


Abstract
Catechol-O-methyltransferase (COMT) is a major enzyme controlling catecholamine levels that plays a central role in cognition, affective mood and pain perception. There are three common COMT haplotypes in the human population reported to have functional effects, divergent in two synonymous and one nonsynonymous position. We demonstrate that one of the haplotypes, carrying the non-synonymous variation known to code for a less stable protein, exhibits increased protein expression in vitro. This increased protein expression, which would compensate for lower protein stability, is solely produced by a synonymous variation (C166T) situated within the haplotype and located in the 5′ region of the RNA transcript. Based on mRNA secondary structure predictions, we suggest that structural destabilization near the start codon caused by the T allele could be related to the observed increase in COMT expression. Our folding simulations of the tertiary mRNA structures demonstrate that destabilization by the T allele lowers the folding transition barrier, thus decreasing the probability of occupying its native state. These data suggest a novel structural mechanism whereby functional synonymous variations near the translation initiation codon affect the translation efficiency via entropy-driven changes in mRNA dynamics and present another example of stable compensatory genetic variations in the human population.
INTRODUCTION
Catechol-O-methyltransferase (COMT) deactivates neurotransmitters and metabolizes catechol-containing structures by methylation of a hydroxyl group (1). The implications of COMT activity are broad and can influence factors such as general cognitive function (2–4), addiction (5), stress response (5) and pain sensitivity (6). Three genetic variants of COMT have been identified in the human population corresponding to low, average and high pain sensitivity haplotypes (LPS, APS and HPS) (6). Higher COMT activity corresponds to lower pain sensitivity and vice versa. A silent mutation differentiates between low (LPS) and high (HPS) pain-sensitive phenotypes via reduced HPS protein levels (7), while APS is characterized by a valine to methionine substitution at amino acid position 108 that reduces its intrinsic activity through lowering protein stability (1,8) (Figure 1a). These haplotypes have also been associated with risk of fibromyalgia (9), temporomandibular joint disorder (TMJD) (6), postsurgical pain (10,11), responses to drugs (12) and development of brain white matter (13).
Figure 1.

Haplotypes of COMT and corresponding expression levels. (a) Organization of SNPs for the three haplotypes along COMT gene. Percent frequency of each haplotype from a cohort of 202 healthy Caucasian females is indicated on the right (6). The LPS designed mutant used for later experiments is also included for comparison. (b) In vitro translation in rabbit reticulocyte lysates. Both LPS and HPS share the same expression levels, while APS exhibits higher expression levels. To test whether this is due to the rs4633 SNP near the start codon, we mutated the LPS variant from 166C to 166T. The resultant LPS-T166 mutant displays expression levels on par with APS.
The ability of highly structured regions of mRNA to inhibit protein expression was recognized for a long time (14–16). However, the exact mechanisms of this inhibition and its relative contributions to regulation of translation efficiency in live cells have only limited examples (17,18). Thus, several in vitro studies have shown that RNA transcripts containing extremely stable stems with melting temperatures higher than 70°C can decrease protein expression at the level of ribosomal translocation (19). The underlying factor preventing translation at highly stable regions is thought to be the ribosome itself. It has been shown that the ribosome contains an intrinsic helicase activity, allowing it to read the individual bases (19). Thus, RNA motifs that are too difficult to unwind cause the ribosome to stall on the transcript.
Protein synthesis is highly regulated at the initiation stage, enabling rapid, reversible and spatial control of gene expression (20–23). Prokaryotic translation of mRNA is regulated at both the 5′ and 3′ ends of a transcript during initiation (24). For eukaryotes, initiation of translation proceeds by the ribosome scanning from the 5′ end of the transcript to the initial start codon (15,25). Scanning through the transcript is facilitated by the eIF4 factor unwinding structured RNA regions through an ATP-dependent process (14), and because of the scanning mechanism ribosomes cannot bind circular mRNA transcripts (26). Earlier work has demonstrated that gene expression can be repressed by increasing the stability of 5′ end mRNA secondary structures (27). Recent experiments with green fluorescent protein (GFP) constructs have also shown that the folding free energy of the 5′ end of an mRNA transcript is most correlated with protein expression, as opposed to a codon bias (28). Furthermore, reduced stability of the mRNA at the translation-initiation site was found to be a common feature for most species (29).
To uncover the translation mechanisms that allelic variants of common COMT haplotypes contribute to variation in COMT activity, we performed a set of molecular and computational studies. We first conducted in vitro translation studies of three haplotypes in rabbit reticulocyte lysates. Unlike the in vivo expression system, we did not observe a difference in an amount of translated COMT protein between LPS and HPS haplotypes, suggesting that rs4818-dependent stem–loop structure (7) requires additional cellular chaperons to affect translation efficiency. However, we observed robust increase in amount of protein of APS haplotype-coded mRNA. Here, we show how APS haplotype-specific T allele of the single-nucleotide polymorphism (SNP) rs4633 located at the 5′ end of mRNA near the ribosomal binding site, rather than non-synonymous met158 variation, modulates protein expression in vitro. We also conduct secondary structural analysis and perform simulations at the 5′ end of each haplotype using discrete molecular dynamics (DMD) to determine the mechanism by which the T allele at rs4633 alters translational efficiency (20,30,31). Our results reveal a novel mechanism by which the dynamics of mRNA structures near the initial start codon may influence efficiency of translation initiation.
MATERIALS AND METHODS
In vitro translation
COMT cDNA coding for three haplotypes and LPS-T166 mutant were cloned into a pCMV-Sport6 vector as described previously (7). The mRNA templates used for translation were generated by first restriction enzyme digestion using HindIII to create a linear plasmid. Digested plasmids were subsequently cleaned up using a PCR purification kit (Qiagen). In vitro transcription was performed by adding SP6 RNA polymerase (Promega) along with rNTPs and incubated in a reaction buffer under conditions provided by the manufacturer. RNA was purified from the mixture using Trizol (Invitrogen) and subsequently dissolved in water. The RNA integrity was evaluated by running the samples on the Bioanalyzer 2100 (Agilent).
The in vitro translation reaction was carried out using 1 µg RNA template, 17.5 µl rabbit reticulolysate, 0.5 µl amino acid mixture (-Met), 1 µl 35S-labeled methionine (1200 Ci/mmol), 0.5 µl RNasin and diluted to a total reaction volume of 25 µl. To denature the RNA we heat up the samples for 3 min at 70°C and immediately place on ice. For RNA secondary structure formation, we heat denature then subsequently add 5 mM MgCl2 and cool at a rate 0.1°C/s to a final temperature of 15°C. Once the RNA template is added to the rabbit lysate mix, we incubate for 1.5 h at 30°C. The reaction is stopped by adding 1× Laemmli buffer and heating for 4 min at 80°C.
We quantified the amount of protein product by separating via sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). The gel is initially placed in fixing solution (50% methanol, 40% water, 10% acetic acid) for 30 min under gentle rotation. Afterwards, the gel is soaked in a rinsing solution (85% water, 7% methanol, 7% acetic acid, 1% glycerol) for 5 min with gentle rotation. The gel is then placed in a drier with vacuum pump for 1.5 h at 80°C. The gel is then placed in a cassette with PhosphorImager screen and later quantified using Storm PhosphorImaging System (Molecular Dynamics).
To verify that our radiolabeled protein product is COMT, we performed immunoprecipitations on several lysate reactions. After in vitro translation reaction, an equal amount of NET buffer (150 mM NaCl, 5 mM EDTA, 50 mM Tris–HCl, pH 7.4) is added. We use Ultralink Protein A/G agarose beads and equilibrated them by washing with 0.5 ml NET buffer twice per 100 µl beads and resuspending in 100 µl NET buffer. For each lysate reaction, 5 µl of primary COMT antibody was added and incubated overnight with rotating at 4°C. Then, 50 µl of equilibrated Protein A/G agarose beads are added and incubated at 4°C for 4 h. Samples are then centrifuged for 5 min to remove the supernatant. The supernatant is saved for further SDS–PAGE analysis. The beads are then subsequently washed with 50 µl NET buffer twice by rotating for 5 min in 4°C. The proteins are removed from the beads by dissolving them with 25 µl of Novex Tris–Glycine SDS solution and boiled for 4 min at 80°C. The supernatant from the boiling reaction contains our immunoprecipitated protein and is analyzed via SDS–PAGE.
Transfection and western blotting
The cDNA clones coding for three COMT haplotypes were transfected into mammalian cell lines as described previously (7). COS-1, Hek-293, HepG2 and MCF-7 cell lines were purchased from ATCC and maintained in media [Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS), 4.5 g/l glucose, L-glutamine and sodium pyruvate for COS-1, Hek-293 and HepG2; RPMI 1640 with 5% FBS and L-glutamine for MCF-7] in accordance with manufacturer's recommendations. The western blotting was performed as described previously (7) using anti-hCOMT antibody derived from rabbit (Chemicon, ab5873).
Secondary structure analysis of COMT allelic variants
COMT allelic variants and randomly generated sequences were computationally ‘folded’ and the predicted minimum free energy of the secondary structure was calculated for different window sizes, using our implementation of the algorithm described by Zuker (31). Energy minimization was performed by dynamic programming method using an improved algorithm for evaluation of internal loops (32).
We estimated the free-energy penalty associated with breaking (opening) of the target's local secondary structure (target structure opening, ΔG kcal/mol), considering local disruption of secondary structure in windows with different lengths. Free-energy changes were approximated with nearest-neighbor free-energy parameters using the program OligoWalk (33). Here, we consider local structure for a set of suboptimal structures (Figure 2a). Each structure contributes to the free-energy penalty for disruption of structure in proportion to a Boltzmann weight; and the summations over all suboptimal structures were provided. Thus, this difference between the free energy of each suboptimal structure and the free energy of the corresponding suboptimal structure without base pairs in the region of complementarity in 30-nt window length is defined as the energy required for target structure opening (Figure 2a). Monte Carlo simulation and analysis of randomized sequences (21,34) was used for estimation of the significant difference between target structure opening, ΔG, of two COMT allelic variants. One-thousand unique random sequences for each allelic variant were generated by shuffling the first 210 nt of the COMT mRNA sequence and iteratively mutating two positions randomly along the sequence (except position 166). Each generated sequence is then checked to verify that the %GC and %AU content remain identical to the original COMT gene. Once the sequences are crosschecked with one another to ensure there are no duplicates, we create two sets of sequences where position 166 is occupied by either a C or U. The free-energy penalty associated with opening of the target's local secondary structure (ΔG kcal/mol) for all random sequences, considering local disruption of secondary structure in windows with 30-nt length was calculated. P-values for randomizations and for difference between the C and T alleles were determined by paired t-tests.
Figure 2.

Secondary structure analysis of 166C and 166T-allelic variants of COMT by Mfold. (a) Free energy profile (ΔG) for target structure opening in the vicinity of the rs4633 SNP and the start codon for 166C and 166T-allelic variants determined using OligoWalk. (b) The most probable RNA secondary structural predictions for 166C and 166T-allelic variants near start codon. We zoom in toward the regional differences between the allelic variants. The start codon is colored in green and the varying SNP is colored in red. The regions of difference are within the motifs labeled Loop I, Stem I and Stem II. The two stems leading to the Loop I and Stem II (capped at base pairs 46–110 and 29–178, respectively) are identical as are the rest of the nucleotides within the 210-nt window (Supplementary Data, Figures S1–S3). Both DMD and Mfold predict identical secondary structures for the 166C-allelic variant and 166T-allelic variant.
Three-bead RNA model and DMD
Traditional molecular dynamics simulate the motions of particles by solving Newton's equations of motion for a defined system using an integration algorithm. In DMD, simulations proceed according to the conservation laws of energy, momentum and angular momentum and are evaluated as a series of two-body interactions. The efficiency of the engine is based on an algorithm that searches through an event table, where velocities are only modified as necessary. Here, we classify an event as the instance in which two particles are within a defined interaction range as defined by their potential. The potentials used in DMD are discretized to accommodate the discontinuous nature of the simulations. Further details of the DMD algorithm can be found elsewhere (35,36).
We perform the RNA folding simulations using a simplified three-bead model (freely available on the web at http://ifoldrna.dokhlab.org/) (30). For each nucleotide, each bead represents a phosphate, sugar and base. Interactions contained in the model include standard Watson–Crick base pairing, G-U base pairing, base stacking, phosphate–phosphate repulsion, hydrophobic interactions and loop entropy.
Replica-exchange simulations and analysis
To ensure adequate sampling of the conformational space of each haplotype, we utilize replica exchange where R replicas are simulated each at a temperature Ti where i represents the index of that particular replica (37). A random walk in temperature space is performed by exchanging the temperatures between two replicas i and j under the probability
where
and Ei is the total energy of the system within the ith replica. Thus, as the macromolecule explores conformational space, the energy changes accordingly. Swapping the temperatures between replicas allows conformations that are stuck in local energy minima to escape and resume exploring other conformations. For our RNA folding simulations, we performed replica-exchange simulations with nine replicas (T = 0.1, 0.15, 0.2, 0.225, 0.25, 0.275, 0.35, 0.4 and 0.5 ε/kB) per allelic variant for 2 × 106 time steps. Energies of each RNA conformation throughout the simulation are evaluated according to parameters published previously (30).
Simulations can be analyzed using the weighted histogram analysis method (WHAM) to determine various thermodynamic quantities by deriving a partition function using the trajectories (38). We compute the specific heat of folding using
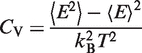 |
where < E > is the average potential energy and < E2 > is the average squared potential energy of the system for each specific temperature (35).
Contact maps provide a useful measure of gauging the frequency of certain conformations over the simulation. For our purposes, we are interested in the frequency of base-pair formations and thus have limited our contacts to the base beads (as opposed to including the sugar and phosphate beads). We define a base pair between two atoms i and j > i + 3 as <6.5 Å. From the contact maps, we can compare structures derived from simulations to secondary structure prediction programs by comparing contact maps of the former to dot plots of the latter. We can also deduce the secondary structures from the tertiary structures by calculating base pairs formed for each trajectory according to the parameters in the force field and determine which secondary structures are most probable (30).
We have clustered conformations using the OC hierarchical clustering package (available at http://www.compbio.dundee.ac.uk/downloads/oc). RNA structures derived from our simulation trajectories were clustered according to RMSD. The lower the RMSD between two RNA structures, the less distant (in terms of clustering) they are from one another. The OC algorithm works by first taking two structures that have the minimum distance and assigning them as a cluster, and then search across all other structures and comparing their distances among other structures. Structures that have been clustered together are considered a single entity. Representative tertiary structures were derived from three clustering methods: single (where the minimum distance between the two clusters is taken as the distance), complete (where the maximum distance between the clusters is taken as the distance) and means (where the average distance between clusters is taken as the distance). Structures that were found to be most dominant for all three clustering methodologies were considered most representative.
RESULTS
In vitro translation of COMT haplotypes
To study the precise molecular mechanism(s) whereby the mRNA of COMT haplotypes (Figure 1) produce different protein levels, we first employed an in vitro translation approach that is very effective in isolating putative mechanisms involving differential effects at the ribosomal level. We performed an endpoint kinetics assay using rabbit reticulocyte lysates. The advantage of this in vitro system is that external biological factors that regulate protein synthesis are absent. We found that the APS haplotype demonstrates higher protein expression levels compared to both the LPS and HPS haplotypes while LPS and HPS haplotypes have equivalent expression levels in vitro (Figure 1b).
There are two unique alleles an APS haplotype carries within its transcribed region – a T allele of the SNP rs4633 at the 5′ end of the mRNA (+32 nt downstream from the start codon) and an A allele of the SNP rs4680 within the second exon of the gene (Figure 1a). As we previously showed, the structural mRNA differences within the second exon are the most pronounced between LPS and HPS haplotypes (7), not the APS haplotype. Consequently, we concluded that it is unlikely that SNP rs4680 contributes significantly to the protein levels in the in vitro translation experiment. In contrast, SNP rs4633 is situated at the 5′end of the COMT mRNA near the start codon; a region which showed the strongest association between stability of mRNA folding and the rates of translation initiation expression levels of individual genes (28,39). To test the individual contribution of SNP rs4633 in the increase in protein levels observed for APS haplotypes, we created a C to T mutant at position 166 for LPS haplotype (LPS-T166). Our results show that mutating LPS at position 166 from C to T recapitulates high expression levels characteristic for APS (Figure 1b). Thus, the determining factor for translation efficiency of COMT resides in the SNP rs4633 alone.
Secondary structure prediction of 5′ region
We then studied local RNA secondary structures contributing to effect of SNP rs4633. It was shown that free-energy stability of the 5′ region of an mRNA transcript is correlated with translational efficiency (21,22,28,39–41). Transcripts that have less stable RNA structural elements near the 5′ end have higher translation rates (28), presumably because tight binding to the initial start codon becomes difficult for the ribosome initiation machinery (20,23,25,28). To test if the T allele of rs4633 specific for APS haplotype results in a change in free energy, we initially utilize secondary structure prediction programs to calculate the free energy of the 5′ end for different respective RNA.
We predicted mRNA local secondary structures in the vicinity of the SNP rs4633 and the start codon by employing different algorithms for both variants (30,32,42). Comparison of the optimal structures for both C and T-allelic variants shows that the main structural differences due to the SNP rs4633 lies within the structural regions of Loop I, Stem I and Stem II (Figure 2a; Supplementary Data, Figures S1–S3). Loop I of the 166T variant (nt 119–126) is more flexible with an additional 2 nt (nucleotides 126 and 127) comprising this single-stranded region. The two nucleotides present in Loop I of the 166T variant causes Stem I to lose two Watson–Crick base pairs and lower the stem's stability. Consideration of these two regions alone, the T allele-carrying mRNA is less stable. However, both free-energy calculations predict that its structure at Stem II downstream from the start codon is ∼1 kcal/mol more stable than the wild type. This is primarily due to an additional A-U base pair formed unique to the T allele-carrying mRNA in Stem II between nucleotides 156 and 165 that increases its stability from an enthalpic standpoint. From visual inspection, it can also be seen that the additional unpaired bases found in the hairpin loop of Stem II for 166C variant are entropically disfavorable due to the number of single-stranded nucleotides comprising the terminal loop compared to only four base pairs within the stem.
To verify whether this structural region is stable independent of the surrounding sequence, we truncated the sequence near the neighboring junctions (nucleotides 119 through 171) and refolded the structures using Mfold, Afold and RNAstructure (31,32,42). All programs predict the same optimal local structures as the 210-nt length transcripts (Figure 2a). Furthermore, we predicted all suboptimal structures for both C and T allelic variants when percent suboptimality is set to 30 (when only folding within 30% from the minimum free energy will be computed). Identical Stem I loops surrounding AUG codons were found in two most stable RNA local structures with the total free energies (ΔG) of −14.8 and 8.63 kcal/mol in the 166C-allelic variant, respectively (Supplementary Data, Figure S4). The 166T-allelic variant produced four structures with the energies ranging from ΔG = −16.6 kcal/mol to ΔG = −10.05 kcal/mol, only one of those identical at Stem I loop (Supplementary Data, Figure S5). Thus, there are not only differences in the secondary structures between 166C and 166T allelic variants, but also 166T-allelic variant also produces a higher diversity of suboptimal structures.
We then estimated the level of pairing and free energy of target breaking (opening) for the 166C-allelic variant mRNA and the 166T variant using full-length transcripts and truncated transcript sequences of different lengths starting from 210 nt, where approximately a half of the sequence length is located in the 5′UTR and the second half is in the coding region of the COMT gene. We modeled the dynamic process of transcript folding and target breaking using 30-nt windows (33,43) (see ‘Materials and Methods’ section). Profiles of the free energy of target breaking for 166C and 166T variants (Figure 2b) show that mRNA secondary structures in the vicinity of the start codon (30-nt length window) are less stable and the free energy of target breaking is significantly (P = 0.0016) higher for the 166T variant of rs4633 (specific for APS haplotype) relative to the 166C variant of rs4633 (specific for LPS and HPS haplotypes). On the other hand, Monte Carlo simulation of the sequences in the vicinity of the start codon showed that the differences in free energy of mRNA secondary structure target opening for the 166T variant and the 166C variant of rs4633 were not random (P < 0.05). Consistent with these findings, our secondary structural analysis using Mfold, Afold and RNAstructure (31,32,42) also demonstrates that the allelic variation of rs4633 directly affects the RNA structure surrounding the start codon (Figure 2b). Thus, our secondary structure predictions therefore provide an important insight that there is an independent motif in which there are structural differences for each haplotype.
Simulations predict different folding barriers
Our folding prediction and analysis of mRNA local secondary structures revealed that the 166T allele promotes base pair disruption near the vicinity of the start codon (Figure 2b). The lower free-energy stability of the 166T-allelic variant implies that there exists a lower energy barrier separating the folded and unfolded states. The eukaryotic initiation factor eIF4a facilitates translation in an ATP-dependent manner by unwinding RNA secondary structure to enable ribosomal translocation (14). Thus, if there are differences in the energetic barriers between the folded and unfolded states for the haplotypes, then structures with a lower energy barrier height would undergo more efficient translation since there would be a higher probability for the structure to exist in an unfolded conformation (44,45). To test this hypothesis, we generate tertiary structures of the RNA motifs by simulating the dynamics (Figure 3a and b) of each allelic variant using discrete molecular dynamics (DMD) (30).
Figure 3.

Tertiary structure analysis by DMD simulations indicates that the unfolding energy barrier is lower for 166T allele, thus enhancing its conformational flexibility to explore higher energy states. (a and b) Structures of variants as predicted using DMD. Leftmost structures represent the native tertiary structure for each allelic variant. The middle structures are the secondary structural elements as predicted by DMD. The rightmost structures are represented in sausage format where thicker backbones represent larger root mean squared fluctuations within the native ensemble (see ‘Materials and Methods’ section). Regions of the RNA are color-coded to correspond to different motifs: Loop I (blue), Stem I (green) and Stem II (red). (a) Structures of 166C-allelic variant. (B) Structures of 166T-allelic variant. (c) The folding transition temperature for 166T is lower than 166C, as denoted by the peak in specific heat of folding. Specific heats were determined using weighted histogram analysis method. The vertical droplines represent the standard deviation for each plot point. (d) Histogram of free energies derived from replica-exchange simulations. The native ensembles that correspond to the structural motifs as predicted by Mfold are denoted on the plot. (e and f) Maps illustrating frequency of contacts at a temperature of 0.10 ε/kB. (e) Frequency of contacts for 166C-allelic variant. The most frequent contacts shown are representative of the native state alone, where the first lower left diagonal line is representative of Stem I (nucleotides 5 through 32 of the simulation correspond to nucleotides 124 through 151 on the transcript). The second smaller diagonal line on the upper right of the plot represents the contacts formed from the Stem II (nucleotides 34 through 51). (f) Frequency of contacts for 166T-allelic variant. Even at the lowest simulation temperature, the 166T SNP creates an ensemble of states where many different enthalpic contacts are made throughout its folding simulation but are entropically unfavorable.
Simulations were performed for both 166C and 166T-allelic variations of rs4633 of COMT transcripts at the 5′ region between nucleotides 119 through 171 using an RNA three-bead model (30). Since we observe multiple transitions between the folded and unfolded states, there is adequate sampling to enable determination of the thermodynamics of the folding transition using the weighted histogram analysis method. A comparison between the 166C and 166T alleles of mRNA transcripts shows that the peak denoting the folding transition temperature is slightly higher for the 166C allele, demonstrating its increase in thermodynamic stability (Figure 3c).
We find that both alleles adopt a native conformation (Figure 3a and b) that is in line with secondary structural predictions (Figure 2a). Both the C and T alleles fold into their respective native conformations at −25.1 kcal/mol and lower (Figure 3d). Notably, the 166C allele has a higher probability in existing in this low energy state. The most stable structures that are unique to the 166C allele are formed due to transient base pair formations in the loops of Stem I and Stem II (Figure 3d). In contrast, the 166T allele-carrying mRNA is more likely to adopt conformations at higher energies along its folding pathway (Figure 3d), a consequence of its lower folding transition temperature (Figure 3c).
Since we know that the T allele is responsible for disrupting the local secondary structure near the start codon (Figure 2b), we wanted to deduce the destabilizing effects on the overall tertiary structure of that region. From our simulations, we determined the flexibility of each allelic variant's tertiary structure by calculating the root mean square fluctuation (RMSF) of the native ensemble (as highlighted in Figure 3d). We find that the dynamics of 166T allele-carrying mRNA are highly entropy driven (Figure 3b; right most structure) with RMSFs up to 11 Å for a single nucleotide (Supplementary Data, Table S1). In contrast, the 166C allele-carrying mRNA has an RMSF <6.5 Å for all nucleotides (Figure 3a; Supplementary Data, Table S1). The contact map highlights the attempts made by 166T allele-carrying mRNA to fold into its native structure (Figure 3e and f). The plethora of contacts demonstrates the competing states that lead to a rugged energy landscape (Figure 3d). Thus, examining the dynamics of 166T allele-carrying mRNA suggests that the free-energy barrier height between folded and unfolded states is smaller (Figure 3c) and therefore likely to adopt higher energy states. Consequently, the 166T allele exists in an unfolded intermediate state more frequently than the 166C allele-carrying mRNA (Figure 3d) (44–46).
Haplotype-dependent COMT protein expression in mammalian cell lines
To examine if the higher efficiency of protein translation contributed by the 166T allele plays a substantial role at the cellular level, we carried out a series of transfection studies. As reported previously, in PC12 rat pheochromacytoma cells, COMT protein expression levels is reduced 25-fold in the HPS haplotype; however, LPS and APS haplotypes display comparable protein levels (7). To determine whether this effect is a general feature of COMT protein expression in mammalian cells or specific to the PC12 cell line, we transfected expression vectors with the three COMT haplotypes in a number of different cell line with divergent tissue origin: COS-1 monkey kidney cells, HEK-293 human embryonic kidney cells, HepG2 human liver cells and MCF-7 human breast cancer cells. We find that all transfected cell types consistently exhibited the same qualitative trend in protein expression (Figure 4), where LPS exhibits higher protein expression than HPS, and HPS shows the most reduced protein expression. Notably, APS showed comparable protein levels to LPS in COS-1 and HepG2 cell lines but not in HEK-293 and MCF-7 where APS showed the highest protein level (Figure 4). Thus, it appears that an increase in translation efficiency of the APS haplotype does contribute to cellular protein levels and this contribution is tissue specific.
Figure 4.

Protein expression levels of COMT constructs that code for three COMT haplotypes in the range of mammalian cell lines. The constructs were transiently transfected into indicated cell lines and its protein expression were analyzed via western blotting. Top band: β-actin. Lower band: COMT. Cell lines: COS-1 monkey kidney cells, HEK-293 human embryonic kidney cells, HepG2 human liver cells, and MCF-7 human breast cells. Among these cell lines, APS protein expression is equivalent or higher than LPS, while HPS exhibits significantly lower levels compared to the two haplotypes.
DISCUSSION
166T mutation contributes to higher translation rates
Our in vitro translation data demonstrate that the APS haplotype of COMT has a higher rate of translation compared to LPS and HPS, while LPS and HPS haplotypes show similar level of expression (Figure 1b). Furthermore, our results suggested that the upstream 5′-end structure in allelic-dependent manner largely controls the in vitro translation rate. The APS-specific 166T allele of rs4633 was hypothesized to drive the difference in protein translation efficiency. The 166T allele of rs4633 was also a strong candidate for the translational ‘switch’ because of its unique location near the start codon, the RNA area known to contribute the most to the RNA structure-dependent translation initiation rate (28). To test this hypothesis, we created a LPS mutant carrying the 166T allele of rs4633 (LPS-T166) specific for APS haplotype. This mutant has similar protein expression levels in comparison to APS (Figure 1b), suggesting that only the SNP rs4633 is necessary for determining expression rates in vitro. Therefore, in the case of translation in vitro, we can rule out the possibility that the other downstream SNPs of three major COMT haplotypes play a role in determining efficiency of in vitro translation.
Computational analysis reveals structural differences
To investigate the structural mechanism in which SNP rs4633 affects in vitro translation of COMT RNA, we employed an array of computational approaches. We initially utilize Mfold to generate secondary structures of each haplotype. By examining the folds of COMT mRNA at various sequence lengths, we observe which type of structures predominate at the 5′ end. We find that much of the structure toward the 5′ end is identical for all haplotypes with the exception near the vicinity of the SNP rs4633 and start codon (Figure 2b). The observation that a single SNP affects the 5′ end structure in the vicinity of the start codon supports the view that this region might regulate COMT translation.
LPS and HPS haplotypes exhibit equivalent in vitro protein expression levels, which can be attributed to identical secondary structures near the start codon due to sharing the 166C allele. However, the 166T allele structure (carried by the APS haplotype) has some unique structural rearrangements in comparison with the 166C-allelic variant that influences structure stability in the vicinity of the start codon (Figure 2a). Furthermore, consistent with these observations, thermodynamic analysis from OligoWalk suggests that the 166T allele structure is less stable near the vicinity of the start codon compared to the 166C allele (Figure 2b). Local secondary structures are extremely conserved in the vicinity of the start codon for the 166C allelic variant, exemplified in the optimal and suboptimal structures predicted by Mfold where Stem I is identical for both (Supplementary Data, Figure S4). Contrarily, local secondary structures in the vicinity of the start codons differ between optimal and suboptimal structures in T-allele and display more structural diversity.
It is suggestive that the region in the vicinity of the start codon may potentially play a more significant role with regard to translational efficiency (28,40). Since we know that the alternative 166T allele yields a unique structure in the vicinity of the start codon, it is possible to fold this sequence using DMD simulations and seek whether tertiary interactions may play an integral role. We find that both 166C allele and 166T allele predicted structures derived from DMD simulations are identical to secondary structures predicted by Mfold and RNAstructure (Figures 2a and 3a and b). However, the native ensemble of 166T allele is less probable in comparison to the 166C allele. The conformational entropy for the 166T allele is higher than the 166C allele, resulting in high flexibility and exploration of higher energy states (Figure 3d). Consequently, this may enable facilitated initiation of translation as displayed by the higher expression levels by this haplotype. Our thermodynamic analysis reveals that this is a strong possibility given that the folding transition temperature is lower for 166T allele's structure than 166C.
Codon bias and mRNA structure as factors in translation efficiency
Two main mechanisms are thought to be important factors in determining the efficiency of mRNA translation: (i) the ease of unwinding the mRNA structure at the 5′ end and (ii) codon usage. The extent to which one mechanism plays a dominating factor over the other is dependent upon the individual genes in question (27,28,47,48). Our simulations in this work have focused on the structural contributions that can lead to increased protein expression from the 166T allele. Here, we consider the possibility of codon bias and its effect on translational regulation of COMT. Both the 166C allele and 166T allele at rs4633 code for histidine through the synonymous codons CAC and CAT, respectively. It has been reported that the CAC allele is nearly twice as efficient compared to the CAT allele (49). Within this context, it is unlikely that the CAT codon is the contributing factor for increased protein expression. However, it has also been reported that use of low-efficiency codons near the initiation site can aid the efficiency of translation as this scenario prevents ribosomal traffic jams near the initiation site (50). It is uncertain to what extent the CAT codon increases efficiency of translation given these two competing scenarios. These explanations are in agreement with previously published data on the role of mRNA structure and codon usage in the vicinity of the start codon for translation efficiency (41). Nevertheless, reduced ribosomal traffic may also play a role in the enhancement of 166T allele protein expression.
Role of mRNA stability and dynamics in translation initiation
Previous reports have only suggested a correlation of free-energy stability and translational efficiency (15,28). Since it is the stability of the 5′ end that is the most determining factor for translation efficiency, it is presumed that the limiting factors are unwinding by the eIF4a factor and ribosomal binding to the start codon. The relationship between free energy and translation initiation is not immediately apparent. The stability of a particular region alone would not necessarily render a recognition site inaccessible. Thus, we proposed that the folding pathway might play an essential role in regulating initiation factor access. Specifically, if the energetic barriers between conformational states of an RNA are low, then the RNA can easily explore conformations that are outside its native ensemble (46). These conformations can have a reduced number of structural elements and therefore this flexibility can facilitate sequence recognition by translational machinery.
The results presented here support this model. Substitution of 166C to 166T at rs4633 in COMT mRNA increases the number of favorable isoenergetic conformational states for its mRNA transcript. The dynamics of the 166T-carrying allele become entropy driven such that the native conformation becomes less populated as the RNA explores conformations at higher energies. Consequently, there are large fluctuations in the positions of each nucleotide, thereby enhancing its flexibility. Further exploration of this model by studying the dynamics for a wide variety of RNA structures would be required to prove its fundamental significance.
Implications for cellular expression levels
Our current results in several transfected mammalian cell lines re-enforce the conclusion from our previous studies (7) that the in vivo expression of COMT is strongly dictated by RNA structures formed by SNPs rs4818 and rs4680 (Figure 4) yielding the lowest expression levels for haplotype HPS. However, the in vitro translation rate seems to be independent from rs4818 and rs4680 interactions and driven solely by local structures near the start codon that is dependent on allelic variants of rs4633 (Figure 1b). Furthermore, in two out of four cell lines we also observed that the APS haplotype produced the highest protein level, consistent with in vitro translation results that are rs4633-dependent. Thus, we observed differential input of three SNPs into two distinct structural mechanisms, apparently contributing to translation regulation at different levels.
These results are in line with the observed mRNA structure-dependent differences in efficiency and rate of translation in vivo and in vitro reported previously (18). Since the rate of translation is much slower than the rate of RNA folding, it is thought that RNA begins to fold locally during the translation process and that the final structure oftentimes is the metastable product of local folding. Thus, the upstream structures dominate folding outcomes in vitro, suggesting that folding occurs sequentially. However, when studied in vivo, upstream and downstream structures are presented equally and folding outcomes reflect the relative stability of alternative structures, probably facilitated by cellular chaperone proteins associated with nascent RNAs (18).
The variation in COMT expression levels across different systems could potentially be explained by the experimental observation that RNA can adapt specific structures in cells due to rapid exchanging of states facilitated by proteins bound to nascent RNAs (18) in contrast to in vitro translation conditions. Because the rate of RNA folding is on the scale of microseconds and thus much faster than the rate of transcription, there is a preference for local folds as opposed to long-ranged base pairs. This preference may be diminished in cells by specific RNA-binding proteins that allow exchange of secondary structures through branch migration (18). Our results suggest that the contribution of these cellular proteins is tissue specific, such that in some cell lines the overall cellular protein expression is almost exclusively controlled by these factors, while other cell lines recapitulate the results found in vitro using rabbit reticulocyte lysates (Figure 4).
It is also plausible that in some cell lines, other factors regulating translation are more strongly contributing to protein expression. The abundance of transfer RNAs (tRNAs) with synonymous codons are known to vary in the cell up to 10-fold across different human tissues (51). The availability of tRNAs during translation could also contribute to the relative speed at which the protein is synthesized.
Alternatively, it is possible that structural modulation of RNA itself is not the sole explanation for differences in protein expression, and there may be additional mechanisms contributing to translational regulation. These downstream structural motifs may potentially be recognized by external biological factors and subject to further regulation. For example, the Fragile X Mental Retardation Protein (FMRP), an established regulator of translation, is known to bind to specific structural RNA motifs (52,53) and can downregulate their expression by association with the RNA-induced silencing complex (54,55). This cascade of structural and cellular mechanisms at the mRNA level is likely to be defined by other specific cellular components and thus contribute to differences in COMT protein expression levels in a tissue-dependent manner.
From a broader perspective, since the APS haplotype carries a nonsynonymous met158 variation known to create a thermolabile mutant and thus display lower enzymatic activity (1,8) in comparison with wild-type val158, it is remarkable that its protein expression level can be significantly higher than wild-type LPS haplotype. Thus, our results represent a potential compensatory mechanism of APS haplotypes to overcome lower enzymatic activity via overexpression in specific cell lines.
CONCLUSIONS
The results presented here demonstrate a new molecular mechanism, thereby synonymous substitution of a known functional human COMT haplotype contributes to translation efficiency, thus representing an exciting example of evolutionary selection of an RNA-structure destabilizing allele to compensate for a destabilizing amino acid substitution within a mutant protein structure. Importantly, this change did not only affect the stability of RNA structure but rather its dynamics, suggesting that increased conformational flexibility enhances translational efficiency. This mechanism by which the destabilizing allele facilitates translation provides a new perspective in functional genomics and requires further investigation to determine the extent of its fundamental applicability for common genetic variations in human population.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The US National Institutes of Health grant (R01GM080742 to N.V.D.); American Recovery and Reinvestment Act supplements (GM080742-03S1, GM066940-06S1 to N.V.D.); National Institute of Dental and Craniofacial Research and National Institute of Neurological Disorders and Stroke grants (RO1-DE16558, UO1-DE017018, PO1 NS045685 to L.D.); and Intramural Research Programs of National Center Biotechnology Information a National Library of Medicine (to S.A.S.). Funding for open access charge: National Institutes of Dental and Craniofacial Research and National Institute of Neurological Disorders and Stroke grants (5-U01-DE017018-04-06 and 2-P01-NS045685-06A1 to L.D.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr Sergei Romanov for his aid in developing the in vitro translation assay.
REFERENCES
- 1.Lotta T, Vidgren J, Tilgmann C, Ulmanen I, Melen K, Julkunen I, Taskinen J. Kinetics of human soluble and membrane-bound catechol O-methyltransferase: a revised mechanism and description of the thermolabile variant of the enzyme. Biochemistry. 1995;34:4202–4210. doi: 10.1021/bi00013a008. [DOI] [PubMed] [Google Scholar]
- 2.Barnett JH, Heron J, Goldman D, Jones PB, Xu K. Effects of catechol-O-methyltransferase on normal variation in the cognitive function of children. Am. J. Psychiatry. 2009;166:909–916. doi: 10.1176/appi.ajp.2009.08081251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lachman HM. Does COMT val158met affect behavioral phenotypes: yes, no, maybe? Neuropsychopharmacology. 2008;33:3027–3029. doi: 10.1038/npp.2008.189. [DOI] [PubMed] [Google Scholar]
- 4.Voelker P, Sheese BE, Rothbart MK, Posner MI. Variations in catechol-O-methyltransferase gene interact with parenting to influence attention in early development. Neuroscience. 2009;164:121–130. doi: 10.1016/j.neuroscience.2009.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ducci F, Goldman D. Genetic approaches to addiction: genes and alcohol. Addiction. 2008;103:1414–1428. doi: 10.1111/j.1360-0443.2008.02203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diatchenko L, Slade GD, Nackley AG, Bhalang K, Sigurdsson A, Belfer I, Goldman D, Xu K, Shabalina SA, Shagin D, et al. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum. Mol. Genet. 2005;14:135–143. doi: 10.1093/hmg/ddi013. [DOI] [PubMed] [Google Scholar]
- 7.Nackley AG, Shabalina SA, Tchivileva IE, Satterfield K, Korchynskyi O, Makarov SS, Maixner W, Diatchenko L. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science. 2006;314:1930–1933. doi: 10.1126/science.1131262. [DOI] [PubMed] [Google Scholar]
- 8.Rutherford K, Alphandery E, McMillan A, Daggett V, Parson WW. The V108M mutation decreases the structural stability of catechol O-methyltransferase. Biochim. Biophys. Acta. 2008;1784:1098–1105. doi: 10.1016/j.bbapap.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 9.Vargas-Alarcon G, Fragoso JM, Cruz-Robles D, Vargas A, Lao-Villadoniga JI, Garcia-Fructuoso F, Ramos-Kuri M, Hernandez F, Springall R, Bojalil R, et al. Catechol-O-methyltransferase gene haplotypes in Mexican and Spanish patients with fibromyalgia. Arthritis Res. Ther. 2007;9:R110. doi: 10.1186/ar2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.George SZ, Dover GC, Wallace MR, Sack BK, Herbstman DM, Aydog E, Fillingim RB. Biopsychosocial influence on exercise-induced delayed onset muscle soreness at the shoulder: pain catastrophizing and catechol-o-methyltransferase (COMT) diplotype predict pain ratings. Clin. J. Pain. 2008;24:793–801. doi: 10.1097/AJP.0b013e31817bcb65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.George SZ, Wallace MR, Wright TW, Moser MW, Greenfield WH, III, Sack BK, Herbstman DM, Fillingim RB. Evidence for a biopsychosocial influence on shoulder pain: pain catastrophizing and catechol-O-methyltransferase (COMT) diplotype predict clinical pain ratings. Pain. 2008;136:53–61. doi: 10.1016/j.pain.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bialecka M, Kurzawski M, Klodowska-Duda G, Opala G, Tan EK, Drozdzik M. The association of functional catechol-O-methyltransferase haplotypes with risk of Parkinson's disease, levodopa treatment response, and complications. Pharmacogenet. Genomics. 2008;18:815–821. doi: 10.1097/FPC.0b013e328306c2f2. [DOI] [PubMed] [Google Scholar]
- 13.Liu B, Li J, Yu C, Li Y, Liu Y, Song M, Fan M, Li K, Jiang T. Haplotypes of catechol-O-methyltransferase modulate intelligence-related brain white matter integrity. Neuroimage. 2010;50:243–249. doi: 10.1016/j.neuroimage.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 14.Ray BK, Lawson TG, Kramer JC, Cladaras MH, Grifo JA, Abramson RD, Merrick WC, Thach RE. ATP-dependent unwinding of messenger RNA structure by eukaryotic initiation factors. J. Biol. Chem. 1985;260:7651–7658. [PubMed] [Google Scholar]
- 15.Kozak M. Circumstances and mechanisms of inhibition of translation by secondary structure in eucaryotic mRNAs. Mol. Cell. Biol. 1989;9:5134–5142. doi: 10.1128/mcb.9.11.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Somogyi P, Jenner AJ, Brierley I, Inglis SC. Ribosomal pausing during translation of an RNA pseudoknot. Mol. Cell. Biol. 1993;13:6931–6940. doi: 10.1128/mcb.13.11.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansen TM, Reihani SN, Oddershede LB, Sorensen MA. Correlation between mechanical strength of messenger RNA pseudoknots and ribosomal frameshifting. Proc. Natl Acad. Sci. USA. 2007;104:5830–5835. doi: 10.1073/pnas.0608668104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahen EM, Watson PY, Cottrell JW, Fedor MJ. mRNA secondary structures fold sequentially but exchange rapidly in vivo. PLoS Biol. 2010;8:e1000307. doi: 10.1371/journal.pbio.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takyar S, Hickerson RP, Noller HF. mRNA helicase activity of the ribosome. Cell. 2005;120:49–58. doi: 10.1016/j.cell.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 20.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell. Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shabalina SA, Ogurtsov AY, Spiridonov NA. A periodic pattern of mRNA secondary structure created by the genetic code. Nucleic Acids Res. 2006;34:2428–2437. doi: 10.1093/nar/gkl287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shabalina SA, Ogurtsov AY, Rogozin IB, Koonin EV, Lipman DJ. Comparative analysis of orthologous eukaryotic mRNAs: potential hidden functional signals. Nucleic Acids Res. 2004;32:1774–1782. doi: 10.1093/nar/gkh313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Pomeroy-Cloney L, Bjerknes M, Tam J, Jay E. The influence of adenine-rich motifs in the 3′ portion of the ribosome binding site on human IFN-gamma gene expression in Escherichia coli. J. Mol. Biol. 1994;240:20–27. doi: 10.1006/jmbi.1994.1414. [DOI] [PubMed] [Google Scholar]
- 25.Kozak M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene. 2005;361:13–37. doi: 10.1016/j.gene.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 26.Kozak M. Inability of circular mRNA to attach to eukaryotic ribosomes. Nature. 1979;280:82–85. doi: 10.1038/280082a0. [DOI] [PubMed] [Google Scholar]
- 27.Griswold KE, Mahmood NA, Iverson BL, Georgiou G. Effects of codon usage versus putative 5′-mRNA structure on the expression of Fusarium solani cutinase in the Escherichia coli cytoplasm. Protein Expr. Purif. 2003;27:134–142. doi: 10.1016/s1046-5928(02)00578-8. [DOI] [PubMed] [Google Scholar]
- 28.Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu W, Zhou T, Wilke CO. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput. Biol. 2010;6:e1000664. doi: 10.1371/journal.pcbi.1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding F, Sharma S, Chalasani P, Demidov VV, Broude NE, Dokholyan NV. Ab initio RNA folding by discrete molecular dynamics: from structure prediction to folding mechanisms. RNA. 2008;14:1164–1173. doi: 10.1261/rna.894608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogurtsov AY, Shabalina SA, Kondrashov AS, Roytberg MA. Analysis of internal loops within the RNA secondary structure in almost quadratic time. Bioinformatics. 2006;22:1317–1324. doi: 10.1093/bioinformatics/btl083. [DOI] [PubMed] [Google Scholar]
- 33.Mathews DH, Burkard ME, Freier SM, Wyatt JR, Turner DH. Predicting oligonucleotide affinity to nucleic acid targets. RNA. 1999;5:1458–1469. doi: 10.1017/s1355838299991148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kondrashov AS, Shabalina SA. Classification of common conserved sequences in mammalian intergenic regions. Hum. Mol. Genet. 2002;11:669–674. doi: 10.1093/hmg/11.6.669. [DOI] [PubMed] [Google Scholar]
- 35.Dokholyan NV, Buldyrev SV, Stanley HE, Shakhnovich EI. Discrete molecular dynamics studies of the folding of a protein-like model. Fold Des. 1998;3:577–587. doi: 10.1016/S1359-0278(98)00072-8. [DOI] [PubMed] [Google Scholar]
- 36.Tsao D, Dokholyan NV. Macromolecular crowding induces polypeptide compaction and decreases folding cooperativity. Phys. Chem. Chem. Phys. 2010;12:3491–3500. doi: 10.1039/b924236h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;314:141–151. [Google Scholar]
- 38.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 39.de Sousa Abreu R, Penalva LO, Marcotte EM, Vogel C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009;5:1512–1526. doi: 10.1039/b908315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seo SW, Yang J, Jung GY. Quantitative correlation between mRNA secondary structure around the region downstream of the initiation codon and translational efficiency in Escherichia coli. Biotechnol. Bioeng. 2009;104:611–616. doi: 10.1002/bit.22431. [DOI] [PubMed] [Google Scholar]
- 41.Zhang F, Saha S, Shabalina SA, Kashina A. Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science. 2010;329:1534–1537. doi: 10.1126/science.1191701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matveeva O, Nechipurenko Y, Rossi L, Moore B, Saetrom P, Ogurtsov AY, Atkins JF, Shabalina SA. Comparison of approaches for rational siRNA design leading to a new efficient and transparent method. Nucleic Acids Res. 2007;35:e63. doi: 10.1093/nar/gkm088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morgan SR, Higgs PG. Barrier heights between ground states in a model of RNA secondary structure. J. Phys. A: Math. Gen. 1998;31:3153–3170. [Google Scholar]
- 45.Hyeon C, Thirumalai D. Can energy landscape roughness of proteins and RNA be measured by using mechanical unfolding experiments? Proc. Natl Acad. Sci. USA. 2003;100:10249–10253. doi: 10.1073/pnas.1833310100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Solomatin SV, Greenfeld M, Chu S, Herschlag D. Multiple native states reveal persistent ruggedness of an RNA folding landscape. Nature. 2010;463:681–684. doi: 10.1038/nature08717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tuller T, Waldman YY, Kupiec M, Ruppin E. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl Acad. Sci. USA. 2010;107:3645–3650. doi: 10.1073/pnas.0909910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonzalez de Valdivia EI, Isaksson LA. A codon window in mRNA downstream of the initiation codon where NGG codons give strongly reduced gene expression in Escherichia coli. Nucleic Acids Res. 2004;32:5198–5205. doi: 10.1093/nar/gkh857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waldman YY, Tuller T, Sharan R, Ruppin E. TP53 cancerous mutations exhibit selection for translation efficiency. Cancer Res. 2009;69:8807–8813. doi: 10.1158/0008-5472.CAN-09-1653. [DOI] [PubMed] [Google Scholar]
- 50.Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010;141:344–354. doi: 10.1016/j.cell.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 51.Dittmar KA, Goodenbour JM, Pan T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006;2:e221. doi: 10.1371/journal.pgen.0020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–499. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- 53.Darnell JC, Fraser CE, Mostovetsky O, Stefani G, Jones TA, Eddy SR, Darnell RB. Kissing complex RNAs mediate interaction between the Fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev. 2005;19:903–918. doi: 10.1101/gad.1276805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Y, Lin L, Jin P. The microRNA pathway and fragile X mental retardation protein. Biochim. Biophys. Acta. 2008;1779:702–705. doi: 10.1016/j.bbagrm.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Plante I, Davidovic L, Ouellet DL, Gobeil LA, Tremblay S, Khandjian EW, Provost P. Dicer-derived microRNAs are utilized by the fragile X mental retardation protein for assembly on target RNAs. J. Biomed. Biotechnol. 2006;2006:64347. doi: 10.1155/JBB/2006/64347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.