

Abstract
The membrane-proximal region spanning residues 649-684 of the HIV-1 envelope protein gp41 (MPR649-684) is an attractive vaccine target for humoral immunity that blocks viral transcytosis across the mucosal epithelia. However, induction of high-titer MPR649-684-specific antibodies remains a challenging task. To explore potential solutions for this challenge, we tested a new translational fusion protein comprising the plague F1-V antigen and MPR649-684 (F1-V-MPR649-684). We employed systemic immunization for initial feasibility analyses. Despite strong immunogenicity demonstrated for the immunogen, repeated systemic immunizations of mice with F1-V-MPR649-684 hardly induced MPR649-684-specific IgG. In contrast, a single immunization with F1-V-MPR649-684 mounted a significant anti-MPR649-684 IgG response in animals that were primed with another MPR649-684 fusion protein based on the cholera toxin B subunit. Additional boost immunizations with F1-V-MPR649-684 recalled and maintained the antibody response and expanded the number of specific antibody-secreting B cells. Thus, while F1-V-MPR649-684 alone was not sufficiently immunogenic to induce detectable levels of MPR649-684-specific antibodies, these results suggest that prime-boost immunization using heterologous antigen-display platforms may overcome the poor humoral immunogenicity of MPR649-684 for the induction of durable humoral immunity. Further studies are warranted to evaluate the feasibility of this strategy in mucosal immunization. Lastly, our findings add to a growing body of evidence in support of this strategy for immunogen design for poorly immunogenic epitopes besides the MPR of HIV-1's transmembrane envelope protein.
Keywords: Plague F1-V antigen, HIV/AIDS, fusion protein, subunit vaccine, prime boost immunization, gp41 membrane proximal external region
1. Introduction
HIV/AIDS remains a leading cause of death worldwide. The long-standing high rate of new infections, estimated to be 2.6 million in 2009 alone, indicates that the epidemic is still out of control [1]. Therefore, ongoing efforts in AIDS prevention campaigns and anti-retroviral drug therapies must be combined with effective microbicides and vaccines. However, there remain fundamental challenges for development of an HIV-1 vaccine. A growing consensus in the field appears to be that successful prophylactic vaccination should protect the mucosa, which is where HIV-1 transmission and initial replication take place [2-4]. Because the immune correlates of protection against HIV/AIDS remain elusive, strategies for the development of an effective vaccine should encompass broad aspects of viral transmission and early stages of infection.
Previously, Bomsel and co-workers showed that the trimeric envelope glycoprotein gp41 of HIV-1 plays a crucial role in viral epithelial transcytosis [5, 6]. Specifically, MPR649-684 initiates the process by binding to the glycosphingolipid galactosylceramide and the heparan sulfate proteoglycan agrin on the apical surface of epithelial cells. A synthetic peptide corresponding to MPR649-684 retains the oligomerization capacity like gp41 and thereby exerts the above biological function associated with viral transcytosis [7-9]. Recently, we reported a series of studies demonstrating that a chimeric protein of the cholera toxin B subunit (CTB) displaying MPR649-684 is capable of inducing antibodies (Abs) to this otherwise poorly immunogenic peptide upon mucosal and systemic immunization in mouse and rabbit models. Furthermore, the mouse mucosal IgAs and serum IgGs as well as the rabbit serum IgGs efficiently blocked transcytosis of B and D clade primary HIV-1 isolates in a human tight epithelial model [10-13]. These results supported our hypothesis that the induction of MPR649-684-specific Abs may contribute to a prophylactic strategy to block mucosal transmission of HIV-1 in humans. Our results further demonstrated that the response to CTB-MPR649-684 alone was not as robust as would probably be required to protect the mucosa. In particular, repeated boosting with CTB-MPR649-684 resulted in a progressively dampened anti-MPR Ab response. This indicated a need for an improved MPR649-684-based immunogen, possibly in combination with CTB-MPR649-684. A candidate platform for such an immunogen must be highly immunogenic and able to provide T cell epitopes for the induction of memory responses.
The fraction 1 capsular antigen (F1), the V antigen, and a fusion of the two (F1-V) derived from Yersinia pestis are known to elicit strong immune responses, particularly of the humoral arm. A number of studies have demonstrated that such immune responses are protective against a virulent Y. pestis challenge in various animal models (reviewed in: [14, 15]). Thus, these recombinant proteins are currently undergoing advanced clinical trials toward a new plague vaccine for human use. Interestingly, recent studies have indicated that F1-V may possess direct immunostimulatory activity on antigen-presenting cells [16], potentially as a Toll-like receptor 2 (TLR2)-agonist [17]. These findings suggest that F1-V might serve as a novel vaccine platform/adjuvant for heterologous antigens. To test this possibility, we constructed a translational fusion protein of F1-V and MPR649-684. Because the efficacy of F1-V fusion in mucosal immunization is elusive, in this short report, we investigated the immunogenicity of the F1-V-MPR649-684 fusion protein in a mouse model in light of an anti-MPR649-684 IgG response upon systemic immunization. Our data showed that a prime-boost immunization using heterologous MPR649-684-displaying immunogens, i.e., F1-V-MPR649-684 and CTB- MPR649-684, may provide a solution to overcome the poor humoral immunogenicity of MPR649-684. The present study may provide an important insight into immunogen design and immunization strategies towards HIV-1 vaccine development.
2. Materials and Methods
2.1. Bacterial expression vectors
A Novagen pET-26b(+) E. coli expression vector containing the gene encoding F1-V fusion protein from Y. pestis (pTM356) was a kind gift from Dr. Hugh Mason (Arizona State University, Tempe, AZ; [18]. Because our preliminary experiments using this vector resulted in poor expression of F1-V (data not shown), we decided to test intracellular protein accumulation by removing the signal peptide. To this end, the DNA fragment corresponding to the mature F1-V (without the pelB signal peptide)-coding sequence was amplified with polymerase chain reaction (PCR) using primers containing NdeI (for the forward side) and BlpI (for the reverse side) and cloned into the PCR-cloning vector Topo-TA (Invitrogen, Carlsbad, CA). Following sequence verification, the NdeI-BlpI fragment was cloned into a pET-26b(+) vector digested with the same restriction enzymes to yield pTM555. To construct an expression vector for F1-V-MPR649-684, a DNA fragment encompassing the MPR649-684-coding sequence was PCR-amplified from the CTB-MPR649-684 gene [13] with primers containing a XhoI site, which was inserted within a XhoI site downstream of the F1-V gene and upstream of a hexahistidine tag to yield the plasmid pTM537. The coding sequence of the entire fusion gene was verified. A Gly-Pro-Gly-Pro peptide linker was placed between F1-V and MPR649-684 to facilitate the surface exposure of the peptide on the fusion molecule.
2.2. Bacterial Expression of Recombinant Proteins
For expression of recombinant proteins we used the E. coli strain BL21(DE3) (EMD Chemicals Inc., Gibbstown, NJ). Single colonies from bacteria transformed with the F1-V-MPR649-684 or F1-V expression vector were inoculated in 3 ml of LB medium supplemented with 50 μg/ml kanamycin and 1% (w/v) glucose. After culturing at 37°C for 16 h, cells were washed in LB medium once to remove glucose. Cells were then inoculated into a larger volume of fresh LB medium supplemented with 50 μg/ml kanamycin and incubated further under the same conditions until the culture reached OD600 of 0.6-1.0. The cells were induced with 0.3 mM Isopropyl-1-thio-L-D-galactopyranoside (IPTG) and the culture was incubated for an additional 2 h under the same conditions. Recombinant protein induction was confirmed by SDS-PAGE and immunoblotting. After electrophoresis, gels were stained, or proteins were electro-transferred to a polyvinylidene difluoride (PVDF) membrane. For western blot analysis, 0.2 μg of purified F1-V and F1-V-MPR649-684 were used (see below for purification and protein quantification). Blots were probed with human monoclonal (m)Abs 2F5 (100 ng/ml; kind gift of Dr. Morgane Bomsel at Institut Cochin, Paris, France) or 4E10 (100 ng/ml; obtained from National Institute of Health [NIH] AIDS Research and Reference Reagent Program) as primary Abs, horseradish peroxidase (HRP)-conjugated anti-human IgG (1:10,000; EMD Chemicals) as secondary to detect MPR649-684, and rabbit polyclonal Abs against F1 or V (kind gift from Dr. Mason) and HRP-conjugated anti-rabbit IgG (1:20,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) as the secondary to detect F1-V. Abs were detected using chemiluminscence (Santa Cruz Biotechnology).
2.3. Protein Purification
Following induction as described above, bacterial cells were harvested by centrifugation at 10,000 ×g for 1 min, washed with PBS, and resuspended in 20 ml of ice-cold Equilibration Buffer (20 mM Tris, pH 8.0, 2 M NaCl). Phenylmethylsulphonyl fluoride (PMSF: final concentration 1 mM) was added to the cell suspension just before lysis. Cells were homogenized by French press (18,000 psi, 2-3 times), and the lysate was centrifuged 30,000 ×g for 20 min. The supernatant was subjected to purification by metal affinity chromatography using TALON metal affinity resin (Clontech, Mountain View, CA). Following equilibration of the resin with Equilibration Buffer, the clarified cell lysate was applied to the column, which was washed with 6 bed volumes of Equilibration Buffer and then with 10 bed volumes of Wash Buffer (20 mM Tris, pH 8.0, 500 mM NaCl, 5 mM imidazole). Finally, F1-V- MPR649-684 was eluted with 2.5 bed volume of Elution Buffer (20 mM Tris, pH 8.0, 500 mM NaCl, 150 mM imidazole). The eluate was concentrated by using 10K Microsep Omega (PALL Corporation, Port Washington, NY) and dialyzed against PBS overnight (10 kDa cutoff membrane; Thermo Fisher Scientific, Inc., Rockford, IL). CTB- MPR649-684 was produced in E. coli and prepared as described previously. The purified proteins were quantitated by determining the A280 values using a DU-640 spectrophotometer (Beckman, Brea, CA). In calculating their concentrations, the extinction coefficient values were used (calculated using Chris Putnam's PROTEIN CALCULATOR v3.3, (http://www.scripps.edu/∼cdputnam/protcalc.html) with an algorithm based on [19]): 0.8944 (mg/ml)-1cm-1 for F1-V-MPR649-684, 0.4587 (mg/ml)-1cm-1 for F1-V and 2.2351 (mg/ml)-1cm-1 for CTB- MPR649-684.
2.4. Mouse Immunization
The experimental protocol involving animals was approved by the Institutional Animal Care and Use Committee of Arizona State University (Protocol 770-R). One milliliter of F1-V-MPR649-684 or CTB-MPR649-684 in PBS was mixed with Pre-liposomes Formulation 4 (Sigma-Aldrich Corp., St. Louis, MO) and sonicated. Immunogens were then diluted to a concentration of 200 μg/ml with PBS and kept on ice until administration. Just prior to administration samples were warmed to room temperature and thoroughly mixed. Seven-week-old female BALB/c mice were separated into two groups. Both groups (n=7 each) were immunized by intraperitoneal (i.p.) injections at weeks 0, 1 and 2. Animals were then injected with 10 μg of F1-V-MPR649–684 (for Group 1) or CTB-MPR649-684 (for Group 2). Both groups were further i.p. injected with an additional dose of F1-V-MPR649-684 on week 7, 16 and 41.
2.5. Detection of antigen-specific IgG by enzyme-linked immunosorbent assay (ELISA)
ELISA plates were coated with 2 μg/ml of synthetic MPR649-684, CTB (Sigma-Aldrich), or F1-V. Threefold serially diluted duplicate samples starting from 1:100 for serum diluted in PBS containing 1% dry milk and 0.05% Tween-20 were applied onto the plates and incubated for 1 h at 37°C. Serum IgGs were detected by HRP-conjugated anti-mouse IgG (1:5,000; SouthernBiotech, Birmingham, AL) and Sigma FAST ophenylenediamine dihydrochloride (OPD) substrate (Sigma-Aldrich) and quantitated by reading the absorbance at 490nm using a plate reader (SpectraMax 340PC; Molecular Devices, Sunnyvale, CA). Samples were considered positive if their mean OD490 values exceeded the reading of 0.1 absorbance units after subtracting the mean value of control (no Ig sample) wells in each plate. End point titers were determined by extrapolation using power series curve fitting to the OD490 values from at least 3 serum dilution points and expressed as the reciprocal of the sample dilution factor giving the OD490 value equal to that of control.
2.6. IgG-secreting cell enzyme-linked immunospot (ELISPOT) assay
One week after the final i.p. immunization with F1-V- MPR649-684 (week 42), spleens were isolated from mice, and splenocytes were prepared via standard method. Cells from the same group were pooled. Control splenocytes were prepared from non-immunized mice (n=5). Recombinant F1-V (1 μg/well), CTB (2 μg/well), or synthetic MPR649-683 peptide (10 μg/well) diluted in PBS were used as capture reagents in 96-well PVDF microtiter plates (Millipore, Billerica, MA). The plates were blocked with RPMI medium containing 10% fetal bovine serum. Five-fold serially diluted splenocytes, starting from 107 cells/well, were added and incubated at 37°C in a humidified 5% CO2 atmosphere for 4 h. After removing cells, plates were extensively washed with PBS containing 0.05% Tween-20. HRP-conjugated anti-mouse IgG (1:5,000; SouthernBiotech) was added and incubated overnight at 4°C. Spots were developed by 3-Amino-9-ethyl-carbazole (AEC Substrate Set; BD Biosciences, San Jose, CA) and analyzed using the CTL ImmunoSpot plate reader and counting software (Cellular Technology Ltd., Shaker Heights, OH).
2.7. Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA). Two-way repeated measures analysis of variance (ANOVA) followed by Bonferroni posttests were used to compare anti-MPR649-684 IgG levels between groups at each time point. Differences of Ab endpoint titers were analyzed by one-way ANOVA followed by the Newman-Keuls multiple comparison test for comparing responses to three antigens (MPR649-683, F1-V and CTB) within a group, and by unpaired, two-tailed Student's t-test for comparing responses to each immunogen between Group 1 and 2. Differences of ELISPOT results between groups were evaluated by unpaired, two-tailed Student's t-test. A p value of <0.05 was considered significant.
3. Results
3.1. F1-V-MPR649-684 expression in E. coli
In our earlier investigation, we found that F1-V can be expressed at a high level in a soluble form within E. coli cytosol (data not shown). The expression of F1-V-MPR649-684 fusion was therefore tested under the same conditions. At 2 h post-induction, a strong protein accumulation was observed, which accounted for 21% of total E. coli proteins in a densitometric analysis (Fig. 1A). Fortuitously, most of the expressed F1-V-MPR649-684 protein was PBS-soluble, which could be easily separated by centrifugation from insoluble proteins and cell debris following cell lysis. Using single-step metal affinity chromatography, F1-V-MPR649-684 was purified to near homogeneity (>85%, as estimated by densitomety; Fig 1A, lane 4). The apparent size of the protein concurred well with the theoretical mass of F1-V-MPR649-684 (59.4 kDa). Immunoblot analysis using polyclonal anti-F1 and -V Abs, and two MPR649-684-specific mAbs 2F5 and 4E10 confirmed that the protein was indeed F1-V-MPR649-684 (Fig. 1B).
Fig. 1.
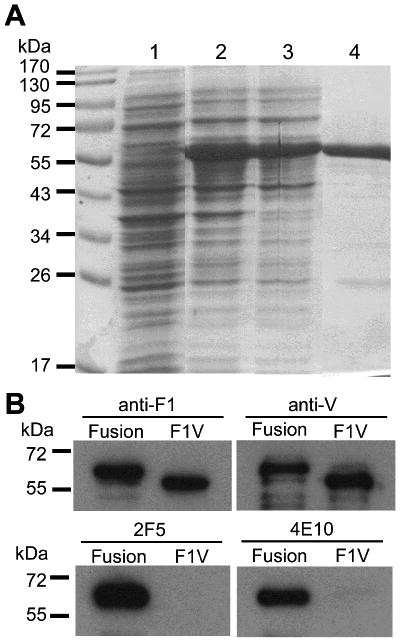
Analysis of F1-V-MPR649-684 expressed in E. coli. (A) SDS-PAGE analysis of F1-V-MPR649-684 expression in E. coli. Lane 1, pre-induction control E. coli proteins; lane 2: total cell proteins 2 h post-induction; lane 3: soluble cell proteins after disruption of cells; and lane 4: F1-V-MPR649-684 purified by metal affinity chromatography. (B) Immunoblot analysis of E. coli-expressed F1-V and F1-V-MPR649-684. “Fusion” represents F1-V-MPR649-684. Polyclonal rabbit Abs to F1 (upper left) or V (upper right), or gp41-specific human mAbs 2F5 (bottom left) or 4E10 (bottom right) were used for detection. F1-V-MPR649-684 was detected by all Abs, whereas F1-V was recognized by anti-F1 and -V Abs only.
F1-V was prepared using the same expression and purification procedures (data not shown). In a sandwich ELISA, F1-V-MPR649-684, but not F1-V, was detected by 2F5 and 4E10 upon capture by F1-V-specific Abs (data not shown), indicating that the MPR649-684 domain is likely surface-accessible for Ab recognition. Taken together, we have successfully constructed F1-V-MPR649-684, which was then used in the subsequent immunization study.
3.2. Immunogenicity of F1-V-MPR649-684
We evaluated the immunogenicity of F1-V-MPR649-684 in Balb/c mice. Mice were i.p. immunized with 10 μg of F1-V-MPR649-684 at weeks 0, 1, and 2 (Group 1). Serum samples were analyzed for antigen-specific IgG induction. As shown in Figure 2A, hardly any IgG response to MPR649-684 was detected at any time points through 5 weeks after the third immunization. A boost immunization induced a marginal level of anti-MPR649-684 IgG in two of seven animals at two weeks after the boost, with geometric mean titers (GMTs) being 1.41×103 and 6.35×102 (Fig. 2B). However, the response rapidly fell below the detection limit. Another boost immunization at week 16 failed to recover the response (Fig. 2A). Despite the response to the MPR hapten, all seven mice showed very strong anti-F1-V IgG responses, with a GMT of 3.91×105 at week 9 (Fig. 2B). Thus, these results demonstrate that the F1-V fusion protein serving as a carrier was sufficiently immunogenic and capable of stimulating a strong Ab response, albeit with a poor immune response to MPR649-684. Alternatively, these results highlight that MPR649-684 has unusually poor antigenicity and/or immunogenicity, a notion that has been suggested in our earlier investigation with another MPR649-684-based fusion protein, CTB-MPR649-684 [11, 12].
Fig. 2.
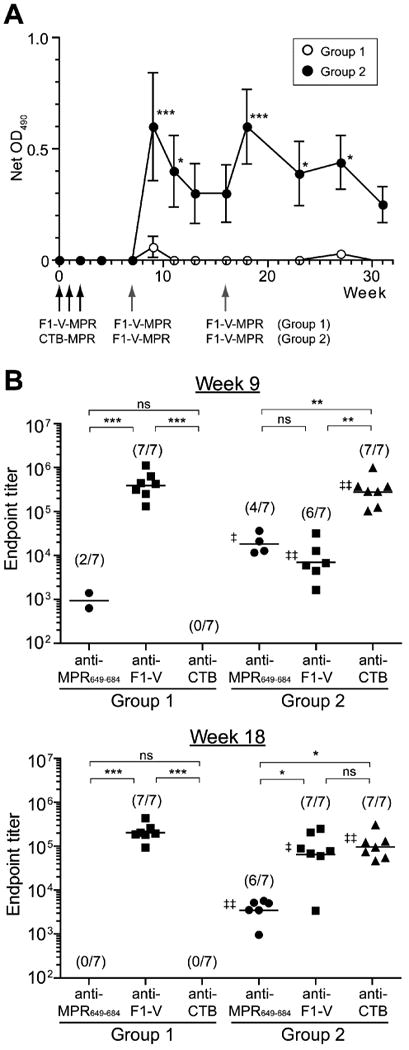
Serum IgG responses. Mice (7 per group) were immunized by three weekly i.p. injections of F1-V-MPR649-684 (for Group 1) or CTB-MPR649-684 (for Group 2), and boosted i.p. with F1-V-MPR649-684 at weeks 7 and 16 (group 1), as indicated by arrows (black for priming and grey for boost) in (A). Ten μg of immunogens were used for all immunizations. (A) Anti-MPR649-684 IgG levels in serum samples at 1:100 dilution were assessed by ELISA at the indicated weeks. Net OD values are shown as arithmetic mean ± S.E.M. Asterisks indicate statistical significance between groups at the indicated weeks, as analyzed by two-way repeated measures ANOVA followed by Bonferroni post-tests (* p < 0.05; *** p < 0.001). (B) Endpoint titers of anti-MPR649-684 (circles), anti-F1-V (squares) and anti-CTB (triangles) IgGs at two weeks after each boost (i.e., weeks 9 and 18). Symbols represent values of individual mice. Horizontal bars indicate GMTs. Parentheses represent numbers of responding mice. Double dagger signs (‡) represent statistical significance between Group 1 and 2 for each antigen, as assessed by a two-way unpaired t-test (‡ p < 0.05; ‡‡ p < 0.01), whereas asterisks indicate statistical significance among different antigens within each group, analyzed by one-way ANOVA followed by Bonferroni post-tests followed by the Newman-Keuls multiple comparison test (* p < 0.05; ** p < 0.01; *** p < 0.001; ns: not significant).
3.3. Significant anti-MPR649-684 IgG response following a prime/boost regimen with CTB-MPR649-684/F1-V-MPR649-684
Recently, we reported that cross-immunization of rabbits with synthetic MPR649-683 peptides chemically linked to keyhole limpet hemocyanin followed by CTB-MPR649-684 induced higher serum anti-MPR649-684 IgG responses than repeated immunizations with CTB-MPR649-684 alone [11]. This raised the hypothesis that prime-boost immunizations with different MPR649-684-displaying immunogens may help overcome the peptide's seemingly poor immunogenicity and thereby induce a strong anti-MPR649-684 Ab response. To test this hypothesis, a group of mice (Group 2) was first immunized with CTB-MPR649-684 and then boosted with F1-V-MPR649-684 using the same immunization scheme as Group 1 (i.e., i.p. immunized weekly with 10 μg of CTB- MPR649-684 for three consecutive weeks, boosted with 10 μg of F1-V- MPR649-684 at weeks 7 and 16). As observed with Group 1, the initial three weekly immunizations with CTB-MPR649-684 minimally induced serum anti-MPR649-684 IgGs. Markedly, however, a single i.p. boost immunization with F1-V-MPR649-684 raised a significant serum anti-MPR649-684 IgG response in four of seven animals (GMT: 1.83×104; Fig. 2B). The Ab level slowly declined after the boost, but remained detectable for 9 weeks (Fig. 2A). A second i.p. boost with F1-V-MPR649-684 at week 16 recovered the anti-MPR649-684 response. Although the GMT of the response was lower (3,473.8) than that of response following the first boost, the Ab response was observed in two additional mice, i.e., six of seven animals. The Ab level declined again, but remained statistically significant as compared to that of Group 1 over 11 weeks (Fig. 2A).
Meanwhile, as in Group1, priming immunizations with CTB-MPR649-684 raised a very high IgG response to the carrier domain CTB in all 7 mice, with a GMT of 2.79×105 at week 9 (Fig. 2B). Even though the mice did not receive any further immunization with CTB after week 3, the anti-CTB IgG response remained high at week 18 (GMT: 9.66×104; Fig. 2B). Likewise, a single immunization at week 7 with F1-V-MPR649-684 in CTB-MPR649-684-primed animals raised a significant F1-V-specific IgG response at week 9 in six of seven mice.
To confirm the above observations at the cellular level, antigen-specific Ab-secreting cells were examined by ELISPOT assay on splenocytes of Groups 1 and 2 mice (Fig. 3A). One week before analysis (i.e., at week 41), both Groups 1 and 2 were re-immunized i.p. with 10 μg of F1-V-MPR649-684 to recall the immunological memory. This boost immunization, as in the earlier time points, did not raise any detectable level of serum anti-MPR649-684 IgG in Group 1, whereas Group 2 showed a significant rebound in the Ab level with a GMT of 35,414.3 (Fig. 3B). On the other hand, both groups showed high serum IgG titers against carrier proteins (GMTs: 1.76×106 for F1-V in Group 1; 7.17×105 for F1-V and 5.01×105 for CTB in Group 2; Fig. 3B). Consistent with the Ab GMTs, both groups showed a high frequency of F1-V-specific IgG-secreting cells (541.7 ± 166.7 in Group 1 and 625.0 ± 72.2 in Group 2 per 107 total splenocytes). As expected, CTB-specific IgG-secreting cells were only detected in Group 2. However, the frequency was considerably lower than that of F1-V-specific cells (40.0 ± 13.2 cells per 107 splenocytes). This was not unexpected because the group had not received a CTB immunogen for as long as 40 weeks after the initial set of immunizations with CTB-MPR649-684. Group 2 showed a significantly higher number of MPR649-684-specific IgG-secreting cells than Group 1, which is in good agreement with the serum IgG levels. The number was nonetheless relatively low in spite of frequent antigen stimulations in the contexts of F1-V-MPR649-684 and CTB-MPR649-684. Taken together, these results confirmed that MPR649-684 possesses an unusually low humoral immunogenicity, but prime-boost immunization using heterologous MPR649-684-displaying immunogens can, at least partially, overcome the problem.
Fig. 3.
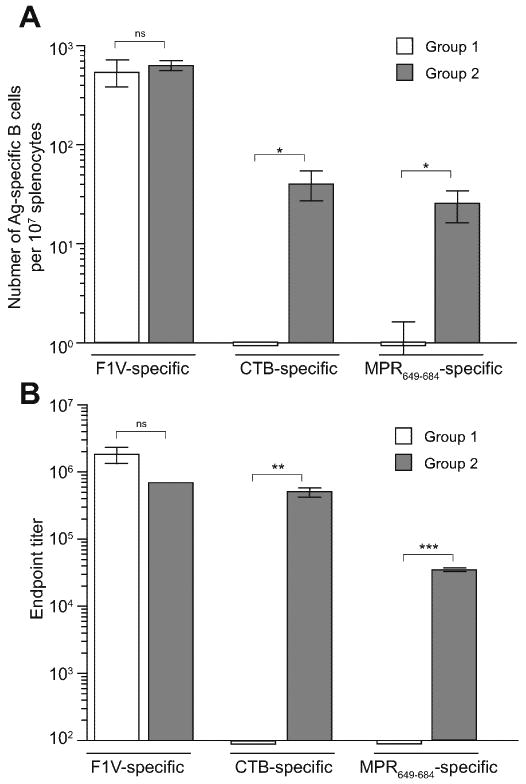
ELISPOT analysis of Ab-secreting cells in splenocytes (A) and determination of serum IgG titers (B) after the third boost immunization with F1-V-MPR649-684. One week prior to analysis, Groups 1 and 2 mice were boosted i.p. with 10 μg of F1-V-MPR649-684 (at week 41). As a control, splenocytes and serum samples prepared from unimmunized mice (N=5) were used, which did not show any detectable spots or antibody responses (not shown). (A) Splenocytes were pooled from the same group of animals and analyzed for Ab-secreting cells specific to F1-V, CTB, and F1-V-MPR649-684. See Materials and Methods for detail. (B) Endpoint titers of anti-MPR649-684, anti-F1-V, and anti-CTB IgGs. Bars represent arithmetic means of triplicate samples (±S.E.M.) of pooled splenocyte cultures and sera. Symbols indicate statistical significance between groups, as analyzed by unpaired two-tailed Student's t-test (* p < 0.05; ** p < 0.01; *** p < 0.01; ns: not significant).
4. Discussion
To develop a vaccine component that specifically blocks HIV-1 mucosal transmission, we have focused on MPR649-684, due to its pivotal role in viral epithelial transcytosis [8, 9]. Our previous studies showed that the translational fusion protein CTB-MPR649-684 can induce MPR649-684-specific Abs in small animal models upon mucosal and systemic immunizations and that the Abs were capable of blocking HIV-1 transcytosis in a human tight epithelial model [10-13]. However, the Ab titers induced by CTB-MPR649-684 could only reach a modest level in spite of repeated boosting, necessitating an alternative strategy to induce a robust anti-MPR649-684 Ab response. In our search for a new vaccine scaffold for MPR649-684, the present study examined a new translational fusion protein based on Y. pestis-derived F1-V antigen, a vaccine candidate against plague that was suggested to possess a potential adjuvant effect (e.g., as a TLR2 agonist) [16, 17]. The production of recombinant F1-V-MPR649-684 in a standard E. coli-based expression system was straightforward; a very high level of the protein was expressed in soluble form within a few hours (Fig. 1). This allowed us to quickly examine whether this new fusion protein could efficiently elicit anti-MPR649-684 Abs in a mouse systemic immunization study.
Contrary to our initial expectation, it was revealed that F1-V-MPR649-684 could not provide an immediate solution for the induction of anti-MPR649-684 Abs. Similar to our previous observations obtained with CTB-MPR649-684, immunization with F1-V-MPR649-684 induced a strong IgG response to the carrier protein but not to the MPR649-684 hapten. Repeated immunizations with the immunogen only augmented the carrier-specific Ab response. These results confirmed the unusually low immunogenicity of MPR649-684. This further suggests that the use of a single immunogen might not help resolve the issue regardless of its overall immunogenicity. However, possibilities remain that have not been fully explored in our present or previous studies. These include the use of powerful adjuvants and/or different antigen display strategies (other than C-terminal fusion) that can enhance the humoral immune recognition of MPR649-684. Corresponding studies are currently underway.
The most important finding in this study was, as demonstrated by ELISA and B-cell ELISPOT assay, that cross-immunizations with CTB-MPR649-684 followed by F1-V-MPR649-684 raised a significant anti-MPR649-684 IgG response, despite the ineffectiveness of repeated immunizations with either of the immunogens alone. We acknowledge the possibility that biological variations within the groups might have been masked in the ELISPOT assay due to sample pooling. Nevertheless, the results are highly congruent with serum Ab titers in ELISA, thereby strongly underscoring the finding. These results have several important implications. First, notwithstanding its poor immunogenicity, MPR649-684 (in the context of CTB and F1-V fusions) appears to possess sufficient antigenicity necessary for humoral immune recognition for priming and boosting an anti-MPR649-684 Ab response. Thus, even if undetectable, an anti-MPR649-684 IgG response could be primed by CTB-MPR649-684, which was successfully restimulated and expanded by the subsequent F1-V-MPR649-684 immunization. Second, the prime-boost immunization appeared to have established immunological memory for the anti-MPR649-684 IgG response, as two subsequent booster immunizations with F1-V-MPR649-684 recovered the Ab level. Third, these suggest that prime-boost immunization with two (or more) different MPR649-684-displaying immunogens may be a solution to overcome poor MPR649-684 humoral immunogenicity and induce a robust and durable anti-MPR649-684 Ab response.
Mechanisms by which the present prime-boost immunization procedure induced a better anti-MPR649-684 Ab response remain to be determined and warrant further investigation. We speculate that such a regimen might have rescued the weakly primed immunity to MPR649-684 from being outcompeted by stronger reactions directed to a more immunogenic portion of the antigens during the secondary immune response upon boost. This in turn might have allowed the establishment of a longer-lasting memory response to MPR649-684. In this context, it is of interest to investigate whether a third MPR649-684-based immunogen could induce an even higher anti-MPR649-684 Ab response. One potential caveat of this approach, however, is the possibility of skewing the fine binding characteristics of anti-MPR649-684 Abs during affinity maturation due to the use of different immunogens. This could affect the function of the Abs. Therefore, future studies should also involve a functional analysis of Abs induced by such an immunization regimen, including transcytosis-blocking, neutralization, and Fc-mediated anti-HIV activities, along with analysis on the structure and key biochemical signatures (e.g., oligomerization capacity, and galactosyl ceramide binding [9]) of the MPR domain.
Given that systemic prime-boost immunization had a strong impact on inducing a serum Ab response to MPR649-684, one of our next interests is whether such an effect is also observed for mucosal immunity. Although we previously showed that MPR649-684-specific IgGs can effectively block HIV-1 transcytosis and it has been shown that IgGs can be effectively transudated into mucosal lumens [20-22], induction of secretory IgA to MPR649-684 at susceptible mucosal sites may be more effective in blocking HIV-1 transmission [6, 23]. This will likely require mucosal immunization. In this context, our preliminary study showed that intranasal immunization with CTB-MPR649-684 followed by i.p. injection of F1-V-MPR649-684 is not effective in boosting a vaginal anti-MPR649-684 IgA response (Matoba & Mor, unpublished observation). This may indicate that the systemic and mucosal immune systems are divergently regulated in response to prime-boost immunization with heterologous immunogens. In this context, a more potent mucosal immunogen, such as virus-like particles (e.g., see [24]), may provide a useful tool for testing this concept.
In summary, the present study demonstrated that F1-V-MPR649-684 does not sufficiently raise an anti-MPR649-684 Ab response but prime-boost immunization with CTB-MPR649-684 and F1-V-MPR649-684 is a possible solution to overcome the poor humoral immunogenicity of the gp41 peptide. Our findings warrant further studies to reveal the feasibility of such an immunization strategy based on heterologous MPR649-684-displaying immunogens in mucosal immunization, and if successful, to evaluate the HIV transmission-blocking effects of mucosal anti-MPR649-684 Abs in relevant models such as humanized mice [25], non-human primates [26], and ex vivo human mucosal explant systems [27].
Acknowledgments
The authors would like to thank Jeffrey Doran, Jonathan Hu and Howard Chen for their assistance in experiments and Jacquelyn Kilbourne for her support in animal immunization. We are grateful to Drs. Kohtaro Fujihashi, Shinich Sekine and Ryoki Kobayashi for their support and useful discussion in ELISPOT assay, Dr. Hugh Mason for sharing E. coli expression vectors and antisera, and Andrew Marsh for editorial assistance. The work was supported in part by a NIH grant to TSM (U19AI62150) and to NM (R03AI073157).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Joint United Nations Programme on HIV/AIDS. 2010. Global report: UNAIDS report on the global AIDS epidemic | 2010. [Google Scholar]
- 2.Haase AT. Targeting early infection to prevent HIV-1 mucosal transmission. Nature. 2010 Mar 11;464(7286):217–23. doi: 10.1038/nature08757. [DOI] [PubMed] [Google Scholar]
- 3.McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol. 2010 Jan;10(1):11–23. doi: 10.1038/nri2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahlers JD, Belyakov IM. Strategies for optimizing targeting and delivery of mucosal HIV vaccines. Eur J Immunol. 2009 Oct;39(10):2657–69. doi: 10.1002/eji.200939269. [DOI] [PubMed] [Google Scholar]
- 5.Bomsel M. Transcytosis of infectious human immunodeficiency virus across a tight human epithelial cell line barrier. Nat Med. 1997 Jan;3(1):42–7. doi: 10.1038/nm0197-42. [DOI] [PubMed] [Google Scholar]
- 6.Alfsen A, Iniguez P, Bouguyon E, Bomsel M. Secretory IgA specific for a conserved epitope on gp41 envelope glycoprotein inhibits epithelial transcytosis of HIV-1. J Immunol. 2001 May 15;166(10):6257–65. doi: 10.4049/jimmunol.166.10.6257. [DOI] [PubMed] [Google Scholar]
- 7.Yu H, Alfsen A, Tudor D, Bomsel M. The binding of HIV-1 gp41 membrane proximal domain to its mucosal receptor, galactosyl ceramide, is structure-dependent. Cell calcium. 2008 Jan;43(1):73–82. doi: 10.1016/j.ceca.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 8.Alfsen A, Yu H, Magerus-Chatinet A, Schmitt A, Bomsel M. HIV-1-infected blood mononuclear cells form an integrin- and agrin-dependent viral synapse to induce efficient HIV-1 transcytosis across epithelial cell monolayer. Mol Biol Cell. 2005 Sep;16(9):4267–79. doi: 10.1091/mbc.E05-03-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alfsen A, Bomsel M. HIV-1 gp41 envelope residues 650-685 exposed on native virus act as a lectin to bind epithelial cell galactosyl ceramide. J Biol Chem. 2002 Jul 12;277(28):25649–59. doi: 10.1074/jbc.M200554200. [DOI] [PubMed] [Google Scholar]
- 10.Matoba N, Kajiura H, Cherni I, Doran JD, Bomsel M, Fujiyama K, et al. Biochemical and immunological characterization of the plant-derived candidate human immunodeficiency virus type 1 mucosal vaccine CTB-MPR(649-684) Plant Biotechnol J. 2009 Feb;7(2):129–45. doi: 10.1111/j.1467-7652.2008.00381.x. [DOI] [PubMed] [Google Scholar]
- 11.Matoba N, Griffin TA, Mittman M, Doran JD, Hanson CV, Montefiori D, et al. Transcytosis-blocking Abs elicited by an oligomeric immunogen based on the membrane proximal region of HIV-1 gp41 target non-neutralizing epitopes. Curr HIV Res. 2008;6(3):218–29. doi: 10.2174/157016208784324994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matoba N, Geyer BC, Kilbourne J, Alfsen A, Bomsel M, Mor TS. Humoral immune responses by prime-boost heterologous route immunizations with CTB-MPR(649-684), a mucosal subunit HIV/AIDS vaccine candidate. Vaccine. 2006 Jun 5;24(23):5047–55. doi: 10.1016/j.vaccine.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 13.Matoba N, Magerus A, Geyer BC, Zhang Y, Muralidharan M, Alfsen A, et al. A mucosally targeted subunit vaccine candidate eliciting HIV-1 transcytosis-blocking Abs. Proc Natl Acad Sci U S A. 2004 Sep 14;101(37):13584–9. doi: 10.1073/pnas.0405297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feodorova VA, Corbel MJ. Prospects for new plague vaccines. Expert Rev Vaccines. 2009 Dec;8(12):1721–38. doi: 10.1586/erv.09.129. [DOI] [PubMed] [Google Scholar]
- 15.Williamson ED. Plague. Vaccine. 2009 Nov 5;27 4:D56–60. doi: 10.1016/j.vaccine.2009.07.068. [DOI] [PubMed] [Google Scholar]
- 16.Kingston R, Burke F, Robinson JH, Bedford PA, Jones SM, Knight SC, et al. The fraction 1 and V protein antigens of Yersinia pestis activate dendritic cells to induce primary T cell responses. Clin Exp Immunol. 2007 Sep;149(3):561–9. doi: 10.1111/j.1365-2249.2007.03452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Del Prete G, Santi L, Andrianaivoarimanana V, Amedei A, Domarle O, DE MM, et al. Plant-derived recombinant F1, V, and F1-V fusion antigens of Yersinia pestis activate human cells of the innate and adaptive immune system. Int J Immunopathol Pharmacol. 2009 Jan-Mar;22(1):133–43. doi: 10.1177/039463200902200115. [DOI] [PubMed] [Google Scholar]
- 18.Santi L, Giritch A, Roy CJ, Marillonnet S, Klimyuk V, Gleba Y, et al. Protection conferred by recombinant Yersinia pestis antigens produced by a rapid and highly scalable plant expression system. Proc Natl Acad Sci U S A. 2006 Jan 24;103(4):861–6. doi: 10.1073/pnas.0510014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989 Nov 1;182(2):319–26. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 20.Hocini H, Barra A, Belec L, Iscaki S, Preud'homme JL, Pillot J, et al. Systemic and secretory humoral immunity in the normal human vaginal tract. Scand J Immunol. 1995 Aug;42(2):269–74. doi: 10.1111/j.1365-3083.1995.tb03653.x. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida M, Claypool SM, Wagner JS, Mizoguchi E, Mizoguchi A, Roopenian DC, et al. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity. 2004 Jun;20(6):769–83. doi: 10.1016/j.immuni.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 22.Yoshida M, Masuda A, Kuo TT, Kobayashi K, Claypool SM, Takagawa T, et al. IgG transport across mucosal barriers by neonatal Fc receptor for IgG and mucosal immunity. Springer Semin Immunopathol. 2006 Dec;28(4):397–403. doi: 10.1007/s00281-006-0054-z. [DOI] [PubMed] [Google Scholar]
- 23.Wright A, Lamm ME, Huang YT. Excretion of human immunodeficiency virus type 1 through polarized epithelium by immunoglobulin A. J Virol. 2008 Dec;82(23):11526–35. doi: 10.1128/JVI.01111-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain S, Patrick AJ, Rosenthal KL. Multiple tandem copies of conserved gp41 epitopes incorporated in gag virus-like particles elicit systemic and mucosal antibodies in an optimized heterologous vector delivery regimen. Vaccine. 2010 Oct 8;28(43):7070–80. doi: 10.1016/j.vaccine.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Denton PW, Garcia JV. Novel humanized murine models for HIV research. Curr HIV/AIDS Rep. 2009 Feb;6(1):13–9. doi: 10.1007/s11904-009-0003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Genesca M, Miller CJ. Use of nonhuman primate models to develop mucosal AIDS vaccines. Curr HIV/AIDS Rep. 2010 Feb;7(1):19–27. doi: 10.1007/s11904-009-0035-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merbah M, Introini A, Fitzgerald W, Grivel JC, Lisco A, Vanpouille C, et al. Cervico-Vaginal Tissue Ex Vivo as a Model to Study Early Events in HIV-1 Infection. Am J Reprod Immunol. 2011 Mar;65(3):268–78. doi: 10.1111/j.1600-0897.2010.00967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]