month period examined. While the authors pointed out similarities between these aging-related changes and those that had been described to be characteristic of activated microglia after acute CNS lesions, they also discussed differences, such as the composition and appearance of inclusion bodies which was indicative of slow accumulation and condensation. Because they were unable to observe any pathological changes in neurons the authors came to the final conclusion that morphological and numerical changes in microglia were a normal response to aging. These initial observations by Vaughan and Peters in the rat were corroborated a couple of years later by Samorajski in the human brain stating that, “ Among the neuroglial cells, the microglia undergo the most significant changes with age.” [9].
Observations on microglial morphology and cell numbers in the rat were confirmed and extended by Perry et al. [10] who provided a first account of the changes occurring in microglial immunophenotype with aging. Using antibodies directed against various immune system antigens, Perry and colleagues showed that a number of antigens, most notably those of the major histocompatibility complex (MHC), become increasingly expressed on microglia with aging. They concluded that with aging microglia assume more of a phagocyte-like phenotype marked by upregulation of macrophage and other immunologically important surface antigens. Their findings are consistent with the aforementioned ultrastructural studies showing accumulation of intracytoplasmic inclusion bodies, and they have been corroborated by others [11,12]. Our own work in the aged rat brain has also shown both microglial hypertrophy and lipofuscin accumulation (Figure 1), suggesting that aging-related hypertrophy of the microglial cytoplasm is due to aging-related accumulation of lipofuscin. Thus, even though senescent microglia may resemble the brain macrophages that appear at any age during an acute neuroinflammatory response, there is a fundamental difference between the two microglial phenotypes: senescent cells are enlarged because they accumulate nondegraded waste material gradually during their life-long “careers” as scavengers, whereas microglia-derived macrophages appearing acutely in response to a lesion are enlarged because they ingest large amounts of tissue debris in a very short period of time. Another difference is that acquisition of the phagocyte-like phenotype during aging seems to affect many, although not all, microglial cells more or less uniformly across different CNS regions, whereas injury-associated brain macrophages are, of course, limited to the immediate vicinity of a lesion. The important corollary of all of this is that aging-related changes are to be distinguished from inflammatory changes, which arise in response to injury. Normal brain aging is accompanied by little, if any apparent injury to neurons or other cells and thus aging-associated morphological, immunophenotypical, and biochemical changes occurring in microglia should not be designated as neuroinflammation or microglial activation. Paradoxically, if with aging the strength of peripheral immune defenses declines, why at the same time would immunological responsiveness increase within the CNS?
Figure 1.

Morphological comparison of microglial cells in the young (A) and old (B) rat brain. Microglia (fluorescently immunolabeled by antibody Iba1 in green, arrow head), along with neurons (arrows) in the aged rat brain accumulate lipofuscin which is autofluorescent (red). 28 day-old (A) and 39 month-old rat cerebral cortex with DAPI counterstaining (blue).
How can one explain the documented increase in microglial cell numbers with aging? Microglia differ from other mature glial cells in that they retain a remarkable capacity to undergo mitosis in adult mammals, and their replicative potential has been demonstrated in numerous in vivo DNA labeling studies [13–17]. While a burst of proliferative activity occurs during microglial activation following a CNS lesion, it is likely that steady- state mitosis of microglia occurs continuously and throughout life within the CNS at rather low levels. Lawson et al. [16] found that endogenous microglial proliferation occurs randomly and sporadically throughout the normal mouse CNS not showing predilection for any particular region, and these authors and others [15] concluded that replenishment of microglia in the normal CNS occurs via mitosis of resident microglia as well as through immigration of blood borne precursor cells. Based on their observation that microglia had a lower DNA labeling index than most resident macrophage populations in other organs, Lawson et al. concluded further that microglia form an extremely stable and long-lived cell population. Our own DNA labeling studies in the normal rodent CNS have confirmed presence of BrdU-labeled microglia in the subventricular and hippocampal neurogenic niches showing that new microglial cells are being generated in adult animals (unpublished). Assuming that such endogenous microgliogenesis continues over a lifetime and that it exceeds the level of microglial cell death, it could lead to an accumulation of aged microglial cells and thus offer an explanation for the higher numbers of microglia encountered in the aged rodent brain. Our studies of microgliosis following acute injury of motoneurons in aged rats support this idea by showing that the rate of microglial proliferation is greater than the rate of microglial programmed cell death [13]. In the same study, we also found that the acute proliferative burst of microglia after motoneuron axotomy is greater in aged than in young animals, which at first glance and with regard to expectations regarding cell senescence seemed counterintuitive. However, this unexpected finding may have an explanation in the possibility that enhanced microglial proliferation after acute injury in aged animals is necessary to compensate for a presumptive aging-associated decline in the vigor of individual cells. Thus, in order for activated microglia to still be able to aid in the regeneration of axotomized motoneurons in aged animals, greater numbers of microglia may be required than in young animals where microglia have a higher level of vitality. In other words, an enhanced proliferative response in aged animals may well be a reflection of cell senescence.
Although the proliferative potential of microglia is clearly advantageous in terms of the cells’ renewal potential, this replicative ability of microglial cells may come at a price if they are forced to divide repeatedly, namely, that microglia are subject to replicative senescence. Replicative senescence is also known as the Hayflick limit; in 1961 Hayflick and Moorhead [18] discovered that diploid cells maintained in vitro were not immortal and that there exists a finite limit to the cultivation period of diploid cells. They found that cells gradually lose mitotic potential and degenerate after about 50 subcultivations, and they formulated the hypothesis that this phenomenon was due to intrinsic factors which are expressed as senescence at the cellular level. Later on it was discovered that cell senescence is accompanied by telomere shortening, a process by which the non-coding ends of chromosomes become shortened progressively with each cell cycle ultimately resulting in cell death [19], unless they are re-elongated by telomerase enzyme [20]. As it turns out, microglial cells maintained in vitro proliferate spontaneously, but when their proliferation rate is accelerated through addition of growth factors (e.g. GM-CSF) forcing them to rapidly go through many cell cycles they will undergo significant telomere shortening and become senescent [21]. Interestingly, this is not the case for cultured astrocytes, likely because astrocytes are better able to re-extend shortened telomeres through upregulating telomerase activity. As an aside, it is interesting to speculate that an increased ability of astrocytes to generate telomerase activity could explain the fact that most brain tumors are astrocytomas, whereas microgliomas represent an exceedingly rare and rather controversial neuropathological entity commonly grouped under primary CNS lymphomas [22].
In order to demonstrate and measure microglial replicative senescence in vivo, we devised an injury paradigm that triggers repeated rounds of microglial cell division. The paradigm involves repeated axotomy of the facial nerve and our findings obtained thus far have shown that the number of cells incorporating BrdU after four axotomies is significantly lower than the number of BrdU-positive cells after just one axotomy [23], thus lending further credence to the idea that declining mitotic capacity is part of the cellular senescence affecting microglial cells. We have not yet studied biochemical changes that may be associated with altered microglial biology in this repeated injury paradigm, such as cytokine and growth factor production, but studies of cultured or acutely isolated cells have shown that cytokine production in microglia changes significantly with chronological aging [24–28]. While these studies vary in their results with regard to differential upregulation of pro- and anti-inflammatory cytokines, a consistent finding across the board is the increased expression of interleukin-6 at both mRNA and protein levels [29]. Interleukin-6 has long been known as a growth factor for B cells and early bone marrow hematopoietic stem cells [30], but its role within the CNS is not entirely clear since it appears that various cell types can express it, including astrocytes, neurons, and microglia [29,31]. However, with reference to the aforementioned aging-associated increase in microglial cell numbers an interesting possibility is that IL-6 serves as a microglial growth factor and mitogen. Thus, a possible reason why IL-6 increases with aging could be that it represents a mechanism by which the system is trying to compensate for microglial senescence by generating new and viable microglia via proliferation of resident cells. Several independent lines of evidence support this idea: a) IL-6 mRNA undergoes dramatic upregulation shortly after neuronal injury and precedes the proliferative burst of microglia [32]; b) IL-6 stimulates microglial mitosis in vitro [31]; c) IL-6 knockout mice show decreased microglial proliferation after neuronal injury [33]; d) IL-6 overexpressing mice show an increase in ramified microglial cells [34]; e) viral vector-mediated overexpression of IL-6 in APP mice produces robust microgliosis concomitant with a reduction in Aβ deposition [35]. It appears therefore that continued studies on IL-6 expression could provide new insights into how the microglial population is maintained naturally during aging, and how IL-6 mimetic drugs could perhaps be used to further facilitate, or ameliorate this process if it becomes defective.
Features of microglial senescence in humans
Since experimentation in humans is obviously limited, most of what we know regarding microglial senescence is derived from post-mortem histopathological studies or from the analysis of bodily fluids, i.e. CSF and blood. However, it is clear and quite important to realize that aging-related changes in microglia present differently in humans than they do in aged laboratory rats and mice. There is direct histopathological evidence in human brain for microglial senescence found in aging-related morphological changes. These structural changes have been termed microglial dystrophy and they are characterized by some rather unique alterations of the microglial cytoplasm and its processes [36]. Some of these morphological changes are quite subtle while others are striking and unambiguous, and clearly indicative of degeneration. Of particular interest is microglial cytorrhexis, fragmentation of the microglial cytoplasm, which probably represents the most advanced stage of microglial dystrophy (senescence) and likely coincides with microglial cell death (Figure 2). In AD and in Down’s syndrome brain we have found fragmented microglia to be co-localized very consistently and in great abundance with neurons undergoing neurofibrillary degeneration [37]. Together with the additional observation that microglial cytorrhexis precedes the onset of neurofibrillary degeneration in humans, these findings offer strong support for the idea that aging-related neurodegeneration is due in large part to microglial deterioration and associated loss of microglial neuroprotection. A very intriguing aspect of microglial cytorrhexis is that it does not occur in normally aged wild type rodents, and so it comes perhaps as no surprise that aged rodents do not develop neurofibrillary degeneration and dementia. Indeed, one might say that this absence of fully developed microglial degeneration in conjunction with an absence of neurofibrillary degeneration in wild type rodents strongly supports the microglial dysfunction hypothesis. Interestingly, inbred senescence-accelerated mice (SAMP-10) do develop more extensive microglial pathology than controls (SAMR1), but even these animals do not develop widespread microglial cytorrhexis and, perhaps consequentially, no neurofibrillary degeneration [38] However, SAMP-10 mice do exhibit some neuronal loss and atrophy [39]. Microglial degeneration also has not been reported to occur in transgenic mouse models of Alzheimer’s, which do not develop neurofibrillary degeneration. It is somewhat unfortunate that most research on AD pathogenesis over the past twenty years has been disproportionately focused on an alleged key role of amyloid-beta protein (Aβ) as the main cause of neurofibrillary degeneration rather than on aging-related brain changes. It is troubling to see that these efforts on the perceived central pathogenic role of Aβ continue with seemingly undiminished zeal despite the fact that Aβ-overexpressing mice fail to develop neurofibrillary degeneration and dementia, findings that essentially refute the amyloid cascade hypothesis. In light of such major differences in brain pathology between mice and humans it makes sense that much of the transgenic mouse work is untranslatable to humans [40].
Figure 2.
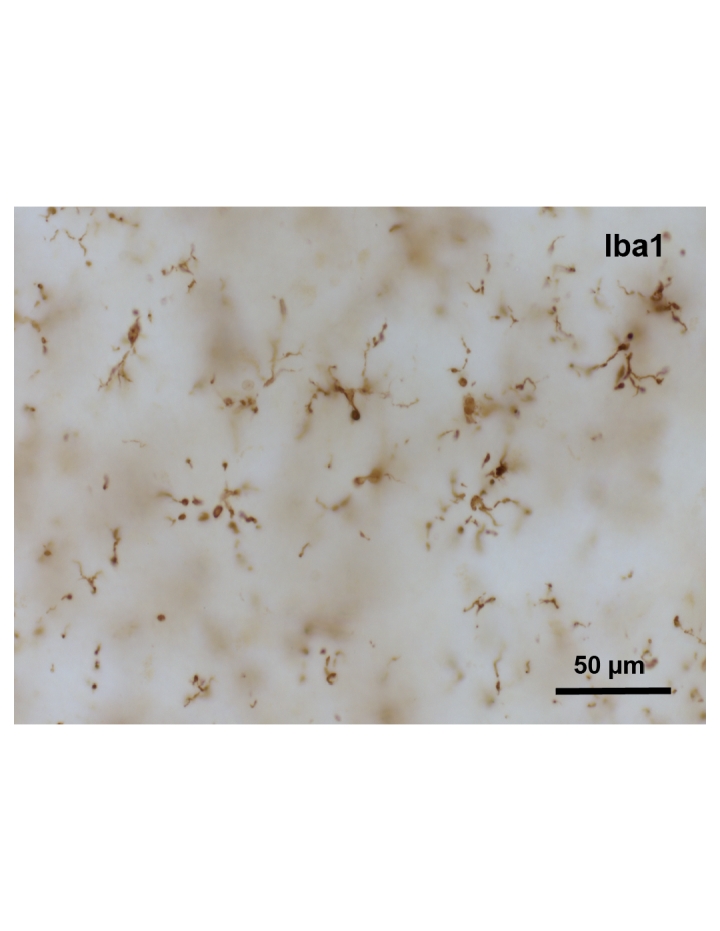
Microglial cytorrhexis is apparent in the cerebral cortex of a human with Alzheimer’s disease. Subject was an 87 year-old male with Alzheimer’s disease, Braak stage V. Iba1 immunohistochemistry.
Another difference between rodents and humans is the heterogeneity of microglial morphology that is apparent in the human but not so much in the rodent brain, and this difference is most likely due to the vast difference in lifespan between mice and men. In humans, assuming that there is indeed some level of microglial turnover, renewal, and death occurring over a lifetime, the most plausible explanation for microglial heterogeneity is a difference in the ages of individual cells. Thus, one could anthropomorphically liken a healthy microglial cell population to a normally distributed human population comprised of some very young and some very old individuals with the majority of individuals being distributed over a bell curve in between. What appears to happen to the microglial cell population during AD development is that, due to aging and possibly other yet unknown factors, the bell curve becomes skewed towards the right resulting in a disproportionate increase in “elderly” microglia. Thus major challenges for future research in AD pathogenesis would be to develop a system by which one could quantify old microglia through further refinements in phenotypic characterization, and to identify potential risk factors that accelerate microglial aging.
In addition to the direct histopathological evidence for microglial senescence, there are a number of other, more indirect lines of evidence derived from blood and CSF analyses that are consistent with the idea that microglial functional deterioration and degeneration is an important component of AD. Ineffective phagocytosis of amyloid-beta protein has been shown in monocytes derived from AD blood samples [41]. These authors found that compared to monocytes derived from age-matched control patients, which internalized and degraded Aβ, the monocytes of AD patients showed only surface uptake of Aβ and were prone to undergo cell death clearly pointing towards a malfunction in macrophage linage cells that can be associated with AD. These findings in human subjects seem to be consistent with in vitro studies using rodent microglia which show that there is an age-related decrease in the ability of microglia to internalize Aβ [27,42]. Further evidence in favor of the idea that AD may be associated with generalized immune dysfunction and/or immunosenescence comes from studies showing that there is significant telomere shortening occurring in peripheral blood mononuclear cells in AD versus control subjects [43], and that antimicroglial antibodies can be found in the CSF of AD subjects [44]. This latter study is of special interest because the extent to which such antimicroglial antibodies are present in the CSF apparently correlates well with disease severity, a finding that matches with histopathological observations, i.e. presence of degenerating microglia increases with more advanced Braak stages [37]. Thus, the extent of neurodegeneration can be directly correlated with the extent of microglial degeneration, exactly as predicted by the microglial dysfunction hypothesis.
Conclusions
We may have reached a turning point in terms of our understanding of the neuroimmunology of AD, and perhaps other neurodegenerative diseases, by realizing that neurodegeneration is not the result of an overactive immune response, but rather occurs because of a decline in immunological functions. Because in the CNS microglia are both immunological defenders and neuroprotective glia, the consequences of microglial senescence include both increased susceptibility to brain infections, as well as an increased susceptibility to neurodegeneration. The challenge is to identify factors or conditions that exacerbate normal microglial aging and then devise appropriate interventions. The hope is that this strategy may facilitate healthy aging for a larger percentage of the human population thereby helping to curb the ever increasing incidence of neurodegenerative diseases.
Acknowledgments
Supported by NIH grant AG023665.
References
- [1].Streit WJ. Microglial activation and neuroinflammation in Alzheimer's disease: a critical examination of recent history. Front Aging Neurosci. 2010;2:22. doi: 10.3389/fnagi.2010.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Streit WJ. Microglia and Alzheimer's disease pathogenesis. J Neurosci Res. 2004;77:1–8. doi: 10.1002/jnr.20093. [DOI] [PubMed] [Google Scholar]
- [3].Nakajima K, Kohsaka S. Microglia: neuroprotective and neurotrophic cells in the central nervous system. Curr Drug Targets Cardiovasc Haematol Disord. 2004;4:65–84. doi: 10.2174/1568006043481284. [DOI] [PubMed] [Google Scholar]
- [4].Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- [5].Streit WJ. Microglia and neuroprotection: implications for Alzheimer's disease. Brain Res Brain Res Rev. 2005;48:234–239. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- [6].Streit WJ. Microglial senescence: does the brain's immune system have an expiration date? Trends Neurosci. 2006;29:506–510. doi: 10.1016/j.tins.2006.07.001. [DOI] [PubMed] [Google Scholar]
- [7].Banati RB, Graeber MB. Surveillance, intervention and cytotoxicity: is there a protective role of microglia? Dev Neurosci. 1994;16:114–127. doi: 10.1159/000112098. [DOI] [PubMed] [Google Scholar]
- [8].Vaughan DW, Peters A. Neuroglial cells in the cerebral cortex of rats from young adulthood to old age: an electron microscope study. J Neurocytol. 1974;3:405–429. doi: 10.1007/BF01098730. [DOI] [PubMed] [Google Scholar]
- [9].Samorajski T. How the human brain responds to aging. J Am Geriatr Soc. 1976;24:4–11. doi: 10.1111/j.1532-5415.1976.tb03246.x. [DOI] [PubMed] [Google Scholar]
- [10].Perry VH, Matyszak MK, Fearn S. Altered antigen expression of microglia in the aged rodent CNS. Glia. 1993;7:60–67. doi: 10.1002/glia.440070111. [DOI] [PubMed] [Google Scholar]
- [11].Ogura K, Ogawa M, Yoshida M. Effects of ageing on microglia in the normal rat brain: immunohistochemical observations. Neuroreport. 1994;5:1224–1226. doi: 10.1097/00001756-199406020-00016. [DOI] [PubMed] [Google Scholar]
- [12].Sheffield LG, Berman NE. Microglial expression of MHC class II increases in normal aging of nonhuman primates. Neurobiol Aging. 1998;19:47–55. doi: 10.1016/s0197-4580(97)00168-1. [DOI] [PubMed] [Google Scholar]
- [13].Conde JR, Streit WJ. Effect of aging on the microglial response to peripheral nerve injury. Neurobiol Aging. 2006;27:1451–1461. doi: 10.1016/j.neurobiolaging.2005.07.012. [DOI] [PubMed] [Google Scholar]
- [14].Kreutzberg GW. [Autoradiographic studies on perineuronal microgliocytes] Acta Neuropathol (Berl): Suppl. 1968;4:141–145. [PubMed] [Google Scholar]
- [15].Ladeby R, Wirenfeldt M, Dalmau I, Gregersen R, Garcia-Ovejero D, et al. Proliferating resident microglia express the stem cell antigen CD34 in response to acute neural injury. Glia. 2005;50:121–131. doi: 10.1002/glia.20159. [DOI] [PubMed] [Google Scholar]
- [16].Lawson LJ, Perry VH, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience. 1992;48:405–415. doi: 10.1016/0306-4522(92)90500-2. [DOI] [PubMed] [Google Scholar]
- [17].Svensson M, Mattsson P, Aldskogius H. A bromodeoxyuridine labelling study of proliferating cells in the brainstem following hypoglossal nerve transection. J Anat. 1994;185(Pt 3):537–542. [PMC free article] [PubMed] [Google Scholar]
- [18].Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- [19].Olovnikov AM. [Principle of marginotomy in template synthesis of polynucleotides] Dokl Akad Nauk SSSR. 1971;201:1496–1499. [PubMed] [Google Scholar]
- [20].Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- [21].Flanary BE, Streit WJ. Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia. 2004;45:75–88. doi: 10.1002/glia.10301. [DOI] [PubMed] [Google Scholar]
- [22].Hochberg FH, Miller DC. Primary central nervous system lymphoma. J Neurosurg. 1988;68:835–853. doi: 10.3171/jns.1988.68.6.0835. [DOI] [PubMed] [Google Scholar]
- [23].Miller KR, Streit WJ. The effects of aging, injury and disease on microglial function: a case for cellular senescence. Neuron Glia Biol. 2007;3:245–253. doi: 10.1017/S1740925X08000136. [DOI] [PubMed] [Google Scholar]
- [24].Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. 2007;55:412–424. doi: 10.1002/glia.20468. [DOI] [PubMed] [Google Scholar]
- [25].Xie Z, Morgan TE, Rozovsky I, Finch CE. Aging and glial responses to lipopolysaccharide in vitro: greater induction of IL-1 and IL-6, but smaller induction of neurotoxicity. Exp Neurol. 2003;182:135–141. doi: 10.1016/s0014-4886(03)00057-8. [DOI] [PubMed] [Google Scholar]
- [26].Ye SM, Johnson RW. Increased interleukin-6 expression by microglia from brain of aged mice. J Neuroimmunol. 1999;93:139–148. doi: 10.1016/s0165-5728(98)00217-3. [DOI] [PubMed] [Google Scholar]
- [27].Njie EG, Boelen E, Stassen FR, Steinbusch HW, Borchelt DR, et al. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function Neurobiol Aging, [Epub ahead of print] 2010 doi: 10.1016/j.neurobiolaging.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ye SM, Johnson RW. An age-related decline in interleukin-10 may contribute to the increased expression of interleukin-6 in brain of aged mice. Neuroimmunomodulation. 2001;9:183–192. doi: 10.1159/000049025. [DOI] [PubMed] [Google Scholar]
- [29].Godbout JP, Johnson RW. Interleukin-6 in the aging brain. J Neuroimmunol. 2004;147:141–144. doi: 10.1016/j.jneuroim.2003.10.031. [DOI] [PubMed] [Google Scholar]
- [30].Wolvekamp MC, Marquet RL. Interleukin-6: historical background, genetics and biological significance. Immunol Lett. 1990;24:1–9. doi: 10.1016/0165-2478(90)90028-o. [DOI] [PubMed] [Google Scholar]
- [31].Streit WJ, Hurley SD, McGraw TS, Semple-Rowland SL. Comparative evaluation of cytokine profiles and reactive gliosis supports a critical role for interleukin-6 in neuron-glia signaling during regeneration. J Neurosci Res. 2000;61:10–20. doi: 10.1002/1097-4547(20000701)61:1<10::AID-JNR2>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- [32].Kiefer R, Lindholm D, Kreutzberg GW. Interleukin-6 and transforming growth factor-beta 1 mRNAs are induced in rat facial nucleus following motoneuron axotomy. Eur J Neurosci. 1993;5:775–781. doi: 10.1111/j.1460-9568.1993.tb00929.x. [DOI] [PubMed] [Google Scholar]
- [33].Klein MA, Moller JC, Jones LL, Bluethmann H, Kreutzberg GW, et al. Impaired neuroglial activation in interleukin-6 deficient mice. Glia. 1997;19:227–233. doi: 10.1002/(sici)1098-1136(199703)19:3<227::aid-glia5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- [34].Fattori E, Lazzaro D, Musiani P, Modesti A, Alonzi T, et al. IL-6 expression in neurons of transgenic mice causes reactive astrocytosis and increase in ramified microglial cells but no neuronal damage. Eur J Neurosci. 1995;7:2441–2449. doi: 10.1111/j.1460-9568.1995.tb01042.x. [DOI] [PubMed] [Google Scholar]
- [35].Chakrabarty P, Jansen-West K, Beccard A, Ceballos-Diaz C, Levites Y, et al. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 24:548–559. doi: 10.1096/fj.09-141754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Streit WJ, Sammons NW, Kuhns AJ, Sparks DL. Dystrophic microglia in the aging human brain. Glia. 2004;45:208–212. doi: 10.1002/glia.10319. [DOI] [PubMed] [Google Scholar]
- [37].Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hasegawa-Ishii S, Takei S, Chiba Y, Furukawa A, Umegaki H, et al. Morphological impairments in microglia precede age-related neuronal degeneration in senescence-accelerated mice Neuropathology, [Epub ahead of print] 2010 doi: 10.1111/j.1440-1789.2010.01126.x. [DOI] [PubMed] [Google Scholar]
- [39].Kawamata T, Akiguchi I, Maeda K, Tanaka C, Higuchi K, et al. Age-related changes in the brains of senescence-accelerated mice (SAM): association with glial and endothelial reactions. Microsc Res Tech. 1998;43:59–67. doi: 10.1002/(SICI)1097-0029(19981001)43:1<59::AID-JEMT9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- [40].Geerts H. Of mice and men: bridging the translational disconnect in CNS drug discovery. CNS Drugs. 2009;23:915–926. doi: 10.2165/11310890-000000000-00000. [DOI] [PubMed] [Google Scholar]
- [41].Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer's disease patients. J Alzheimers Dis. 2005;7:221–232. doi: 10.3233/jad-2005-7304. [DOI] [PubMed] [Google Scholar]
- [42].Floden AM, Combs CK. Beta-amyloid stimulates murine postnatal and adult microglia cultures in a unique manner. J Neurosci. 2006;26:4644–4648. doi: 10.1523/JNEUROSCI.4822-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Panossian LA, Porter VR, Valenzuela HF, Zhu X, Reback E, et al. Telomere shortening in T cells correlates with Alzheimer's disease status. Neurobiol Aging. 2003;24:77–84. doi: 10.1016/s0197-4580(02)00043-x. [DOI] [PubMed] [Google Scholar]
- [44].McRae A, Martins RN, Fonte J, Kraftsik R, Hirt L, et al. Cerebrospinal fluid antimicroglial antibodies in Alzheimer disease: a putative marker of an ongoing inflammatory process. Exp Gerontol. 2007;42:355–363. doi: 10.1016/j.exger.2006.10.015. [DOI] [PubMed] [Google Scholar]