


Abstract
Bacterial Rho-independent terminators (RITs) are important genomic landmarks involved in gene regulation and terminating gene expression. In this investigation we present RNIE, a probabilistic approach for predicting RITs. The method is based upon covariance models which have been known for many years to be the most accurate computational tools for predicting homology in structural non-coding RNAs. We show that RNIE has superior performance in model species from a spectrum of bacterial phyla. Further analysis of species where a low number of RITs were predicted revealed a highly conserved structural sequence motif enriched near the genic termini of the pathogenic Actinobacteria, Mycobacterium tuberculosis. This motif, together with classical RITs, account for up to 90% of all the significantly structured regions from the termini of M. tuberculosis genic elements. The software, predictions and alignments described below are available from http://github.com/ppgardne/RNIE.
INTRODUCTION
Transcription termination in bacteria is accomplished by two different mechanisms, both dependent upon RNA polymerase (RNAP) pause sites. The first relies upon the interaction of a protein called Rho with RNAP. The second, often called ‘intrinsic termination’, depends on the presence of short genomically encoded motifs called Rho-independent terminators (RITs). These are characterized by short G + C-rich hairpin loops containing ∼30 nt followed by a polyuridine tail of typically three to seven consecutive uridines which precede the stop site (Figure 1). These motifs bind to components of RNAP causing the bacterial polymerase to stall, releasing the nascent transcript resulting in transcription termination (1).
Figure 1.

(A) Rho-independent termination: the RNA polymerase traverses the DNA template strand from 3′ to 5′, synthesizing the nascent RNA molecule. (B) As the polymerase nears a termination site, a G + C-rich terminator stem sequence (highlighted in blue) is transcribed. (C) Formation of a hairpin structure causes the polymerase to pause, and together with a string of unstable rU-dA bonds causes the polymerase to release from the template.
An early bioinformatic analysis of complete genomes implied that there is a broad range in the degree of dependence on intrinsic versus Rho-dependent termination (2). This result is supported by mutagenesis experiments showing that the terminator protein Rho is essential in some organisms such as Salmonella enterica yet non-essential in others such as Bacillus subtilis (3,4). Indeed, Rho itself is the only protein known to depend on Rho-dependent termination in B. subtilis. Consequently, there appears to be competition between the two termination systems across bacterial species, resulting in clade-specific skews in usage for one or the other (5–7). For example, in Eschericha coli just 171 genes have experimentally characterized RITs, whereas in B. subtilis there are 891 confirmed terminations by RITs.
As the number of bacterial genome sequences grows (1194 in the June 2010 release of EMBL version 104), gene annotation is increasingly reliant on automated approaches. This will accelerate as new sequencing technologies are targeted towards sparse and poorly covered corners of the bacterial tree of life (8). Therefore, it is of utmost importance to ensure that this annotation is as accurate as possible. Transcription terminators, along with promoters, Shine–Dalgarno sequences and the start and stop codons form important landmarks in the bacterial genomic landscape. By evaluating each landmark in the context of neighbouring landmarks, we can start to improve the depth of genome annotations and support predictions of biological significance (9). Furthermore, some researchers have successfully inferred and subsequently verified the existence of non-coding RNA (ncRNA) genes using promoter and terminator annotation tools (10,11). Historically, these ncRNAs have been difficult to infer. This approach has now been automated using the sRNApredict and sRNAscanner bioinformatic tools (12,13).
The task of predicting RITs has been tackled previously but no existing method results in highly reliable predictions. Typically, these approaches use a free energy-based approach to infer a secondary structure and either an ad hoc score for the characteristic poly-U tail or a further energy-based approach computing the affinity for the 3′ RIT tail with the template DNA strand (14–16). Some of the methods also give bonuses to, or are biased towards, predictions that occur in the typical genetic context e.g. immediately 3′ to an annotated protein coding sequence (CDS) (16,18).
In this work, we have implemented a covariance model (CM) based approach for annotating RITs (17,20). We show that the CM-based approach is more accurate than existing methods, despite the fact that we are not using additional information such as proximity to the 3′-end of an annotated gene. After noticing a paucity of predicted RITs in the Mycobacterium tuberculosis genome, we investigated the existence of significantly structured regions in the proximity of 3′-ends of annotated genes relative to shuffled controls. We were surprised to discover the presence of a previously unknown abundant hairpin motif with a well-conserved sequence.
MATERIAL AND METHODS
The approach for RIT prediction that we have developed makes use of CMs (19,20). CMs are powerful statistical models for identifying homologues to a family of related RNAs. This is done by comparison to a ‘seed’ alignment of representative RNAs that have been annotated with a consensus secondary structure. In recent years, CM methods have become increasingly practical due to dramatic improvements in the memory requirements (21), computation time (22–24) and accuracy (25). For the following work, we have used the Infernal package, version 1.0.2 (26).
This approach avoids the awkward issue of combining unrelated measures of free energy and sequence composition. Instead, the primary sequence and predicted secondary structure of target sequences are scored within a unified statistical framework. The probability that each subsequence of a target database was generated by the CM is computed and compared with the probability that is generated by a background model of random sequence, resulting in a log-odds score called a ‘bit score’. In general, positive scores indicate a given sequence that looks more like the seed sequences than a random sequence; conversely, negative scores look more like random sequences than seed sequences. More specifically, a bit score of x bits indicates the sequence is 2x times more likely to have been generated from the CM than from the random background model.
Building a RIT alignment
We obtained 171 and 891 experimentally validated terminators from the Gram-negative Proteobacteria E. coli and the Gram-positive Firmicute B. subtilis, respectively (5–7). These were made available by the ECDC (E. coli database collection) and a well annotated set of B. subtilis RITS from the supplementary materials of de Hoon et al. (7). The evidence for the RITs was carefully checked and 981 sequences with experimental evidence for RIT activity were used for further work.
We ran iterative rounds of alignment, structure prediction, refinement and homology search on this dataset to produce a large alignment of RITs. The alignments and structures were inferred and refined using a combination of the computational methods WAR (28), CMfinder (29), MLocarna (30) and Infernal (26) followed by manual refinement using the RALEE alignment editor (31). Searches of the EMBL database were performed using the Rfam annotation pipeline (32). Carefully selected predicted terminators, variant from existing seed sequences, were incorporated into the seed. These selected sequences were required to fulfil the following criteria: (i) the maximum similarity to an existing seed sequence had to be 95% and the minimum 60%, (ii) the minimum fraction of canonical basepairs had to be 75%, (iii) the sequence annotation should not contain terms like contaminant, pseudogene, repeat or transposon and (iv) they must score above a bit score threshold of 20. These criteria have been found in other work to produce candidate sequences with useful levels of variation for extending RNA families (32). Finally, the selected sequences were manually checked for the typical position 3′ to a gene annotation. This resulted in a total of 1117 aligned sequences (the Rfam pipeline produces a multiple alignment). We then split the sequences into two groups based on bit score by building a CM from the alignment and using it to rescore all 1117 sequences. If a sequence scored 14 bits or higher it was placed into group A, else it was placed into group B. Each group's alignment was then iteratively refined by re-aligning the group's sequences to a CM built from the current alignment until no major changes in bit score were observed (using the –refine option in Infernal's cmbuild program). The resulting two alignments were then manually refined and used to build the final two RNIE CMs that were used for all subsequent annotations and benchmarks.
Two major RNIE modes
We have built two major modes into the RNIE program. The default mode, dubbed ‘genome mode’, is optimized for the task of high-throughput genome annotation. This mode employs parameters that ensure a rapid search (∼43 kb/s) with a very low false positive rate (∼1.7 FP/M). The sensitivity, positive predictive value and Matthews' correlation coefficient for this mode is 0.70, 0.79 and 0.74, respecively. The second major mode, dubbed ‘gene mode’, is optimized for the task of individually annotating the downstream regions of genes. Typically, these are smaller datasets and a higher sensitivity (0.83) is desirable, while a slower search is tolerable (∼1 kb/s). The false positive rate, positive predictive value and Matthews' correlation coefficient for this mode is ∼9.6 FP/MB, 0.45 and 0.61, respecively.
The genome and gene modes are launched with the following respective cmsearch parameters:
cmsearch -T 16 -g --fil-no-qdb
--fil-T-hmm 2 --cyk --beta 0.05
CM query_sequence.fasta
cmsearch -T 14 -g --fil-no-qdb
--fil-no-hmm --no-qdb --inside
CM query_sequence.fasta
Benchmarks
In order to evaluate the performance of our method relative to comparable tools, we have conducted two independent benchmarks.
First, we discuss some caveats to this benchmark. In any bioinformatic setting, the ideal is to seperate one's training and test data in order to avoid problems due to over-training. However, in this situation we had relatively few examples for training or testing purposes; these were from just two organisms. Furthermore, it was impossible to remove the training data from the alternative methods that we test here TransTermHP (16), RNAmotif (14) and Rnall (33). Therefore, we have had to include a biased test (the alpha benchmark) using the training data for testing. The results of this can be considered an upper bound on the likely true performance of these algorithms. To alleviate the worst of these concerns, we took the two best algorithms from the alpha benchmark and added a ‘beta benchmark’ which is independent of the training data. This test considered the correlation of whole genome annotations with gene ends on both native and shuffled genome sequences. These genome sequences were selected from a broad range of bacteria spanning all the main bacterial phyla and specifically avoided either E. coli or B. subtilis.
Alpha benchmark
The first benchmark used 485 previously established E. coli and B. subtilis terminators. The 144 E. coli RITs were derived from the ECDC (6). These RITs have little associated annotation. Consequently, the provenance of the data is difficult to establish and therefore some of these RITs might not be biological. This contrasts with the 341 B. subtilis RITs that are derived from a screen by de Hoon et al. (7). This data set has an excellent annotation of the evidence for each RIT. We manually selected those with good experimental evidence for function.
These RITs were embedded in 1000 bases of randomly selected and then permuted bacterial genomic sequence (see Table 1 for the sources of genomic sequences). The permuted genomic sequences were shuffled using a dinucleotide frequency preserving procedure that preserves some of the strongest statistical signals in the genome such as CpG content and the stacking signals that are important to control for when investigating RNA secondary structure (34). An additional set of 100 decoy sequences were generated for each known terminator. These were also embedded in 1000 bases of randomly selected permuted genomic sequence. The decoy sequences were generated using a first-order Markov process with nucleotide transition rates estimated from the known terminators. This method was used rather than shuffling since short terminator sequences may have a limited number of permuted conformations with an identical dinucleotide content. TransTermHP (16), the Lesnik et al. RNAmotif descriptor (14) and Rnall (33) and RNIE were used to predict RITs in these datasets. Since the TransTermHP algorithm requires annotated genic features, we artificially generated sets of 2, 4, 9 and 10 features for each sequence. In each set, one of the features had a 3′-end corresponding to the start of a known terminator or a decoy terminator sequence. Each terminator prediction for each algorithm was classified as either a true positive or a false positive depending upon whether the prediction overlapped a known terminator sequence by 1 nucleotide or more. The score (or scores) for each prediction were also stored for each terminator prediction and the receiver operator characteristic (ROC) and sensitivity versus positive predictive value (PPV) plots were generated for each tool and score combination (Figure 2).
Table 1.
Control genomes
| Species | EMBL accession | Phylum | Genome size | Number CDSs | G + C content | Number predictions |
|||
|---|---|---|---|---|---|---|---|---|---|
| (Mb) | Genome |
Gene |
|||||||
| Native | Shuffled | Native | Shuffled | ||||||
| Mycobacterium tuberculosis | AE000516 | Actinobacteria | 4.40 | 4189 | 0.66 | 19 | 0 | 111 | 3 |
| Streptomyces griseus | AP009493 | Actinobacteria | 8.55 | 7138 | 0.72 | 72 | 0 | 353 | 2 |
| Bacteroides thetaiotaomicron | AE015928 | Bacteroidetes | 6.26 | 4778 | 0.43 | 783 | 2 | 1470 | 44 |
| Chlamydophila pneumoniae | AE001363 | Chlamydiae | 1.23 | 1052 | 0.41 | 61 | 3 | 135 | 19 |
| Prochlorococcus marinus | AE017126 | Cyanobacteria | 1.75 | 1882 | 0.36 | 81 | 5 | 131 | 22 |
| Deinococcus radiodurans | AE000513 | Deinococcus-Thermus | 2.65 | 2579 | 0.67 | 283 | 0 | 506 | 2 |
| Bacillus subtilis | AL009126 | Firmicutes | 4.22 | 4245 | 0.44 | 1851 | 4 | 2540 | 54 |
| Clostridium difficile | AM180355 | Firmicutes | 4.29 | 3777 | 0.29 | 431 | 8 | 1152 | 58 |
| Fusobacterium nucleatum | AE009951 | Fusobacteria | 2.17 | 2067 | 0.27 | 155 | 1 | 457 | 34 |
| Thermodesulfovibrio yellowstonii | CP001147 | Nitrospirae | 2.00 | 2033 | 0.34 | 78 | 6 | 176 | 41 |
| Escherichia coli | U00096 | Proteobacteria | 4.64 | 4321 | 0.51 | 601 | 6 | 1058 | 35 |
| Helicobacter pylori | AE000511 | Proteobacteria | 1.67 | 1566 | 0.39 | 28 | 12 | 128 | 61 |
| Salmonella enterica | AE014613 | Proteobacteria | 4.79 | 4323 | 0.52 | 537 | 4 | 980 | 32 |
| Leptospira interrogans | AE016823 | Spirochaetes | 4.28 | 3394 | 0.35 | 164 | 18 | 375 | 132 |
| Ureaplasma parvum | AF222894 | Tenericutes | 0.75 | 611 | 0.26 | 54 | 0 | 163 | 5 |
| Fervidobacterium nodosum | CP000771 | Thermotogae | 1.95 | 1750 | 0.35 | 409 | 3 | 588 | 28 |
| Methylacidiphilum infernorum | CP000975 | Verrucomicrobia | 2.29 | 2472 | 0.45 | 50 | 7 | 157 | 52 |
Figure 2.

Alpha benchmark. The accuracy of RNIE compared to existing methods of terminator prediction. The left figure shows a ROC plot for four independent methods. The middle figure compares the sensitivity and PPV for the four methods. The figure on the right shows the speeds for each algorithm in kilobases per second.
The sensitivity, PPV and false-positive rate (FPR) metrics that were used to generate Figure 2 are defined as:
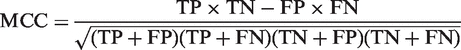 |
For each method, the true-positive (TP) values were computed by counting all the predicted terminators that overlapped a known terminator by at least one nucleotide. All other predicted terminators on the positive strand were counted as false positives (FP). The known terminators that were not detected contributed to the false negative (FN) count. In this context, the true negative (TN) values can be computed in many different ways: one could conservatively count the regions that remain unclassified and one could liberally count every possible substring that remains unclassified. We chose the middle ground by using the number of nucleotides that remained unclassified.
Beta benchmark
The second benchmark relies upon the correlation of predicted terminators with annotated genic elements on a range of native and shuffled genome sequences. For this test, we were able to exclude all the training data from the test set by taking a selection of 14 representative bacterial genomes that are widely distributed throughout the better characterized portions of bacterial phylogeny (Table 1). The annotations for each genome were extracted from the EMBL nucleotide database (35) and supplemented with ncRNA annotations from Rfam 10.0 (32). We took all the unique genic features for each test genome and computed the minimum distance between these and each terminator prediction on the same strand for each method. The pooled results are shown in Figure 3.
Figure 3.

Beta benchmark. Ideal terminator predictors will generally produce predictions that are immediately 3′ to annotated genes on native sequence and no predictions on shuffled controls. For all the test genomes in Table 1 (excluding E. coli and B. subtilis), we computed the distance to the nearest 3′ genic element, including CDSs, ncRNAs and riboswitches. This was done for both native sequences and dinucleotide shuffled control sequences with corresponding gene annotation transferred to the controls. The figure on the left shows the distribution of distances for RNIE genome and gene modes and for the TransTermHP method. Inset is a barplot showing the total number of predictions for each method on native and shuffled genomes. The figures on the right show the percentage of genes that have a predicted RIT in the region −50 to +150 from an annotated 3′-end of a CDS or ncRNA across all the genome sequences described in Table 1. The upper panel illustrates the results for the native genomes, while the lower panel illustrates results for the permuted genomes.
RESULTS
The results of the alpha and beta benchmarks are presented in Figures 2 and 3. In the following sections, we discuss these results in more detail.
Alpha benchmark
The alpha benchmark illustrates some interesting features of the terminator predictors (Figure 2). Many of the energy-based methods employ scoring schemes based on the free energy of the RIT hairpin and the free energy of the RIT tail disassociation with the template DNA strand (Figure 1). These two score types show characteristic trajectories through the ROC and sensitivity versus PPV plots. We noted in particular that the disassociation energies reported by RNAMotif (dG), RnaII (hbG) and RnaII-Brkr (hbG) show very little potential for discriminating between true and false RITs based on their atypical trajectories through these plots. However, the RIT hairpin energies from RNAMotif (struct), RnaII (dG) and RnaII-Brkr (dG) show some discriminatory potential; in particular, the RNAMotif descriptor by Lesnik et al. (14) performed well. In fact, this was the only method of this class to reach over the y = 1-x threshold on the sensitivity versus PPV plot (see the red dotted line in Figure 2). This line is an indicator whether a method is doing better or worse than a ‘random’ predictor.
For the TransTermHP method, we were forced to provide fictional gene annotations in order to get the method to run. In order to assess dependence on the number of features, we ran four tests using 2, 4, 9 and 10 regularly spaced genes. In each case, one annotation was terminated by either a native or decoy RIT. There was little consistent influence on the performance of TransTermHP based on the number false gene annotations. The maximum Matthews' correlation coefficient was acheived by the run with 10 annotations [max(MCC)=0.50], the minimum was with 4 annotations [max(MCC)=0.44].
The RNIE method we are presenting in this work performed very well compared to the alternative tools on this particular benchmark. The highest maximum Matthews' correlation coefficient was attained by RNIE run in genome mode [max(MCC) = 0.75], followed by the run in gene mode [max(MCC) = 0.74]. We used these results to identify thresholds for each mode that optimally balanced the number of true and false positives. A too high threshold will mean we miss a lot of real terminators: a too low threshold will mean we are swamped in noisy predictions. For both genome and gene modes, we selected thresholds (16.00 and 14.00 bits, respectively) slightly lower than those suggested by the optimal MCC-based threshold (16.45 and 19.09 bits, respectively). The lower thresholds accepted a few more FP but generally researchers are more forgiving of these than FN in genomics work. Furthermore, most FP can be discounted by their genomic context.
The speed of the RNIE algorithm, in genome mode, is comparable to the alternative methods (Figure 2). While CMs have long been known to be computationally intensive, for this work we use an optimized CM approach that employs several methods to increase the computational efficiency (24,26). In genome mode, RNIE can scan >43 kb/s, in gene mode it scans just 1 kb/s. This is comparable to TransTermHP which scans 74–186 kb/s; however, for this tool there is a linear relationship between the number of annotations and speed for this tool i.e. the more annotations the slower it scans. The RNAmotif descriptor scans 602 kb/s and is the fastest tool we encountered. Finally, Rnall scans ∼1 kb/s; however, the Rnall speed had to be estimated by computing a CPU factor as the only version we had access to runs on an outdated computer architecture.
Beta benchmark
The beta benchmark illustrates that RNIE can accurately detect terminators across a broad range of bacterial genomes outside of E. coli and B. subtilis genomes and without requiring gene annotation information (Figure 3). Figure 3A shows that in genome annotation mode there is an excess of RIT predictions near the 3′-end of CDS and ncRNA annotations. The dotted lines show that RNIE, in genome annotation mode, makes a negligible number of predictions in the permuted genomes, verifying that the FPR for this approach is very low. The results for RNIE in gene annotation mode show similar results, with an expected higher number of predictions in the correct context to gene annotation but with a correspondingly higher FPR. The results for TransTermHP show the worrying result that RIT predictions are enriched in the 3′-ends of genes for both the native and permuted genome sequences. This suggests that a significant fraction of predictions by TransTermHP are false even though they appear in a genomic context associated with genic termini.
The Figure 3B plot shows the fraction of genes with RIT predictions associated with genic termini for all the species in Table 1 for each of the three prediction approaches. Again, these generally illustrate the high sensitivity of RNIE and low FPR relative to TransTermHP. This plot also illustrates the diverse degree of RIT usage across bacterial species which does not follow traditional lines of bacterial classification. For example, the Gram-positive species from the phyla Firmicutes and Actinobacteria have a mixture of exemplars from both ends of the usage spectrum. That is, B. subtilis makes substantial use of RITs, whereas neither of the Actinobacteria M. tuberculosis and Streptomyces griseus make substantial use of RITs. Even within phyla, there can be a lot of variation of RIT usage. For example, the Proteobacteria E. coli and S. enterica clearly employ high levels of RITs for transcriptional termination, whereas Helicobacter pylori does not.
Case study: M. tuberculosis termination
In M. tuberculosis, the number of predicted RITs was very low. However, other researchers have suggested that M. tuberculosis do employ a rho-independent terminator mechanism (2,18,36–38). Therefore, we chose to analyse genic termini in more detail within this species. There is published evidence that M. tuberculosis genes are enriched for stable secondary structures near the coding terminus (2). However, further analysis has shown that these do not fit the canonical terminator model. A method has been developed that attempts to classify predicted secondary structures from coding termini sequence into any of five different classes (18,36–38), with bonuses given for ‘correct’ genomic context. The predominant form of these are ‘i-shaped’ structures (>90%) or a short stem loop, that have <3 U's in the 10 nt stretch following the stem loop.
To illustrate the flexibility of our approach, we took all the annotated coding sequences from the M. tuberculosis CDC1551 complete genome and extracted subsequences from −20 to +80 nt around all annotated gene termini. These were each folded using the RNAfold routine from the Vienna package (39) and then subjected to a permutation test, where native minimum free energies (MFEs) were compared to the pooled distribution of MFEs for 1000 permuted sequences with the same dinucleotide content for each termini (34). The regions that had a P < 0.001 where subsequently fed into the alignment and folding algorithm CMfinder (29). This alignment was manually refined using the RALEE alignment editor (31). A covariance model was built for the resulting alignment (26) and then RNIE was deployed for annotating the entire M. tuberculosis CDC1551 genome with the more specific covariance model.
We were surprised to discover a previously unpublished well-conserved motif (Figure 4) that we have called the ‘Tuberculosis Rho-independent terminator’ or TRIT. The TRITs account for 72% (59/82) of the significantly stable terminator sequences (P < 0.001), the standard models add a further 7%. This ratio increases to 81% (29/36) when we use a more stringent threshold of P < 0.0001 plus a further 8% from the standard models (Figure 4B). Consequently, the RIT and TRIT models account for 80−90% of all the highly structured regions near gene termini in M. tuberculosis. There are 147 copies of TRIT scattered throughout the M. tuberculosis genome (EMBL accession: AE000516). Given the palindromicity of these sequences, the bulk are bidirectional. The TRITs are closely associated with the terminal regions of annotated genic features (Figure 4A).
Figure 4.

(A) The frequency of TRITs and RITs near the terminal regions of M. tuberculosis (EMBL accession: AE000516) genic features. (B) The distribution of structural stability derived P-values for the most significant M. tuberculosis terminal regions coloured by TRIT (red), RIT (black) or unclassified (blue). (C) The secondary structure and sequence conservation of the TRIT motif as displayed by R2R (27). (D&E) Sequence logos generated for the 5′ (D) and 3′ (E) halves of an alignment of the 147 copies of TRIT in the M. tuberculosis genome.
A unique TRIT feature that we observed is that the distribution of TRIT sequences relative to the nearest annotated stop codon is very narrow. These are largely positioned within the coding sequence, around the −8 position, whereas the RIT sequences are much more broadly distributed and are predominately located further downstream between +10 and +20. Scans of the public sequence databases for other TRITs show that this terminator type is restricted to Mycobacterium. The TRIT utilizing species that we identified include M. abscessus, M. avium, M. bovis, M. gilvum, M. intracellulare, M. kansasii, M. marinum, M. smegmatis, M. ulcerans and M. vanbaalenii.
DISCUSSION
There are two major modes researchers want from RIT prediction software. The first, that has been targeted by existing methods, is to investigate the possibility of a RIT for a specific gene or cis-regulatory element. For this a high-sensitivity approach is desirable with an acknowledged cost to specificity, where the context of the prediction should add some specificity. The second major mode is to screen entire bacterial genomes for RITs in the hope of identifying short ORFs, sRNAs and cis-regulatory elements such as riboswitches (10,40). These can also be used to validate, provide strand information and otherwise improve the annotation of transcripts from transcriptome data such as RNA-seq (41). The benchmarks have shown that the ‘gene’ and ‘genome’ modes implemented in RNIE both provide complementary features that are both accurate and computationally efficient.
Our further investigation of gene termini in M. tuberculosis identified a terminator motif with both strong sequence and structure conservation. This TRIT motif, together with our RIT predictions, account for 80−90% of all the most highly structured regions near gene termini in M. tuberculosis. The high sequence conservation of the TRITs implies that further cellular machinery may be involved in termination in this organism; possibly a factor that binds the double stranded RNA sequence motif.
We tried the same approach with two other species with a paucity of RIT predictions; these were H. pylori and Fervidobacterium nodosum, but could not identify any obvious terminator motif. Fervidobacterium nodosum did show some enrichment of structured elements; however, the bulk of these fit the traditional RIT motif with a lower G + C content than the other well-characterized examples, where a lower threshold for this species identified the missing RITs.
In conclusion, this work has shown that covariance models can be deployed to predict Rho-independent terminators with an accuracy that has not been available previously. The method we propose is slightly slower than some competing approaches; however, the boost in prediction accuracy is worth the sacrifice.
FUNDING
Wellcome Trust [grant number WT077044/Z/05/Z] (to P.P.G., L.B. and A.B.); Howard Hughes Medical Institute support to Sean R. Eddy (to E.P.N.) and to Ronald R. Breaker (to Z.W.). Funding for open access charge: The Wellcome Trust.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors are grateful to Dave Ecker from Ibis Biosciences for providing the Lesnik et al. (14) RNAmotif descriptor that after a decade still performs well.
REFERENCES
- 1.Wagner R. Transcription Regulation in Prokaryotes. Oxford, UK: Oxford University Press; 2000. [Google Scholar]
- 2.Washio T, Sasayama J, Tomita M. Analysis of complete genomes suggests that many prokaryotes do not rely on hairpin formation in transcription termination. Nucleic Acids Res. 1998;26:5456–5463. doi: 10.1093/nar/26.23.5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langridge GC, Phan MD, Turner DJ, Perkins TT, Parts L, Haase J, Charles I, Maskell DJ, Peters SE, Dougan G, et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 2009;19:2308–2316. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quirk PG, Dunkley EA, Lee P, Krulwich TA. Identification of a putative Bacillus subtilis rho gene. J. Bacteriol. 1993;175:647–654. doi: 10.1128/jb.175.3.647-654.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.d'Aubenton Carafa Y, Brody E, Thermes C. Prediction of rho-independent Escherichia coli transcription terminators. A statistical analysis of their RNA stem-loop structures. J. Mol. Biol. 1990;216:835–858. doi: 10.1016/s0022-2836(99)80005-9. [DOI] [PubMed] [Google Scholar]
- 6.Kröger M, Wahl R. Compilation of DNA sequences of Escherichia coli K12: description of the interactive databases ECD and ECDC. Nucleic Acids Res. 1998;26:46–49. doi: 10.1093/nar/26.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Hoon MJ, Makita Y, Nakai K, Miyano S. Prediction of transcriptional terminators in Bacillus subtilis and related species. PLoS Comput. Biol. 2005;1:e25. doi: 10.1371/journal.pcbi.0010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462:1056–1060. doi: 10.1038/nature08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naville M, Gautheret D. Transcription attenuation in bacteria: theme and variations. Brief. Funct. Genomic Proteomic. 2010;9:178–189. doi: 10.1093/bfgp/elq008. [DOI] [PubMed] [Google Scholar]
- 10.Livny J, Brencic A, Lory S, Waldor MK. Identification of 17 Pseudomonas aeruginosa sRNAs and prediction of sRNA-encoding genes in 10 diverse pathogens using the bioinformatic tool sRNAPredict2. Nucleic Acids Res. 2006;34:3484–3493. doi: 10.1093/nar/gkl453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fineran PC, Blower TR, Foulds IJ, Humphreys DP, Lilley KS, Salmond GP. The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc. Natl Acad. Sci. USA. 2009;106:894–899. doi: 10.1073/pnas.0808832106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pánek J, Bobek J, Mikulík K, Basler M, Vohradský J. Biocomputational prediction of small non-coding RNAs in Streptomyces. BMC Genomics. 2008;9:217. doi: 10.1186/1471-2164-9-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sridhar J, Narmada SR, Sabarinathan R, Ou HY, Deng Z, Sekar K, Rafi ZA, Rajakumar K. sRNAscanner: a computational tool for intergenic small RNA detection in bacterial genomes. PLoS ONE. 2010;5:e11970. doi: 10.1371/journal.pone.0011970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lesnik EA, Sampath R, Levene HB, Henderson TJ, McNeil JA, Ecker DJ. Prediction of rho-independent transcriptional terminators in Escherichia coli. Nucleic Acids Res. 2001;29:3583–3594. doi: 10.1093/nar/29.17.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wan X-F, Xu D. Intrinsic terminator prediction and its application in Synechococcus sp. WH8102. J. Comput. Sci. Technol. 2005;20:465–482. [Google Scholar]
- 16.Kingsford CL, Ayanbule K, Salzberg SL. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007;8:R22. doi: 10.1186/gb-2007-8-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994;11:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Unniraman S, Prakash R, Nagaraja V. Conserved economics of transcription termination in eubacteria. Nucleic Acids Res. 2002;30:675–684. doi: 10.1093/nar/30.3.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakakibara Y, Brown M, Hughey R, Mian IS, Sjölander K, Underwood RC, Haussler D. Stochastic context-free grammars for tRNA modeling. Nucleic Acids Res. 1994;22:5112–5120. doi: 10.1093/nar/22.23.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eddy SR. A memory-efficient dynamic programming algorithm for optimal alignment of a sequence to an RNA secondary structure. BMC Bioinformatics. 2002;3:18. doi: 10.1186/1471-2105-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinberg Z, Ruzzo WL. Exploiting conserved structure for faster annotation of non-coding RNAs without loss of accuracy. Bioinformatics. 2004;20(Suppl. 1):i334–i341. doi: 10.1093/bioinformatics/bth925. [DOI] [PubMed] [Google Scholar]
- 23.Weinberg Z, Ruzzo WL. Sequence-based heuristics for faster annotation of non-coding RNA families. Bioinformatics. 2006;22:35–39. doi: 10.1093/bioinformatics/bti743. [DOI] [PubMed] [Google Scholar]
- 24.Nawrocki EP, Eddy SR. Query-dependent banding (QDB) for faster RNA similarity searches. PLoS Comput. Biol. 2007;3:e56. doi: 10.1371/journal.pcbi.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolbe DL, Eddy SR. Local RNA structure alignment with incomplete sequence. Bioinformatics. 2009;25:1236–1243. doi: 10.1093/bioinformatics/btp154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weinberg Z, Breaker RR. R2R - software to speed the depiction of aesthetic consensus RNA secondary structures. BMC Bioinformatics. 2011;12:3. doi: 10.1186/1471-2105-12-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torarinsson E, Lindgreen S. WAR: webserver for aligning structural RNAs. Nucleic Acids Res. 2008;36:W79–W84. doi: 10.1093/nar/gkn275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao Z, Weinberg Z, Ruzzo WL. CMfinder–a covariance model based RNA motif finding algorithm. Bioinformatics. 2006;22:445–452. doi: 10.1093/bioinformatics/btk008. [DOI] [PubMed] [Google Scholar]
- 30.Otto W, Will S, Backofen R. In Proceedings of German Conference on Bioinformatics (GCB'2008) Vol. P-136. Bonn, Germany: Lecture Notes in Informatics (LNI), Gesellschaft für Informatik (GI); 2008. pp. 178–188. [Google Scholar]
- 31.Griffiths-Jones S. RALEE–RNA ALignment editor in Emacs. Bioinformatics. 2005;21:257–259. doi: 10.1093/bioinformatics/bth489. [DOI] [PubMed] [Google Scholar]
- 32.Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, et al. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37:D136–D140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wan XF, Lin G, Xu D. Rnall: an efficient algorithm for predicting RNA local secondary structural landscape in genomes. J. Bioinform. Comput. Biol. 2006;4:1015–1031. doi: 10.1142/s0219720006002363. [DOI] [PubMed] [Google Scholar]
- 34.Workman C, Krogh A. No evidence that mRNAs have lower folding free energies than random sequences with the same dinucleotide distribution. Nucleic Acids Res. 1999;27:4816–4822. doi: 10.1093/nar/27.24.4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leinonen R, Akhtar R, Birney E, Bonfield J, Bower L, Corbett M, Cheng Y, Demiralp F, Faruque N, Goodgame N, et al. Improvements to services at the European Nucleotide Archive. Nucleic Acids Res. 2010;38:D39–D45. doi: 10.1093/nar/gkp998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Unniraman S, Prakash R, Nagaraja V. Alternate paradigm for intrinsic transcription termination in eubacteria. J. Biol. Chem. 2001;276:41850–41855. doi: 10.1074/jbc.M106252200. [DOI] [PubMed] [Google Scholar]
- 37.Mitra A, Angamuthu K, Nagaraja V. Genome-wide analysis of the intrinsic terminators of transcription across the genus Mycobacterium. Tuberculosis (Edinb) 2008;88:566–575. doi: 10.1016/j.tube.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 38.Mitra A, Angamuthu K, Jayashree HV, Nagaraja V. Occurrence, divergence and evolution of intrinsic terminators across eubacteria. Genomics. 2009;94:110–116. doi: 10.1016/j.ygeno.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:W70–W74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abreu-Goodger C, Ontiveros-Palacios N, Ciria R, Merino E. Conserved regulatory motifs in bacteria: riboswitches and beyond. Trends Genet. 2004;20:475–479. doi: 10.1016/j.tig.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Perkins TT, Kingsley RA, Fookes MC, Gardner PP, James KD, Yu L, Assefa SA, He M, Croucher NJ, Pickard DJ, et al. A strand-specific RNA-Seq analysis of the transcriptome of the typhoid bacillus Salmonella typhi. PLoS Genet. 2009;5:e1000569. doi: 10.1371/journal.pgen.1000569. [DOI] [PMC free article] [PubMed] [Google Scholar]