


Abstract
Effective translation of the viral genome during the infection cycle most likely enhances its fitness. In this study, we reveal two different strategies employed by cyanophages, viruses infecting cyanobacteria, to enhance their translation efficiency. Cyanophages of the T7-like Podoviridae family adjust their GC content and codon usage to those of their hosts. In contrast, cyanophages of the T4-like Myoviridae family maintain genomes with low GC content, thus sometimes differing from that of their hosts. By introducing their own specific set of tRNAs, they appear to modulate the tRNA pools of hosts with tRNAs that fit the viral low GC preferred codons. We assessed the possible effects of those viral tRNAs on cyanophages and cyanobacterial genomes using the tRNA adaptation index, which measures the extent to which a given pool of tRNAs translates efficiently particular genes. We found a strong selective pressure to gain and maintain tRNAs that will boost translation of myoviral genes when infecting a high GC host, contrasted by a negligible effect on the host genes. Thus, myoviral tRNAs may represent an adaptive strategy to enhance fitness when infecting high GC hosts, thereby potentially broadening the spectrum of hosts while alleviating the need to adjust global parameters such as GC content for each specific host.
INTRODUCTION
It is now well established that viruses infecting cyanobacteria, cyanophages, of the T4-like Myoviridae family (myoviruses) as well as of the T7-like Podoviridae family (podoviruses), may add genes from their hosts to their basic and essential core genomes (1–9). Isolates of both families were found within cyanobacteria of either the Synechococcus or Prochlorococcus genera (23), the latter of which is further divided into high-light (HL) and low-light (LL) adapted clades (10). These hosts differ significantly: Synechococcus genomes are not only larger than those of HL Prochlorococcus, but are also of higher GC content (Figure 1A), whereas LL Prochlorococcus have intermediate GC content (Supplementary Table S2) (11–14). The GC content imposes constraints on the codon usage and thus may indirectly affect the translation process. As the translation process is time consuming and represents a key step in the viral infection cycle, we wondered about the strategies employed by the cyanophages to harness the translation machinery of potential hosts. Failure to translate efficiently a particular coding sequence, or the whole genome, may incur a large cost for the organism due to reduction in gene expression (15–18), an increase in translation errors (19,20), an increased fraction of misfolded proteins (21) or a combination of all three.
Figure 1.

Genome size, GC content and codon usage, of cyanobacteria and cyanophages. Colors distinguish between bacteria (transparent), myoviruses (red) and podoviruses (blue). Symbols distinguish between Synechococcus and Synechococcus-infecting phages (circles), and Procholorococcus and Prochlorococcus-infecting phages (triangles). (A) The tRNA Gene Copy Number of various cyanobacteria and cyanophages is plotted against their GC Content. The tendency of Synechococcus to have high GC content, of myoviruses to have low GC content, and of Synechococcus-infecting myoviruses to favor the inclusion of tRNAs within their genomes, is evident. (B) Average genome sizes of podoviruses, myoviruses, Prochlorococcus and Synechococcus is plotted along the y-axis. Myoviruses have an average size that is about four times that of podoviruses. Error bars depict 1 SD. (C) Stripcharts of the correlation coefficients of the codon frequencies. Symbols are as in panel A; in addition, stars (*) indicate comparisons between all viruses of the indicated group. The range of values is depicted along the x-axis. Top strips: correlation within each group; bottom strips: correlation between phages and hosts. Values between 0.3 and 0.5, in LL-Prochlorococcus stem from bacteria LL Prochlorococcus MIT9303 and MIT9313 which are relatively GC-rich (see Supplementary Table S2). These two species also lead to the variations seen within LL-Prochlorococcus infecting podoviruses and myoviruses versus LL-Prochlorococcus. Podoviruses present two different groups corresponding to the high-GC and low-GC isolates in contrast with the strong correlation within myoviruses. Note: the variation in the vertical position within each group is arbitrary; its purpose is to minimize the overlay of data points.
With the emerging genomes of cyanophages fully sequenced to date, it seems that marine myoviruses have in general larger genomes (14,22) that contain a higher number of open reading frames (ORFs), compared to the genomes of the marine podoviruses (Figure 1B and Supplementary Table S1). It was also reported that whereas myoviruses may have broad host ranges, podoviruses are host specific (23). Furthermore, both myoviruses and podoviruses might bear within their genomes complete or partial tRNA genes (2,24–26). The discovery of tRNA genes within genomes of phages is not new and dates back to 1968, when they were first reported to be found in a genome of the T4 myovirus infecting Escherichia coli (27). Extensive subsequent work have argued for expression and functionality of the T4 tRNAs (28,29).
Since tRNAs have been reported to serve as integration sites of temperate phages into genomes of their hosts, it has been proposed that the tRNA genes found in genomes of phages, mainly partial sequences of tRNAs, may be the result of a concomitant excision with the phage genome (30–33). Nevertheless, many tRNA genes found in genomes of phages are full length, and moreover, a significant positive association between the exact cognate anticodon distribution of the phage and its codon usage was observed (34). It has therefore been suggested that some tRNA genes are selectively retained if they match codons highly used by the phage and poorly used by the bacterial host (34–37). Such traits may explain the previously proposed role of tRNAs to raise the virulence of phages bearing them compared to phages without tRNAs (34). A certain fitness was proposed to be acquired because, for example, deletion of tRNAs of the E. coli T4 phage induced lower burst sizes and rates of protein synthesis (38).
The importance of the tRNA pool in determining the optimality of the codon choice was demonstrated, for example, by Kanaya et al. (39). Using several unicellular organisms, they have shown that genomes tend to tune their codon usage such that it will fit the availability of tRNAs in the hosting cell. They also showed that the tRNA content in the cell correlates well with the tRNA gene copy number (tGCN). The extent at which the codon usage of a given coding sequence is adapted to the cellular tRNA abundance, can be estimated using the tRNA adaptation index (tAI) (40).
In the present research, we postulate that different cyanophages adapt different strategies to enhance the translation of their genomes when confronting different hosts. Podoviruses employ a ‘specialization’ strategy and adjust the GC content and the codon usage of their genomes to the GC content of their hosts, whereas myoviruses employ an ‘adaptation’ strategy and maintain a low GC content genome. Instead of adjusting their GC content to that of high GC potential hosts, myoviruses retain a selective set of tRNA genes that, once expressed, improve the adaptation to their own codon usage.
MATERIALS AND METHODS
Protein, coding sequences, tRNA genes and alignments
For all viral and host species in the analysis, coding sequences were downloaded from GenBank (http://www.ncbi.nlm.nih.gov/Genbank) or the CAMERA database (http://camera.calit2.net/). Full lists of the 12 cyanobacteria (Tables 2 and 3) and 20 cyanophages (Tables 1 and 3) analyzed are detailed in the Supplementary Data. The tGCNs were obtained by applying the tRNAscan-SE software version 1.23 (41).
The tAI for coding sequences
For a thorough discussion in the tAI, and its underlying assumptions—in particular, that the tGCN is a good proxy for the tRNA cellular abundance—we refer the readers to dos Reis et al. (40). Nevertheless, to make the article self-contained we included a description of the tAI in the Supplementary Data.
We used a modified version of the definition of the tAI (40) in which each codon weight was normalized to the genome-wide tAI:
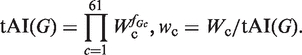 |
For each bacterium-phage pair, we computed two sets of tAI weights: one in which the tGCN equals that of the bacterium genome alone, and a second set in which the copy number is the sum of the bacterium and phage copy numbers. Using this we computed for each bacterial or viral gene its tAI value while accounting for, or ignoring, the contribution of the viral tRNAs, tAI+(g) and tAI(g), respectively.
The ΔΔtAI
Given the two tAI values for each gene, g, we defined the tAI difference of a gene
as the difference in translation efficiency of the gene due to the addition of the viral tRNAs to the tRNA pool. From this definition follows the definition of the separation between the bacterial and phage proteome response to the inclusion of the viral tRNA pool: (gv and gh denote viral and host genes, respectively):
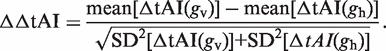 |
Statistical analysis
All the analyses and statistical tests were performed using Matlab, and its statistical tools.
The difference in the impact of Prochlorococcus and Synechococcus tRNA pools on the tAI of the viral genes
The tAI of the same set of viral genes was computed repeatedly using the various hosts’ tRNAs pools excluding the viral tRNAs pools. The Kruskal–Wallis test (42)—a non-parametric analog of the one-way analysis of variance (ANOVA)—was used to test for differences between medians of the tAIs within different hosts. The results were further corrected for multiple hypotheses using Bonferroni correction (43) for 5% significance level.
Clustering of the viral genes tAI difference across species
A matrix containing the tAI ratio of each viral gene (columns) across all the bacteria (rows) was generated. Two-way hierarchical clustering (44) was applied to produce heatmaps. In both clustering along the rows, and along the columns, Euclidean distance was used as the distance measure, and averaged linkage as the linkage measure.
The random tRNA sets
A random combination of a given number of tRNAs, K, was generated by randomly choosing K anticodons with repetitions from a uniform distribution. The three stop codons, and seven codons that are forbidden due to potential interfering wobble interaction, were omitted from the population. This process was repeated 10 000× and only distinct sets were retained. For each such random set of tRNAs, the adaptiveness values, Wc, were recalculated.
We chose to use the entire pool of 54 possible anticodons, as one cannot rule out the possibility of cyanophages carrying tRNA genes from an as yet unknown host. tRNAIle(TAT) (Figure 2A), for example, was found in several myoviruses but not in cyanobacteria analyzed to date. However, we also ran a similar analysis with tRNAs drawn out from the cyanobacterial pool alone.
Figure 2.

A detailed view of codon usage, tRNA gene copy number, and their relationship, in myoviruses and cyanobacteria. (A) Data from representatives of the three cyanobacteria groups, and from the three Synechococcus-infecting myoviruses S-PM2, S-RSM4 and Syn9, is presented. Data are organized according to anticodon identity. Going from the innermost ring, to the third ring, the letters denote the contents of the first anticodon position to the third one. The gray shades in each ring reflect the codon usage (see color bar) of one of the genomes (inner three for hosts and outer three from the viruses). The overlaid green circles reflect the presence of at least one cognate tRNA within the organism's genome (mostly only one cognate tRNA is present—detail of copy numbers can be seen in Table 3 of the Supplementary Data). The outer circles reflect the count of the corresponding tRNAs in additional myoviruses (http://camera.calit2.net/), in green myoviruses infecting Synechococcus and in orange myoviruses infecting Prochlorococcus. Also depicted in red: the anticodons that correspond to the stop codons (‘stop signs’), and to tRNAs that are forbidden due to the wobble interaction (‘X’); see main text. Note the avoidance of A-headed anticodons (T-ended codons), and the preference of phages to T-headed anticodons. (B) Codon usage of myovirus Syn9 is plotted against the codon usage of a potential host, Synechococcus WH8102. The symbols, A, C, G and T, reflect the nucleotide that occupy the first anticodon position (wobble position) of the corresponding codon. Colors reflect the presence of the corresponding exact-matching tRNAs within the genomes of the organisms: (gray) found only in the cyanobacteria, (blue) found in cyanobacteria and Syn9; (red) not found in cyanobacteria or myovirus. The viral and bacterial preference towards AT and CG rich genomes, respectively, is very clear. Also well-evident are: the tendency of Syn9 to carry tRNAs which favor its preferred codons [TTT (Lys) and TTC (Glu), marked in the plot with T1 and T2, respectively, are two outstanding exceptions; see main text], and the tendency of the cyanobacteria to avoid tRNAs that will favor the viral-preferred codons.
Test for the significance of overlap between pathogenicity islands and elevated tAI
The genes of WH8102 were divided in two different ways: first according to whether their tAI improvement is larger than zero; second according to their location—inside or outside pathogenicity islands. A hypergeometric test was performed, using matlab's builtin hypergeometric distribution function, to estimate the significance of the intersection between the genes with improved tAI and genes that reside within pathogenicity islands.
RESULTS
Here we bring diverse evidence to support the hypothesis that viral tRNAs are advantageous inside hosts whose codon usage mismatches significantly the viral codon usage. We analyze the impact that the viral tRNAs may incur on the translation of viral and host genes. We further show that the particular viral tRNA set is among the best that the phage could introduce in terms of promoting the translation of endogenous genes. We begin the ‘Results’ section, though, with a summary of the main relevant genomic properties that distinguish podoviruses from myoviruses.
The presence of tRNAs in cyanophage genomes is associated with a discrepancy in codon usage between phage and host
The purpose of this section is to point out the global genomic properties that led us to propose that myoviruses and podoviruses employ very distinct strategies of co-evolution to their hosts. Podoviruses, with small genome sizes (Figure 1B), match their own GC content to the GC contents of their hosts: those that infect the low GC Prochlorococcus hosts exhibit low GC content, whereas those infecting Synechococcus show corresponding high GC content in their genome, like their hosts (Figure 1A). The similarity between podoviruses and their hosts extends beyond their GC content: they also maintain a highly correlated codon usage (Figure 1C). This may explain why podoviruses show a restricted range of hosts (23) in which they ‘specialize’. In contrast, myoviruses, with up to five times larger genomes (Figure 1B), have constitutive low GC contents (Figure 1A). The codon usage of myoviruses is strongly correlated with Prochlorococcus hosts, but not with Synechococcus hosts (Figure 1C). Interestingly, as seen in Figure 1A, myoviruses that infect Synechococcus tend to carry within their genomes more tRNA genes (between 4 and 23 genes). The presence of tRNA genes in myoviruses that infect Prochlorochoccus is limited (up to four genes, or none at all). Thus, the presence of tRNA genes in genomes of myoviruses appears to be associated with a significant difference in GC content and codon usage between them and their hosts.
We chose to focus on the first three myoviruses that infect Synechococcus for which a full genome was published: Syn9, S-RSM4 and S-PM2. The number of tRNA genes these viruses carry (6, 12 and 23, respectively) spans the full range of tGCN observed in myoviruses. A detailed view of the genome-wide codon usage in the above-mentioned myoviruses and their potential hosts, and of the identity of the available tRNA genes, is presented in Figure 2A. For sake of clarity the plot includes data for the myoviruses and only one representative from each bacterial group and is organized according to the anticodon pool (further details can be found in the Supplementary Table S3 organized by codons). The overall preference of HL-Prochlorococcus and myoviruses for AT-rich codons (codons with AT in their third position), and of Synechococcus and LL-Prochlorococcus for GC-rich codons (codons with GC in their third position) is evident. These observations are in accordance with previously published results (45–48). The reader is reminded that variation at the third codon position (first anticodon position) reflects the redundancy of the genetic code that is made possible through the wobble interaction between the respective positions in codons and anticodons (49).
Further insight into the adaptation of the tRNA pool to the respective genomic context is given by Figure 2A (and Supplementary Table S3) that exhibit the codon and corresponding anticodon pool. Variation in the presence of tRNA genes is seen for anticodons AAG\CAG\GAG (Leu), CGG (Pro), CAC (Val), CGC (Ala) and CCC (Gly) (Figure 2A). Variation in tGCN is observed for anticodons GAT (Ile), GGT (Thr), TGC (Ala), and CAT (Met) (Supplementary Table S3). Also, tRNA genes matching anticodons TGG (Pro), TGT (Thr) and GCC (Gly) were not detected in a single Synechococcus species (MIT9215). As was observed in other Bacteria and Archaea (50), the tRNA repertoire of cyanobacteria (and consequently of cyanophages) lack most adenosine-starting anticodons with the exceptions of two out of sixteen possibilities: anticodons AAG (Leu) and ACG (Arg). Seven out of these 14 tRNAs are avoided in all organisms due to the nature of the wobble interaction (40). Had they existed, the tRNA repertoire would have been over-promiscuous leading to repetitive built-in translation errors. For comparison, the total number of anticodons that start with cytosine, guanine, or thymine, for which exact-matching tRNAs are avoided in cyanobacteria is 10 (out of 48). The latter include the three anticodons, TTA, TCA and CTA, that correspond to the stop codons, and the following anticodons: TAT (Ile), TCG (Arg), CTT (Lys), CTC (Glu), CTG (Gln) and GCG (Arg). Thus, cyanobacteria have a repertoire of 40 tRNAs at most. The GC-rich Synechococcus utilize the full repertoire, whereas GC-poor HL-Prochlorococcus use a subset of only 32 tRNAs avoiding mostly GC-rich anticodons. Figure 2A clearly shows that the myoviral tRNAs tend to belong to the thymine-starting family of tRNAs (top-right quarter). We note that the six tRNAs of Syn9 are a subset of the tRNA sets of S-RSM4 and S-PM2. In fact, as revealed by Figure 2A and Supplementary Table S3, the Syn9 tRNAs are the six most frequently-used tRNAs by myoviruses. They consist of tRNAs for Leu (TTA\TAA), Thr (ACA\TGT), Asn (AAC\GTT), Arg (AGA\TCT), Val (GTA\TAC) and Ala (GCC/GGC) (codon\anticodon). It is important to note that the myoviral tRNAs represent an additional pool of AT-rich anticodons to the already existing cyanobacterial tRNA pool. The change will be in the AT-rich tGCN increasing their weight among the overall pool of tRNAs present in the cell when they are expressed. Therefore, these six viral tRNAs are very likely to offset the gap in codon usage between AT-rich myoviruses and their Synechococcus hosts. This is clearly seen in Figure 2B. First, as was mentioned earlier, the codon usage of phage and host is uncorrelated. Second, all the six Syn9 tRNAs match codons that are used more frequently in Syn9 rather than in its host. This is significant since the repertoire of tRNAs that exactly match codons that are favored in Syn9 is more limited compared to those favored by the host WH8102 (17 versus 24). This finding is in accord with previous reports that phages bear tRNA genes in their genomes with anticodon preference for AT at the wobble position (first anticodon position), corresponding to the higher AT content of their genomes (34,36,37,51). Interestingly, two anticodons that would highly favor translation of viral genes (marked as T1 and T2 in Figure 2B), are only seen in the S-PM2 tRNAs repertoire. Our hypothesis regarding the reason to avoid such tRNAs will be elaborated in the discussion.
The myoviral-borne tRNAs promote efficient translation of viral genes within GC-rich hosts
In order to measure the impact of the myoviral tRNA pools on their own genomes, we computed the translation efficiency of viral genes in various potential hosts using either the host tRNA pool alone, or a combined pool that also includes the viral encoded tRNAs. We measured the association between the codons of the myoviruses Syn9, S-RSM4 and S-PM2 and cyanobacterial anticodon pool (without the additional anticodon pool of the myoviruses), using the tAI (see ‘Materials and Methods’ section). The Prochlorococcus anticodon pools are well correlated to the majority of the myoviral genes, whereas the Synechococcus anticodon pools show a negative correlation (Figure 3A). Hence, most myoviral genes will be efficiently translated within Prochlorococcus hosts and inefficiently translated within Synechococcus hosts, if only the tRNA pool of the host is considered. The difference in viral genes' tAI between these two groups of cyanobacteria hosts is significant according to the Kruskal–Wallis test (52) with P-values <0.05 [at a 95% confidence level (CI)] as detailed in the Material and Methods section. Indeed, as shown in Figure 1C, the codon usage of the three myoviruses reflects the codon usage of Prochlorococcus. With the original pool of each host's tRNAs, it is expected that the matching will be greater in an environment that fits a Prochlorococcus-like codon usage. Thus, we propose that myoviruses deprived of their own set of anticodons will have more difficulty expressing their genes, and perhaps ultimately also propagate within Synechococcus-like hosts.
Figure 3.

The impact of the viral-borne tRNAs on the translation of phage and host genomes. (A) The tAI of all viral genes is computed using only the bacterial tRNAs. Rows depict the outcome using different hosts (black HL-Prochlorococcus, blue LL-Prochlorococcus and red Synechococcus). Data are shown separately for the three myoviruses. The red lines inside each box represent the median. The edges of each box are the 25 and 75% percentiles of the viral genes’ tAI. Red crosses represent outliers. (B) The ratio of tAI values for all viral genes, when computed with or without the viral tRNA is presented. The three panels correspond to the three viruses as in (A). Rows depict the outcome using different hosts as in (A). Columns are the viral genes. Higher positive tAI scores are marked in red and lower negative tAI scores are marked in green. See the color bar, and note the logarithmic scale. (C) Improvement of the tAI of for each Synechococcus WH8102 genes, and its relationship to pathogenicity islands. Improvement is the ratio of tAI values when computed with or without the addition of the viral tRNAs to the bacterial pool. Data using the tRNA pools of Syn9 (blue), S-PM2 (green) and S-RSM4(red) is shown. Gray shadow mark pathogenicity islands locations in association with phage-like integrase genes. Note the natural-log scale of the y-axis.
To estimate the potential impact of tRNAs carried by myoviruses, we computed the viral tRNA contribution to translation efficiency as the logarithm of the ratio between the tAI with the added anticodon pools of the myoviruses versus the tAI without the myoviral tRNAs (Figure 3B). The additional set of tRNAs have greater and positive effect on viral genes when introduced to an anticodon pools with higher GC contents as Synechococcus and LL Prochlorococcus MIT9303 and MIT9313. As the genomes of the cyanophages were already adapted to the HL Prochlorococcus tRNA pools, the tRNA sets of the cyanophages have a negative effect on their translation.
The myoviral-borne tRNAs reduce and enhance the translation efficiency of selected cyanobacterial genes
In the previous section we showed that the viral tRNAs are advantageous to the phage by enhancing its translation efficiency in GC-rich hosts. Similarly, we calculated the effects of the myoviral pool of tRNAs on the cyanobacterial genomes. We computed the genome-wide average change in tAI due to the inclusion of the viral tRNAs. Indeed, we found that the translation efficiencies within high GC content Synechococcus are those that decrease most by the addition of the viral pool of tRNAs. On average, the genomic tAI values of Synechococcus WH8102 and WH7803, as well as of LL Prochlorococcus MIT9303 and MIT9313, decrease, whereas the tAI of other cyanobacterial genes remain unchanged (Supplementary Figure S1).
Further analysis of the virtual response of Synechococcus WH8102 protein coding genes to the different sets of tRNAs encoded by the genome of Syn9, S-PM2 and S-RSM4 is displayed in Figure 3C. Despite the overall lowered tAI scores of the Synechococcus WH8102 genome, some genes had an elevated tAI scores. The loci that show enhanced tAI levels highly overlap with loci previously annotated as ‘pathogenicity islands’ (53). These are regions with low GC content and atypical trinucleotide composition that are associated in Synechococcus WH8102 with putative phage integrases. These regions were suggested to be remains from an horizontal gene transfer event between cyanophages and the cyanobacteria (54). We thus conducted a statistical hypergeometric test to evaluate the significance of the overlap between the set of host genes that show an elevation in tAI due to the addition of the viral tRNAs and the set of genes that reside within the pathogenicity islands. We obtained highly significant overlap (P-values of 6.7e–25, 9.5 e–27 and 3.3 e–33, for Syn9, S-RSM4 and S-PM2, respectively) indicating that typically hosts genes that belong to the pathogenicity islands show enhanced translation efficiency due to incorporation of the viral genes. It thus seems that pathogenicity islands represent low GC contents zones on the cyanobacterial genome to which the myoviral tRNAs are better adapted. This observation supports the hypothesis that pathogenicity islands in the host have their origins in viral infections.
The myoviral-borne tRNAs maximize the gap in translation efficiency between viral and host genes
In line with the results of Bailly-Bechet et al. (34), our data indicate that the viral tRNAs enhance the translation efficiency of the viral genes, and at the same time decrease the translation efficiency of the bacterial genes. To estimate the disparity between the effect of the viral tRNAs on the phage and the host genome-wide translation efficiency, we computed the difference between the average changes in the tAI of viral genes and the average changes in the tAI of bacterial genes. To compensate for the variance in both distributions, we divided that difference by the joint variance of the two genomes (see ‘Materials and Methods’ section). We will refer to this measure as ΔΔtAI. Synechococcus WH8102—one of the GC-rich cyanobacteria—was chosen as subject matter for this analysis. Testing the three tRNA sets carried by myoviruses (Figure 2A), we found ΔΔtAI (Syn9) = 1.357, ΔΔtAI(S-RSM4) = 1.215, ΔΔtAI(S-PM2) = 1.503. These values correspond to an overall increase of the translation efficiency of the myoviral genes with means 0.041, 0.063 and 0.103, and standard deviations (SD) 0.028, 0.052 and 0.058, for adding Syn9, S-RSM4 and S-PM2 tRNAs, respectively. The corresponding values for the cyanobacterial genes depict a slight decrease of the average −0.005, −0.009 and −0.005 with SD 0.018, 0.028 and 0.043, corresponding to the addition of the Syn9, S-RSM4 and S-PM2 tRNAs, respectively. To evaluate the significance of these numbers, we calculated the probability to obtain such ΔΔtAI values from a random set of tRNAs that has the same size as the actual viral set. For that we produced three sets of 10 000 random combinations of 6, 12 and 23 tRNAs (out of 54 possible anticodons), corresponding to the number of tRNAs carried by Syn9, S-RSM4 and S-PM2, respectively. For each such random tRNA set we computed the ΔΔtAI using WH8102 as the host genome (Figure 4A). The means of the resulting ΔΔtAI distributions for sets of 6, 12 and 23 anticodons were −0.128, −0.158 and −0.333 with corresponding SD of 0.604, 0.540 and 0.567, respectively. All three distributions are biased towards negative values, implying that, on average, a randomly chosen tRNA sets will favor the GC-rich host—not the phage. However, the actual tRNA sets carried by the myoviruses are among the best available sets in this sample: both Syn9 and S-RSM4 are more than 2 SD above the mean of the distribution, whereas S-PM2 ΔΔtAI exceeds the mean with more than 3 SD. Very few random sets yield better ΔΔtAI values (21, 34 and 16, out of 10 000 for Syn9, S-RSM4 and S-PM2, respectively—Figure 4A). Similar results were also obtained when random tRNAs were retrieved from a more restricted pool of host-only anticodons (Supplementary Figure S2). We conclude that there is a selective evolutionary advantage to keep the particular set of tRNA genes that each phage carries, in order to increase the separation between the translation efficiency of the phage and bacterial genomes. This result is in agreement with earlier work that investigated—using a different statistical approach and data set—the selection pressure on the acquisition of tRNA genes from host genomes (34).
Figure 4.

Could the viral-borne tRNA sets be improved? Results from randomization tests tackling this question are presented. (A) The distribution of ΔΔtAI (separation) values, based upon 10 000 random tRNA sets, is shown. For each of the three myoviruses in which we are interested, a separate distribution was generated. Synechococcus WH8102 was used as a host. The ΔΔtAI of the actual viral tRNA sets are marked, and seen to be among the best possible sets. (B) The genome-wide average in tAI as function of the size of viral-borne tRNA set is shown. For each tRNA set size we produced 10 000 random sets. The impact of each random set was computed using the genomes of Syn9 and Synechococcus WH8102. As the size of the random tRNA sets increases, mean[ΔtAI(g)], increases in the Synechococcus host (solid line), while it sharply decreases in the phage Syn9 (dotted line).
All the randomly-generated tRNA sets that outperformed the three viral tRNA sets (Supplementary Figure S3), except for one, were distinguished from the viral tRNA sets by at least one of the two following features. First, most of these sets (all of them in the case of SPM2-outperformers) relied on at least one tRNA that is not possessed by any of the cyanobacteria. The rest of the outperforming sets relied only on tRNAs found in one of their hosts. However, all of these, except for one, had a second common feature, namely, they included a tRNA decoding for Lys (AAA). Interestingly, Lys (AAA) is the codon mostly used by HL-Prochlorococcus (Figure 2), hence it seems as additional selective mechanisms are applied on the tRNA genes pool (see Discussion section).
A random increase of the number of viral-borne tRNA will favor the bacterial host
Given the fact that viral tRNAs can confer a significant advantage to the virus when infecting a host with mismatching GC content, one may naively conclude that the more tRNA genes the virus would bear—the better for the virus. However, roughly speaking, this is true only in the case that the tRNAs present an anticodon with A or T at the first position (AT-rich tRNA). Inspection of the tRNA repertoire of the cyanobacteria (Figure 2A) shows that it is biased towards GC-rich tRNAs (27 versus 14 AT-rich tRNAs). This implies that the chance for a randomly-added tRNA to be advantageous to the virus is only one-third, whereas the probability it will be advantageous to the host is about two-third. In evolutionary terms, this means an increasingly difficult search problem for the virus: the fraction of tRNA sets that will be advantageous to the virus decreases with the increase of the number of tRNAs it will carry in its genome. In further testing this hypothesis we generated 10 000 random sets of tRNAs for each number of tRNAs between 1 and 25, and calculated the change in translation efficiency. Our calculations reveal an increasing disadvantage to the virus in parallel with a slight increase in advantage to the host, as the size of the viral-borne tRNA set increases (Figure 4B). These data support and emphasize the dynamics of tRNA acquisition and selection that ultimately maintains in the viral genomes highly optimized sets of tRNAs. This result probably explains why it is not in the interest of the virus to incorporate additional tRNA genes, and it reduces the likelihood that the number of viral tRNA genes is limited due to other causes such as the costs of increasing genome size.
DISCUSSION
This study presents for the first time two different strategies employed by two different families of cyanophages to enhance their translation efficiency during infection of a heterogeneous pool of hosts that differ in their codon usages and corresponding anticodon pools (Figure 5). We speculate that the choice between the two strategies is strongly related to the genome size. Marine podoviruses, with four and up to five times smaller genome sizes compared with myoviruses, adjust their GC content and codon usage to the GC content and codon usage of the host they infect (Figure 1). Although such ‘specialization’ strategy benefits the podovirus with a readily efficient translation of its genes, it may also restrict its ability to infect hosts with different GC contents. Indeed, isolates of podoviruses were reported to be very restricted in their host ranges, typically infecting a single cyanobacterial host and only seldom another closely related strain (23).
Figure 5.

Schematic overview of the podovirus specialization versus the myovirus adaptation to different Hosts. The illustration shows the tradeoff between genome sizes, the GC content of such genomes and the resulting occurrence of tRNA genes. Podoviruses, with up to five times smaller genome sizes when compared with myoviruses, adjust their GC content to their hosts’ GC content, whereas myoviruses maintain low GC content genomes. As long as myoviruses infect Prochlorococcus with lower GC content genomes, no tRNAs are needed. However, when infecting high GC content cyanobacteria, such as Synechococcus, those same myoviruses would face more difficulties to replicate their genome efficiently. Thus, we contend that tRNAs carried by myoviruses represent an adaptive process to enhance their fitness when infecting high GC content hosts unlike the specialization process that podoviruses undergo.
For myoviruses, with larger genomes, the low guanine and cytosine cellular availability and high-energy costs of those nucleotides (55) may restrict them to the use of low GC codons. However, the larger genomes of myoviruses allow them to incorporate tRNA genes into their genomes without much cost but with large payoff in terms of translation efficiency (Supplementary Table S1). As expected, the GC content and codon usage of myoviruses are well correlated to potential Prochlorococcus hosts (Figure 1). Nevertheless, a similar low GC content myoviral genome, is more difficult to efficiently replicate within high GC content cyanobacteria, such as Synechococcus and LL Prochlorococcus, and it is very likely that this process will take more time. Hence, myoviruses have evolved an ‘adaptation’ strategy whereby their constitutively low GC content genomes are supplemented with tRNA genes to, once expressed, enhance translation efficiency when infecting high GC content hosts. We surmise that the burden of bearing few tRNA genes is minimal given the large genomes of myoviruses and the advantage the tRNAs deliver. Interestingly, such strategy might have enabled myoviruses acquire broader host ranges, as some myoviral isolates were reported to be able to infect both Prochlorococcus and Synechococcus strains (23). We thus proceeded with further examination of the relationship between three representatives of myoviruses (Syn9, S-RSM4 and S-PM2) and their corresponding potential hosts.
In this article, we have made several important observations on the structure of cyanobacterial tRNA repertoire that are important to understand the constraints that are imposed on the infecting phages. We reiterate the main finding in this regard as we believe they are important in their own right. Cyanobacteria, as other Bacteria and Archaea (50), almost completely avoid anticodons starting with adenosine (codons ending with thymine). Only seven out of those 14 avoided anticodons are explained directly by restrictions due to the wobble interaction (40). This is to be compared with the pronounced homogeneity in the usage of thymine-starting anticodons as all cyanobacteria use 12 out of 14 possible thymine-starting anticodons. Further restriction of the anticodon pools leaves Synechococcus with a repertoire of up to 40 tRNAs, whereas for HL Prochlorococcus it may reach 32 tRNAs (Supplementary Table S3). The difference in the repertoires is mainly attributed to HL-Prochloroccous with lowest GC content genomes that avoid some of the guanine and cytosine starting anticodons used by Synechococcus and LL-Prochlorococcus. We therefore consider it to represent a GC-poor anticodon pool rather than AT-rich one (Figure 2A and Supplementary Table S3).
Myoviruses with constitutive low GC content genomes are limited in the low GC content anticodon pool available from the cyanobacterial repertoire. Indeed, as can be seen in Figure 2A most myoviral anticodon pools are predominated by thymine-starting anticodons that span the entire cyanobacterial repertoire. Thus, improving the translation within high GC hosts by introducing AT-rich tRNAs may restrain the ability to infect AT-rich hosts that benefit from such additional tRNA pool.
The repertoires of tRNA genes that marine myoviruses Syn9, S-RSM4 and S-PM2, incorporated into their genomes, consist of 6, 12 and 23 tRNA genes, respectively. The Syn9 set is common to all three sets perhaps serving as a minimal anticodon pool necessary for infection. The set consists of tRNA genes for Leu (TTA\TAA), Thr (ACA\TGT), Asn (AAC\GTT), Arg (AGA\TCT), Val (GTA\TAC) and Ala (GCC/GGC) (codon\anticodon). Bailly-Bechet et al. (2007) in their article computed the average difference in codon usage between pairs of phages and their hosts, and found it to be significant (34). Thus they concluded that the viral tRNA genes are retained selectively to compensate for the gap in codon usage between the phage and its host. In other words, viral tRNA genes are selected to enhance the translation of the viral genes on the expense of the host's genes. Our results support the conclusions of Bailly-Bechet with the following important refinement. We found that the viral tRNA genes are selected to enhance the separation in translation efficiency of viral and host genes, only for hosts were indeed the codon usage gap is large. In addition, our methodology has several improvements over the analysis originally carried by Bailly-Bechet et al. First, our statistical analyses consider all codons rather than only codons for which there are tRNA genes present in the viral genomes. Second, and related to this, our analysis considers the effect, and the importance, of the wobble interactions and not only the Watson–Crick complementarities. Third, Bailly-Bechet et al. disregarded the background effect of the contribution of the hosts’ tRNAs when computing the contribution of the viral tRNAs to the separation in host-viral codon usages. This contribution, though, sets the scale for the contribution of the viral tRNAs. Furtheremore, from a biological point of view, one should not ignore the contribution of the bacterial tRNAs as they are necessary for the translation of both viral and host genes.
As expected, those AT-rich anticodon pools enhance translation efficiency of viral genomes mainly within high GC hosts that otherwise would disfavor the translation (Figure 3A and B). At the same time, the translation efficiency of the high GC host genomes slightly decrease as the overall tRNA pool becomes more AT-rich. Further detailed examination of the Synechococcus genes response to the added viral sets of tRNAs has revealed great compatibility of those tRNAs to pathogenicity islands (Figure 3C). Those islands were previously reported to be associated with integrases and suggested to remain from a horizontal gene transfer event (25). It might be remarked that additional genes, outside the pathogenicity islands, have their translation improved by the addition of the viral tRNAs. Those genes, sometimes in clusters, correspond to low GC regions that were also formerly proposed to represent recent horizontal gene transfer (54). Although is tempting to suggest that among those genes several host defense genes might be present, we did not find any evidence for this
In order to test the hypothesis brought by previous studies claiming for the retention of tRNA genes that match the greater difference in codon usage between the host and virus (34), we generated random sets of tRNA pools and examined the differences in genomic translation efficiencies (Figure 4A). As it occurs, the specific sets of anticodons are among the best sets that produce the highest difference between myoviral genome translation enhancement and Synechococcus genome translation deterioration. Nevertheless, we remarked that the outperforming sets of tRNAs are greatly enriched with Lys (TTT) anticodons matching the Lys (AAA) codon (Supplementary Figure S2). As inferred from Figure 2A and B, such anticodon as well as anticodon TTC encoding for Glu(GAA), would have greatly boosted the translation efficiency of the myoviral genome if it would only infect Synechococcus hosts, as it would match the high usage of the respective codon by the virus and low usage of the host. Indeed, myovirus S-PM2, infecting Synechococcus exclusively (26) carry the tRNA that match these codons. Nevertheless, it can be observed that the Lys (AAA) and Glu (GAA) codons are extensively used by Prochlorococcus (Figure 2A). As several myoviruses were previously reported to be able to infect both Synechococcus and Prochlorococcus (23), we propose that tRNA pools of marine myoviruses are influenced by the collective impacts of their different potential hosts. It is also tempting to suggest that other, less obvious hosts may affect the pool of tRNAs of myoviruses, as tRNA genes for Ile (ATA) predicted for several myoviruses (Figure 2A, anticodon TAT) are absent from all cyanobacteria analyzed.
Hosts not only affect the identity of the selected tRNAs, but also the optimal number of tRNA genes that the virus should carry. Using computer simulation we found that upon infection of a Synechococcus host, increased number of additional tRNAs favors the host because such sets will inevitably contain high GC anticodons (Figure 4B). As a result, the larger the set of tRNAs, the fewer the possibilities to obtain better tRNA sets. We therefore conclude that S-PM2, S-RSM4 and Syn9, with pools of 23, 12 and 6 tRNAs respectively, represent different snapshots of the selective process of tRNAs whereby a minimal pool of tRNA genes expressing tRNAs that maintain elevation of the translation efficiency are ultimately retained.
It is known that phages can influence the gene expression of their host, possibly influencing the cellular tRNA pools. Nevertheless, cyanomyoviruses lack the alt, modA and modB genes expressing moderators of the RNA polymerase to increase its affinity to early promoters (56). Furthermore, cyanomyoviruses also lack the motA and asi genes that express proteins responsible for the transition to the middle mode transcription (48,56) and differ in their putative middle mode promoters (56). Thus, expression during at least the early stages of infection was proposed to be driven by an unmodified host RNA polymerase (56). In addition, it has been reported that relative amount of tRNAs remain constant for the first hour after transcriptional inhibition (57). Hence, we assume our findings relevant for at least the early stages of infection.
Our overall data shows that myoviruses maintain economical low GC content genomes that are readily adapted to translation in hosts with similar low GC content genomes. In order to adapt to high GC content genomes, myoviruses supplement the cyanobacterial tRNA pool with their own set of tRNAs consisting of AT-rich anticodons. We show that the process of acquisition and retention of tRNA genes is delicately balanced by the enhancement of the myoviral genes and concomitant impact on translation of the whole range of potential hosts. Such an adaptation strategy may render myoviruses with the ability to infect a wider range of hosts. In contrast, podoviruses that employ the specialization strategy and adjust the GC content of their small genomes to their hosts, are host specific (Figure 5).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The authors thank the European Research Council ‘Ideas’ program (Y.P.); Avron-Minerva foundation for photosynthesis (A.S.); Alternative Sustainable Energy Research Initiative program of the Weizmann Institute (A.S.) for supporting this research. A.S. is the incumbent of the Robert and Yaddele Sklare Professorial Chair in Biochemistry. Funding for open access charge: European Research Council ‘Ideas’ program (Y.P.); Avron-Minerva foundation for photosynthesis (A.S.); Alternative Sustainable Energy Research Initiative program of the Weizmann Institute (A.S.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mrs. Gingold Hila and Ms Navon Sivan for their help in programming and fruitful discussions.
REFERENCES
- 1.Mann NH, Cook A, Millard A, Bailey S, Clokie M. Marine ecosystems: bacterial photosynthesis genes in a virus. Nature. 2003;424:741. doi: 10.1038/424741a. [DOI] [PubMed] [Google Scholar]
- 2.Sullivan MB, Coleman ML, Weigele P, Rohwer F, Chisholm SW. Three Prochlorococcus cyanophage genomes: signature features and ecological interpretations. PLoS Biol. 2005;3:790–806. doi: 10.1371/journal.pbio.0030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindell D, Sullivan MB, Johnson ZI, Tolonen AC, Rohwer F, Chisholm SW. Transfer of photosynthesis genes to and from Prochlorococcus viruses. Proc. Natl Acad. Sci. USA. 2004;101:11013–11018. doi: 10.1073/pnas.0401526101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sullivan MB, Lindell D, Lee JA, Thompson LR, Bielawski JP, Chisholm SW. Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol. 2006;4:1344–1357. doi: 10.1371/journal.pbio.0040234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Millard A, Clokie MRJ, Shub DA, Mann NH. Genetic organization of the psbAD region in phages infecting marine Synechococcus strains. Proc. Natl Acad. Sci. USA. 2004;101:11007–11012. doi: 10.1073/pnas.0401478101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chenard C, Suttle CA. Phylogenetic diversity of sequences of cyanophage photosynthetic gene psbA in marine and freshwaters. Appl. Environ. Microbiol. 2008;74:5317–5324. doi: 10.1128/AEM.02480-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang K, Chen F. Prevalence of highly host-specific cyanophages in the estuarine environment. Environ. Microbiol. 2008;10:300–312. doi: 10.1111/j.1462-2920.2007.01452.x. [DOI] [PubMed] [Google Scholar]
- 8.Zeidner G, Bielawski JP, Shmoish M, Scanlan DJ, Sabehi G, Beja O. Potential photosynthesis gene recombination between Prochlorococcus and Synechococcus via viral intermediates. Env. Microbiol. 2005;7:1505–1513. doi: 10.1111/j.1462-2920.2005.00833.x. [DOI] [PubMed] [Google Scholar]
- 9.Alperovitch-Lavy A, Sharon I, Rohwer F, Aro EM, Glaser F, Milo R, Nelson N, Beja O. Reconstructing a puzzle: existence of cyanophages containing both photosystem-I and photosystem-II gene suites inferred from oceanic metagenomic datasets. Environ. Microbiol. 2011;13:24–32. doi: 10.1111/j.1462-2920.2010.02304.x. [DOI] [PubMed] [Google Scholar]
- 10.Moore LR, Rocap G, Chisholm SW. Physiology and molecular phylogeny of coexisting Prochlorococcus ecotypes. Nature. 1998;393:464–467. doi: 10.1038/30965. [DOI] [PubMed] [Google Scholar]
- 11.Kettler GC, Martiny AC, Huang K, Zucker J, Coleman ML, Rodrigue S, Chen F, Lapidus A, Ferriera S, Johnson J, et al. Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS Genet. 2007;3:2515–2528. doi: 10.1371/journal.pgen.0030231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dufresne A, Garczarek L, Partensky F. Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol. 2005;6:R14. doi: 10.1186/gb-2005-6-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rocap G, Distel DL, Waterbury JB, Chisholm SW. Resolution of Prochlorococcus and Synechococcus ecotypes by using 16S-23S ribosomal DNA internal transcribed spacer sequences. Appl. Environ. Microbiol. 2002;68:1180–1191. doi: 10.1128/AEM.68.3.1180-1191.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dufresne A, Ostrowski M, Scanlan DJ, Garczarek L, Mazard S, Palenik BP, Paulsen IT, de Marsac NT, Wincker P, Dossat C, et al. Unraveling the genomic mosaic of a ubiquitous genus of marine cyanobacteria. Genome Biol. 2008;9:R90. doi: 10.1186/gb-2008-9-5-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stenstrom CM, Jin HN, Major LL, Tate WP, Isaksson LA. Codon bias at the 3'-side of the initiation codon is correlated with translation initiation efficiency in Escherichia coli. Gene. 2001;263:273–284. doi: 10.1016/s0378-1119(00)00550-3. [DOI] [PubMed] [Google Scholar]
- 16.Coleman JR, Papamichail D, Skiena S, Futcher B, Wimmer E, Mueller S. Virus attenuation by genome-scale changes in codon pair bias. Science. 2008;320:1784–1787. doi: 10.1126/science.1155761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Welch M, Govindarajan S, Ness JE, Villalobos A, Gurney A, Minshull J, Gustafsson C. Design parameters to control synthetic gene expression in Escherichia coli. PLoS ONE. 2009;4:e7002. doi: 10.1371/journal.pone.0007002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlini DB, Stephan W. In vivo introduction of unpreferred synonymous codons into the Drosophila Adh gene results in reduced levels of ADH protein. Genetics. 2003;163:239–243. doi: 10.1093/genetics/163.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Archetti M. Selection on codon usage for error minimization at the protein level. J. Mol. Evol. 2004;59:400–415. doi: 10.1007/s00239-004-2634-7. [DOI] [PubMed] [Google Scholar]
- 20.Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou T, Weems M, Wilke CO. Translationally optimal codons associate with structurally sensitive sites in proteins. Mol. Biol. Evol. 2009;26:1571–1580. doi: 10.1093/molbev/msp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clokie MR, Mann NH. Marine cyanophages and light. Environ. Microbiol. 2006;8:2074–2082. doi: 10.1111/j.1462-2920.2006.01171.x. [DOI] [PubMed] [Google Scholar]
- 23.Sullivan MB, Waterbury JB, Chisholm SW. Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature. 2003;424:1047–1051. doi: 10.1038/nature01929. [DOI] [PubMed] [Google Scholar]
- 24.Mann NH, Clokie MRJ, Millard A, Cook A, Wilson WH, Wheatley PJ, Letarov A, Krisch HM. The genome of S-PM2, a “photosynthetic” T4-type bacteriophage that infects marine Synechococcus strains. J. Bacteriol. 2005;187:3188–3200. doi: 10.1128/JB.187.9.3188-3200.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weigele PR, Pope WH, Pedulla ML, Houtz JM, Smith AL, Conway JF, King J, Hatfull GF, Lawrence JG, Hendrix RW. Genomic and structural analysis of Syn9, a cyanophage infecting marine Prochlorococcus and Synechococcus. Environ. Microbiol. 2007;9:1675–1695. doi: 10.1111/j.1462-2920.2007.01285.x. [DOI] [PubMed] [Google Scholar]
- 26.Millard AD, Zwirglmaier K, Downey MJ, Mann NH, Scanlan DJ. Comparative genomics of marine cyanomyoviruses reveals the widespread occurrence of Synechococcus host genes localized to a hyperplastic region: implications for mechanisms of cyanophage evolution. Environ. Microbiol. 2009;11:2370–2387. doi: 10.1111/j.1462-2920.2009.01966.x. [DOI] [PubMed] [Google Scholar]
- 27.Weiss SB, Hsu WT, Foft JW, Scherber Nh. Transfer RNA coded by T4 bacteriophage genome. Proc. Natl Acad. Sci. USA. 1968;61:114–121. doi: 10.1073/pnas.61.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daniel V, Sarid S, Littauer UZ. Amino acid acceptor activity of bacteriophage T4 transfer RNA. FEBS Lett. 1968;2:39–41. doi: 10.1016/0014-5793(68)80095-x. [DOI] [PubMed] [Google Scholar]
- 29.Littauer UZ, Daniel V. The induction of tRNA synthesis following T4 phage infection. J. Cell. Physiol. 1969;74(Suppl. 1):71–80. doi: 10.1002/jcp.1040740406. [DOI] [PubMed] [Google Scholar]
- 30.Canchaya C, Fournous G, Brussow H. The impact of prophages on bacterial chromosomes. Mol. Microbiol. 2004;53:9–18. doi: 10.1111/j.1365-2958.2004.04113.x. [DOI] [PubMed] [Google Scholar]
- 31.Tan YL, Zhang KB, Rao XC, Jin XL, Huang JJ, Zhu JM, Chen ZJ, Hu XM, Shen XD, Wang L, et al. Whole genome sequencing of a novel temperate bacteriophage of P-aeruginosa: evidence of tRNA gene mediating integration of the phage genome into the host bacterial chromosome. Cell. Microbiol. 2007;9:479–491. doi: 10.1111/j.1462-5822.2006.00804.x. [DOI] [PubMed] [Google Scholar]
- 32.Campbell AM. Chromosomal insertion sites for phages and plasmids. J. Bacteriol. 1992;174:7495–7499. doi: 10.1128/jb.174.23.7495-7499.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheetham BF, Katz ME. A role for bacteriophages in the evolution and transfer of bacterial virulence determinants. Mol. Microbiol. 1995;18:201–208. doi: 10.1111/j.1365-2958.1995.mmi_18020201.x. [DOI] [PubMed] [Google Scholar]
- 34.Bailly-Bechet M, Vergassola M, Rocha E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007;17:1486–1495. doi: 10.1101/gr.6649807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daniel V, Sarid S, Littauer UZ. Bacteriophage induced transfer RNA in Escherichia coli. New transfer RNA molecules are synthesized on the bacteriophage genome. Science. 1970;167:1682–1688. doi: 10.1126/science.167.3926.1682. [DOI] [PubMed] [Google Scholar]
- 36.Kunisawa T. Synonymous codon preferences in bacteriophage T4: a distinctive use of transfer RNAs from T4 and from its host Escherichia coli. J. Theor. Biol. 1992;159:287–298. doi: 10.1016/s0022-5193(05)80725-8. [DOI] [PubMed] [Google Scholar]
- 37.Kunisawa T. Functional role of mycobacteriophage transfer RNAs. J. Theoret. Biol. 2000;205:167–170. doi: 10.1006/jtbi.2000.2057. [DOI] [PubMed] [Google Scholar]
- 38.Wilson JH. Function of bacteriophage-T4 transfer-RNAs. J. Mol. Biol. 1973;74:753–754. doi: 10.1016/0022-2836(73)90065-x. [DOI] [PubMed] [Google Scholar]
- 39.Kanaya S, Yamada Y, Kudo Y, Ikemura T. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene. 1999;238:143–155. doi: 10.1016/s0378-1119(99)00225-5. [DOI] [PubMed] [Google Scholar]
- 40.dos Reis M, Savva R, Wernisch L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 2004;32:5036–5044. doi: 10.1093/nar/gkh834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rice J. Mathematical Statistics and Data Analysis. Belmont, CA: Duxbury Press, Wadsworth Publishing Company; 1995. [Google Scholar]
- 43.Hochberg Y, Tamhane AC. Multiple Comparison Procedures. New York: Wiley; 1987. [Google Scholar]
- 44.Azuaje F. Clustering-based approaches to discovering and visualising microarray data patterns. Brief Bioinform. 2003;4:31–42. doi: 10.1093/bib/4.1.31. [DOI] [PubMed] [Google Scholar]
- 45.Partensky F, Hess WR, Vaulot D. Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol. Mol. Biol. Rev. 1999;63:106–127. doi: 10.1128/mmbr.63.1.106-127.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palenik B. Cyanobacterial community structure as seen from RNA polymerase gene sequence analysis. Appl. Environ. Microbiol. 1994;60:3212–3219. doi: 10.1128/aem.60.9.3212-3219.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Partensky F, Hess WR, Vaulot D. Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol. Mol. Biol. Rev. 1999;63:106–127. doi: 10.1128/mmbr.63.1.106-127.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Ruger W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003;67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crick FHC. Codon-anticodon pairing - Wobble hypothesis. J. Mol. Biol. 1966;19:548–555. doi: 10.1016/s0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- 50.Marck C, Grosjean H. tRNomics: analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA. 2002;8:1189–1232. doi: 10.1017/s1355838202022021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cowe E, Sharp PM. Molecular evolution of bacteriophages - discrete patterns of codon usage in T4 genes are related to the time of gene-expression. J. Mol. Evol. 1991;33:13–22. [Google Scholar]
- 52.Wallis WHKWA. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952;47:583–631. [Google Scholar]
- 53.Hacker J, Kaper JB. Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 2000;54:641–679. doi: 10.1146/annurev.micro.54.1.641. [DOI] [PubMed] [Google Scholar]
- 54.Palenik B, Brahamsha B, Larimer FW, Land M, Hauser L, Chain P, Lamerdin J, Regala W, Allen EE, McCarren J, et al. The genome of a motile marine Synechococcus. Nature. 2003;424:1037–1042. doi: 10.1038/nature01943. [DOI] [PubMed] [Google Scholar]
- 55.Rocha EP, Danchin A. Base composition bias might result from competition for metabolic resources. Trends Genet. 2002;18:291–294. doi: 10.1016/S0168-9525(02)02690-2. [DOI] [PubMed] [Google Scholar]
- 56.Clokie MR, Millard AD, Mann NH. T4 genes in the marine ecosystem: studies of the T4-like cyanophages and their role in marine ecology. Virol. J. 7:291. doi: 10.1186/1743-422X-7-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dittmar KA, Mobley EM, Radek AJ, Pan T. Exploring the regulation of tRNA distribution on the genomic scale. J. Mol. Biol. 2004;337:31–47. doi: 10.1016/j.jmb.2004.01.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.