


Abstract
The double-stranded DNA of the genome contains both sequence information directly relating to the protein and RNA coding as well as functional and structural information relating to protein recognition. Only recently is the importance of DNA shape in this recognition process being fully appreciated, and it also appears that minor groove electronegative potential may contribute significantly in guiding proteins to their cognate binding sites in the genome. Based on the photo-chemical probing results, we have derived an algorithm that predicts the minor groove electronegative potential in a DNA helix of any given sequence. We have validated this model on a series of protein–DNA binding sites known to involve minor groove electrostatic recognition as well as on stable nucleosome core complexes. The algorithm allows for the first time a full minor groove electrostatic description at the nucleotide resolution of any genome, and it is illustrated how such detailed studies of this sequence dependent, inherent property of the DNA may reflect on genome organization, gene expression and chromosomal condensation.
INTRODUCTION
Proteins such as transcription factors and histones in nucleosomes are pivotal in correct decoding of the genetic information of the double-stranded DNA helix by binding to specific sequences in the genomes thereby controlling proper gene activation. Sequence-specific protein binding is primarily accomplished through direct reading of the nucleobase sequence via specific protein nucleobase contacts predominantly in the DNA major groove (1). However, the base sequence also controls DNA double helix conformation and specific properties such as minor groove width and electronegative potential (2–4). It has been known for more than a decade that DNA recognition by many small ligands (5) is relying on minor groove shape and electrostatic potential, and it is also recognized that these features of the DNA helix can be critical for protein recognition (6–8). Nevertheless, only recently has a more general and detailed understanding of the importance of variations in minor groove electronegative potential for protein binding been documented (9–11). In particular, it has been suggested based on the crystal structure data that a large number of proteins may recognize and be guided to their binding sites on the DNA helix through specific arginines reading the electronegative potential in the minor groove (9). Thus, knowing the electronegative potential along the DNA of the genome is important for a detailed understanding of the DNA function in terms of protein recognition. However, reliable techniques for directly probing and especially predicting minor groove electronegative potential have hitherto not been available.
Minor groove width and electronegative potential of the DNA helix are clearly interconnected properties as a closer distance of the phosphates across the groove will increase the negative potential in the groove, but the electron distribution within the base pairs at the floor of the groove also plays a decisive role, as does the sugar conformation and thus the exact position of the phosphates relative to the groove (2–4). Therefore, minor groove width and electronegative potential are distinct but not independent helix parameters.
We have previously demonstrated that the sequence dependence of photo-chemical cleavage of double-stranded DNA by the uranyl(VI) ion (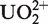 ) reflects these DNA parameters (12–14). Mechanistically, we have presented evidence that the uranyl divalent cation bound to the phosphates of the backbone photo-oxidizes proximal deoxyriboses (15). Thus, we proposed that uranyl photo-probing of duplex DNA in principal could be exploited to semi-quantitatively assess the minor groove width, but presumable more precisely the minor groove electronegative potential, along any DNA helix sequence (12,14). We argued that sensing groove width could be due to bis dentate coordination to opposite phosphates across the groove, while electronegative potential sensing obviously would be caused by electrostatic attraction of the cationic (solvated) uranyl. In this study, we present further validation that uranyl photo-cleavage analysis does indeed to a very large extent reflect DNA minor groove electronegative potential, and we offer an algorithm that with high accuracy allows prediction of an electronegative potential genome map at the nucleotide level. Thus in the present study, we discuss the uranyl probing data only with reference to minor groove electronegative potential, but the close connection to minor groove width as discussed above should be considered throughout.
) reflects these DNA parameters (12–14). Mechanistically, we have presented evidence that the uranyl divalent cation bound to the phosphates of the backbone photo-oxidizes proximal deoxyriboses (15). Thus, we proposed that uranyl photo-probing of duplex DNA in principal could be exploited to semi-quantitatively assess the minor groove width, but presumable more precisely the minor groove electronegative potential, along any DNA helix sequence (12,14). We argued that sensing groove width could be due to bis dentate coordination to opposite phosphates across the groove, while electronegative potential sensing obviously would be caused by electrostatic attraction of the cationic (solvated) uranyl. In this study, we present further validation that uranyl photo-cleavage analysis does indeed to a very large extent reflect DNA minor groove electronegative potential, and we offer an algorithm that with high accuracy allows prediction of an electronegative potential genome map at the nucleotide level. Thus in the present study, we discuss the uranyl probing data only with reference to minor groove electronegative potential, but the close connection to minor groove width as discussed above should be considered throughout.
MATERIAL AND METHODS
The pentamer library, consisting of 7 members (94 nt in length), and the protein binding sites were cloned into the BamHI site of plasmid pUC19 by standard methods. Each clone member of the library contained a subset of all the 1024 possible pentanucleotide sequences (Supplementary Figure S1A–D and Supplementary Table S1). The pentamer library sequences were designed to contain a common internal control sequence (A-tracts) proximal to the terminal BamHI cloning sites, in order to aid data normalization. The uranyl cleavage peak areas of the flanking common BamHI sequences were used to normalize the data for the seven clone members.
The binding sequences of 12 minor groove protein binding sequences were cloned in pUC 19 as two fragments each containing six binding sites. The two Drosophila Hox binding sequences were cloned as a single fragment in pUC 19 (Supplementary Table S1). All DNA fragments used in this study were 32P-labelled at the 3′-end of the EcoR I or Hind III sites of pUC 19 derivatives by use of standard techniques.
The uranyl photo-cleavage was performed as previously described (12,13,16). A Molecular Dynamics Storm 860 phosphorimager was used to collect data from phosphor storage screens. In order to quantify band intensities, we used the software SAFA package (Semi-Automated Footprinting Analysis) (17).
The algorithm is using a sliding window of five bases (Figure 1D). Each score vector of five scores (a–j) is found by matching the DNA pentamer in the uranyl cleavage table (shown as histogram in Supplementary Figure 1A–D), which holds all 1024 possible pentamers. The two first scores are set to zero according to the n + (n − 2) algorithm (Figure 1C). The score vector is placed in a result matrix which has five rows and as many columns as there are bases in the sequence. The sliding window is then moved one base and the next score vector is placed on the second line in the result matrix below its pentamer. When the window has been shifted five times, the row number of the result matrix is reset and the sixth score vector will be inserted at the first line in the result matrix, etc. The final score vector (y1 − y8) is the sum of each column in the result matrix, which holds three scores, divided by three. Thus for prediction of values for the electronegative potential the sum of predicted uranyl cleavage scores for each n + (n − 2) base pairs are averaged and the value is assigned to the n − 1 base (Figure 1E).
Figure 1.

Uranyl cleavage pattern of a pentanucleotide library and model for minor groove potential prediction. (A) Global view of the relative nucleotide cleavage intensities (in arbitrary units) in a DNA fragment containing all 1024 combinations of pentanucleotide sequences. Both strands are represented. (B) Expansion of a region (rectangle) of the 1024 bp library from panel A. (See Supplementary Figure S1 for details of all sequences in the library). (C) Principle of the analysis of uranyl cleavage and minor groove electronegative potential. A pentamer [example GATGC (X-strand)] contains three nx (T, G and C) bases connected with lines with the ny − 2 positions (C, T and A) on the lower y strand. (D) Algoritm used for predicting the uranyl cleavage and electronegative potential in a given DNA sequence (see ‘Materials and Methods’ section for details) (E) In order to obtain a relative value for the minor groove potential defined by the bases nx and ny − 2, the cleavage at the nx and ny − 2 positions were summed and the potential assigned to base nx − 1 in the sequence.
RESULTS AND DISCUSSION
Extensive studies on bent A-tract DNA have shown that sequence-dependent DNA conformation and thus also minor groove width and electrostatic potential require tetra/pentamer regions in order to be defined in terms of sequence dependence (18). Therefore, to derive at an experimentally founded model for sequence based prediction of DNA minor groove electronegative potential, we decided to analyse the uranyl photo-cleavage of all possible pentanucleotide helixes as found in a previously published DNA fragment library of all 1024 possible pentanucleotides (19). As can be seen from the results (Figure 1A) more than 10-fold variation in cleavage intensity is observed along the fragment. In particular A/T rich regions are, as expected, generally most efficiently cleaved (Figure 1B and Supplementary Figure S1A–D (13). However, no direct correlation between the size and simple sequence of the AT region and the cleavage is apparent. For instance sequences containing two or three contiguous A/T base pairs such as GAACT (Supplementary Figure S1A), GAAAC (Supplementary Figure S1B), GAACC (Supplementary Figure S1B) and GATAC (Supplementary Figure S1C and Figure 1B) and even sequences without any A/T dinucleotide steps AGACT (Supplementary Figure S1B) and GGACA (Supplementary Figure S1D) exhibit increased cleavage while other seemingly analogous sequences such as CAATC (Supplementary Figure S1A), GAAGA (Supplementary Figure S1A), CAAAC (Supplementary Figure S1B) and CATAG (Supplementary Figure S1B) do not exhibit hyper-reactivity towards uranyl cleavage. Also in accordance with all previous results, the positions of cleavage maxima and minima on the two DNA strands in general are consistent with binding/cleavage across the minor groove, exhibiting a 2 nt stagger towards the 3′-end between the two strands (the shortest distance across the minor groove) (12–15) (Figure 1B and Supplementary Figure S1A–D). Thus, within each pentamer three such minor groove nx − (ny − 2) ‘base pairs’ are defined (Figure 1C). Consequently, we designed an algorithm that divides a sequence in overlapping pentamers (Figure 1D). For each pentamer, the cleavage values for the first three 3′-bases on each strand is obtained from the experimental pentamer library cleavage data (Supplementary Figure S1). Thus, eventually each base position is assigned the sum of three cleavage values (Figure 1D, example in Supplementary Figure S2). Often a quantitative asymmetry of the uranyl cleavage between the two strands is observed (13). Therefore the prediction of the relative minor groove electronegative potential is obtained as the sum of the (predicted) cleavage at position nx and position ny − 2, and is assigned to base position n − 1 at the centre of an imaginary line between the nx’th and the ny − 2 nd phosphate (Figure 1E).
In order to validate the model both in terms of uranyl cleavage and minor groove electronegative potential prediction, we chose 14 well-described protein binding sites and a thoroughly analysed nucleosome sequence (9,10). All of the analysed protein binding sites involve arginine interactions in regions with a narrow minor groove and enhanced electronegative potential according to calculations on the basis of 3D crystal structures found in the Protein Data Bank (PDB) (9,10). Comparison of the experimentally obtained uranyl photo-cleavage pattern of these DNA regions with that predicted from our sliding window model algorithm, clearly validates the model in terms of cleavage prediction, although minor differences are seen in regions of less intense cleavage (Figure 2A and Supplementary Figure S3). Likewise the relative minor groove electronegative potential map calculated from the data as described above shows very good correspondence between the experimentally based and the algorithm predicted values (Figure 2B). Furthermore, the relative minor groove electronegative potential map obtained from uranyl cleavage data corresponds excellently with that previously calculated from crystal structure data (9,10), and thus with the experimentally determined positions of arginines in the protein–DNA complexes (Figure 2B). Consequently, we conclude that the algorithm provides a very valuable and reliable tool for relative, semi-quantitative prediction of DNA helix minor groove electronegative potential solely based on DNA sequence. Furthermore, because the uranyl probing reflects the properties of free DNA in solution, we conclude that the minor groove features responsible for protein recognition through electrostatic binding via arginines (at least for the protein–DNA complexes analysed here) is a feature of the native DNA helix conformation and is not induced by protein binding. This conclusion is not possible based on crystal data on protein–DNA complexes. Thus these proteins may indeed find their cognate DNA target by electrostatic search of the helix minor groove.
Figure 2.

Experimental and predicted values of uranyl cleavage and minor groove electronegative potential of protein binding sites. (A) Experimental (filled triangle) and predicted (filled square) relative uranyl photo-cleavage (in arbitrary units) at Hox (fkh250 and fkh250con*) (10), MogR (20), UBX-EXD (21), MATa1/Matα2 (22) and Oct1 (23) binding DNA sequences [the difference in intensities between the predicted and experimentally determined values are not meaningful as they depend on the intensities of the bands (exposure) on the gel in the particular analyses]. (B) Predicted and experimental relative electrostatic potential (in arbitrary units) of the six protein binding sites in A and eight additional protein binding sites [Msx-1 (24), Oct-1 POU (25), Pit-1 POU (26), the PhoB response regulator (27), the MATα2/MCM1 complex (28) and the Hap1-18 activator (29), the Tc3 transposase (30) and the phage 434 repressor (31)] based on experimental (filled triangle) and predicted (filled square) uranyl photo-cleavage data. The relative values for electronegative potential determined from the X-ray crystal structure of the Hox-fsh250 complexes (10) are shown as stippled lines (filled diamond). The position of arginine contacts are indicated by arrows.
The strength of the approach is clearly illustrated by the analysis of the two Drosophila Hox binding sequences, the fsh250 and the variant fsh250con* sites, differing only by a single TA/AT base pair change. The two binding sequences, containing the ATTAAT (fsh250) and ATTTAT (fsh250con*) hexamer sequences, respectively, have been shown to mediate functional specificity through small differences in minor groove width and electrostatic potential as calculated from X-ray structures (10). Our prediction clearly reflects this subtle difference in minor groove potential that allows two arginines and one histidine to bind in the broader region of a higher electronegative potential in the ATTAAT sequence, whereas only a single arginine binds in the ATTTAT sequence due to an elevated electronegative potential only in the left part of the hexamer sequence (Figure 2B). Analogously, excellent correspondence between the uranyl data and crystallography results of minor groove potential and arginine positions were found for the motility gene repressor (MogR) (20), which binds to a long A/T region in which a TA step with at local widening of the minor groove produces a bifurcated electronegative potential where two arginines can bind. Indeed our analysis is fully consistent with the data for all 14 protein binding sites including the Drosophila Hox protein, UBX and the cofactor EXD, which binds by inserting an arginine into a very electronegative pocket of the minor groove (21), for the DNA site recognized by the MATa1–MCM1 complex (22) (in which Arg 7 binds within a long electronegative region whereas Arg 4 binds in a region of a less electronegative potential), and for the mammalian OCT1–PORE complex in which two POU domains (23) binds to two A-tract regions possessing electronegative pockets for four arginines. These examples together with the results of the binding sites for Msx-1 (24), OCT1-Pou (25), Pit1 (26), PhoB (27), MATα2-MCM1 (28), HAP1-18 (29), Tc3 transpotase (30) and the 434 repressor (31) (Figure 2B and Supplementary Figure S3) all clearly demonstrate that the predicted minor groove electronegative potential of free DNA provides a powerful prediction for potential positions of arginines in protein–DNA complexes.
Analysis of the symmetrical DNA used for X-ray structure determination of a nucleosome core complex (32) gives a slightly different picture. Although as found for the protein recognition sites, the correlation between experimental and predicted uranyl cleavage (Figure 3A) (and thus the derived minor groove electronegative potential measure) (Figure 3B) is qualitatively very good, only a subset of the regions identified as having highly electronegative (and also narrow) minor groove in free DNA appear so in the nucleosome core particle (32) (Figure 3B). Specifically, we note that there is very good correlation at nucleosome positions 68, 55, 45 and 38 at the outer part of the core DNA (one half of the symmetrical DNA is shown) while very little correlation between the free DNA structure and the nucleosome structure is found in the central region. In particular, the highly electronegative minor grooves at positions 32, 10 and 0 are virtually out of phase with the DNA wrapping around the nucleosome core. Not unexpectedly, this would imply that the conformation of this DNA is not fully predetermined for nucleosome wrapping, but that certain, presumably key regions (in this case positions 68, 55, 45 and 38), are nucleating the process and thus direct the final folding which by induced fit ‘forces’ the remaining DNA into the 10 bp regular phasing around the histone core. Clearly it would be biologically advantageous for evolution to select nucleosomes of intermediate thermodynamic stability or nucleosomes with alternative positioning preferences as this would allow energetically less costly remodelling for gene activation, and sliding and unravelling during replication and transcription. Thus upon genomic wide analyses of minor groove electronegative potential employing dedicated algorithms it may become possible to predict preferred nucleosomal positioning as well as possible remodelled states in the genome. Furthermore, other patterns may signify functional regions such as promoters, enhancers, etc.
Figure 3.

Uranyl cleavage and minor groove electronegative potential of nucleosome forming sequences. Experimental (filled triangle) and predicted (filled square) uranyl photo-cleavage (A) and relative electrostatic potential map (B) of DNA of a nucleosome core particle (32). One half of the symmetrical DNA in the nucleosome core particle is shown with numbers relative to the center of symmetry. The regions of the highest electronegative potential determined from the X-ray crystal structure (9) are shown as underlined in (B). The positions of arginine contacts are indicated by arrows. (C) Predicted minor groove electronegative potential map in the GAL1-10 locus (positions above the graphs) in chromosome II from Saccaromyces cerevisie. The reported (33,34) in vivo positions of four nuclesomes are shown by ellipses and the region of four GAL four binding sites is indicated by a grey rectangle. The two transcription start sites (GAL 1 and GAL10) in the region are indicated.
This latter point is very clearly illustrated by the analysis of the divergent yeast GAL1 and GAL10 promoters, which share a common upstream activating region containing four binding sites for the GAL 4 protein. This region has been thoroughly characterized for nucleosome positioning in vivo (33,34). The minor groove electronegative potential map (Figure 3C) shows a very distinct difference between the GAL4 binding region and the surrounding DNA, which contain specifically positioned nucleosomes. While, the nucleosome regions show major variations in electronegative potential, many within a 10 bp period compatible with nucleosome predisposition, the GAL4 region shows only minimal variation which would indicate much lower propensity for forming nucleosomes. Furthermore analysis of a series of human promoters indicate a similar general pattern in which the regions around transcription start site exhibit significantly lower electronegative potential variation than the regions surrounding the promoters (Supplementary Figure S4).
In view of recent uncertainty of the importance of sequence directing effects on nucleosome positioning (35,36), these results clearly would indicate a very pronounced influence of DNA helix properties (which are sequence instructed) on in vivo nucleosome positioning [at least for the GAL1-10 locus and also for (certain) human promoters]. Obviously, nucleosome positioning is more complicated than merely a question of electronegative potential. Nucleosome positioning and octamer binding to G/C-rich motifs (37,38) will not be predicted by the electronegative potential analysis, possibly because this binding is predominantly driven by DNA flexibility properties. Eventually a combination of two or more algorithms taking into account both groove potential as well as DNA flexibility may be required to fully predict energetically preferred nucelosome positioning. Nonetheless, the present algorithm for the first time allow this type of analyses of the influence and importance of DNA structure in terms of minor groove electronegative potential on genome function and organization, and the results so far on promoters and nucleosome DNA clearly indicate that this parameter is important. Further comprehensive genome analyses will reveal the implications of this approach.
Obviously, regions of high minor groove electronegative potential will in general also be AT-rich because the highest negative potential is found in connection with AT-tracts (12); but AT-rich regions [e.g. short AT (N ≥ 3) runs interrupted by single G/C base pairs] do not necessarily show high minor groove electronegative potential. Therefore, the electronegative potential prediction by the DNA structure—due to the molecular origin of this DNA property—is inherently sequence biased, but a simple sequence analysis without the algorithm will of course not reveal the electronegative potential information at all. It is also important to emphasize that it is not the electronegative potential per se but the pattern along the DNA helix which is the target of such analyses.
An algorithm based on the hydroxyl radical cleavage method has recently been developed for measuring local variations in DNA structure at single nucleotide resolution (19). Modulation in hydroxyl radical cleavage reflects the average helix structure in terms of solvent accessibility of the deoxyribose in the minor groove. Thus the charge neutral hydroxyl radical may sense differences in groove width (and helix conformation) but hardly electronegative potential per se. In contrast, the DNA interaction of the cationic uranyl ion will be directly influenced by the local electrostatic potential of the DNA helix. Therefore as agued previously (13,39), information obtained by the two methods is not equivalent but rather complementary, and may be combined in order to obtain a full description of the DNA helix conformation and properties. Furthermore, variations in uranyl cleavage is much more pronounced than hydroxyl radical cleavage and therefore more sensitively reflects subtle differences in helix structure and property, in casu minor groove electronegative potential. Thus a comparison of the two methods at the genomic level would be interesting in order to deduce the correlation between minor groove width and electronegative potential in genomes, and also to help unravelling the structural and molecular details of the connection between these two parameters of the DNA double helix. Finally, because the algorithm is based on pentamer data we recognize that (special) features of the DNA helix that are dependent on longer stretches of the helix may not be predicted by the algorithm, and that it therefore can be refined by incorporation of more experimental data or by expanding it to a heaxamer or heptamer format.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online. The supplementary algorithm is available at: http://gastro.sund.ku.dk/nar/
FUNDING
Funding for open access charge: University of Copenhagen.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mrs Neel Louv-Jansen for technical assistance and Dr Jesper Thorvald Troelsen for helpful discussions.
REFERENCES
- 1.Rohs R, Jin X, West SM, Joshi R, Honig B, Mann RS. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010;79:233–269. doi: 10.1146/annurev-biochem-060408-091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lavery R, Pullman B, Zakrzewska K. Intrinsic electrostatic properties and base sequence effects in the structure of oligonucleotides. Biophys. Chem. 1982;15:343–351. doi: 10.1016/0301-4622(82)80017-3. [DOI] [PubMed] [Google Scholar]
- 3.Lavery R, Pullman B. The electrostatic field of DNA: the role of the nucleic acid conformation. Nucleic Acids Res. 1982;10:4383–4395. doi: 10.1093/nar/10.14.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lavery R, Pullman B, Corbin S. The molecular electrostatic potential and steric accessibility of poly (dA-dT). poly (dA-dT) in various conformations: B-DNA, D-DNA and 'alternating-B' DNA. Nucleic Acids Res. 1981;9:6539–52. doi: 10.1093/nar/9.23.6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geierstanger BH, Wemmer DE. Complexes of the minor groove of DNA. Annu. Rev. Biophys. Biomol. Struct. 1995;24:463–493. doi: 10.1146/annurev.bb.24.060195.002335. [DOI] [PubMed] [Google Scholar]
- 6.Dalma-Weiszhausz DD, Gartenberg MR, Crothers DM. Sequence-dependent contribution of distal binding domains to CAP protein-DNA binding affinity. Nucleic Acids Res. 1991;19:611–616. doi: 10.1093/nar/19.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gartenberg MR, Crothers DM. DNA sequence determinants of CAP-induced bending and protein binding affinity. Nature. 1988;333:824–829. doi: 10.1038/333824a0. [DOI] [PubMed] [Google Scholar]
- 8.Bailly C, Waring MJ, Travers AA. Effects of base substitutions on the binding of a DNA-bending protein. J. Mol. Biol. 1995;253:1–7. doi: 10.1006/jmbi.1995.0530. [DOI] [PubMed] [Google Scholar]
- 9.Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. The role of DNA shape in protein-DNA recognition. Nature. 2009;461:1248–1253. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joshi R, Passner JM, Rohs R, Jain R, Sosinsky A, Crickmore MA, Jacob V, Aggarwal AK, Honig B, Mann RS. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell. 2007;131:530–543. doi: 10.1016/j.cell.2007.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stella S, Cascio D, Johnson RC. The shape of the DNA minor groove directs binding by the DNA-bending protein Fis. Genes Dev. 2010;24:814–826. doi: 10.1101/gad.1900610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nielsen PE, Møllegaard NE, Jeppesen C. DNA conformational analysis in solution by uranyl mediated photocleavage. Nucleic Acids Res. 1990;18:3847–3851. doi: 10.1093/nar/18.13.3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Møllegaard NE, Lindemose S, Nielsen PE. Uranyl photoprobing of nonbent A/T- and bent A-tracts. A difference of flexibility? Biochemistry. 2005;44:7855–7863. doi: 10.1021/bi0502083. [DOI] [PubMed] [Google Scholar]
- 14.Sönnichsen SH, Nielsen PE. Enhanced uranyl photocleavage across the minor groove of all (A/T)4 sequences indicates a similar narrow minor groove conformation. J. Mol. Recognit. 1996;9:219–227. doi: 10.1002/(sici)1099-1352(199605)9:3<219::aid-jmr242>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 15.Nielsen PE, Hiort C, Sonnichsen SH, Buchardt O, Dahl O, Norden B. DNA binding and photocleavage by uranyl(VI)(UO22+) salts. J. Am. Chem. Soc. 1992;114:4967–4975. [Google Scholar]
- 16.Lindemose S, Nielsen PE, Møllegaard NE. Polyamines preferentially interact with bent adenine tracts in double-stranded DNA. Nucleic Acids Res. 2005;33:1790–1803. doi: 10.1093/nar/gki319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das R, Laederach A, Pearlman SM, Herschlag D, Altman RB. SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA. 2005;11:344–354. doi: 10.1261/rna.7214405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nadeau JG, Crothers DM. Structural basis for DNA bending. Proc. Natl Acad. Sci. USA. 1989;86:2622–2626. doi: 10.1073/pnas.86.8.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenbaum JA, Pang B, Tullius TD. Construction of a genome-scale structural map at single-nucleotide resolution. Genome Res. 2007;17:947–953. doi: 10.1101/gr.6073107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen A, Higgins DE, Panne D. Recognition of AT-rich DNA binding sites by the MogR repressor. Structure. 2009;17:769–777. doi: 10.1016/j.str.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Passner JM, Ryoo HD, Shen L, Mann RS, Aggarwal AK. Structure of a DNA-bound Ultrabithorax-Extradenticle homeodomain complex. Nature. 1999;397:714–719. doi: 10.1038/17833. [DOI] [PubMed] [Google Scholar]
- 22.Li T, Jin Y, Vershon AK, Wolberger C. Crystal structure of the MATa1/MATalpha2 homeodomain heterodimer in complex with DNA containing an A-tract. Nucleic Acids Res. 1998;26:5707–5718. doi: 10.1093/nar/26.24.5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reményi A, Tomilin A, Pohl E, Lins K, Philippsen A, Reinbold R, Schöler HR, Wilmanns M. Differential dimer activities of the transcription factor Oct-1 by DNA-induced interface swapping. Mol. Cell. 2001;8:569–580. doi: 10.1016/s1097-2765(01)00336-7. [DOI] [PubMed] [Google Scholar]
- 24.Hovde S, Abate-Shen C, Geiger JH. Crystal structure of the Msx-1 homeodomain/DNA complex. Biochemistry. 2001;40:12013–12021. doi: 10.1021/bi0108148. [DOI] [PubMed] [Google Scholar]
- 25.Klemm JD, Rould MA, Aurora R, Herr W, Pabo CO. Crystal structure of the Oct-1 POU domain bound to an octamer site: DNA recognition with tethered DNA binding modules. Cell. 1994;77:21–32. doi: 10.1016/0092-8674(94)90231-3. [DOI] [PubMed] [Google Scholar]
- 26.Jacobson EM, Li P, Leon-del-Rio A, Rosenfeld MG, Aggarwal AK. Structure of Pit-1 POU domain bound to DNA as a dimer: unexpected arrangement and flexibility. Genes Dev. 1997;11:198–212. doi: 10.1101/gad.11.2.198. [DOI] [PubMed] [Google Scholar]
- 27.Blanco AG, Sola M, Gomis-Ruth FX, Coll M. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure. 2002;10:701–713. doi: 10.1016/s0969-2126(02)00761-x. [DOI] [PubMed] [Google Scholar]
- 28.Tan S, Richmond TJ. Crystal structure of the yeast MATalpha2/MCM1/DNA ternary complex. Nature. 1998;391:660–666. doi: 10.1038/35563. [DOI] [PubMed] [Google Scholar]
- 29.King DA, Zhang L, Guarente L, Marmorstein R. Structure of HAP1-18-DNA implicates direct allosteric effect of protein-DNA interactions on transcriptional activation. Nat. Struct. Biol. 1999;6:22–27. doi: 10.1038/4893. [DOI] [PubMed] [Google Scholar]
- 30.Watkins S, van Pouderoyen G, Sixma TK. Structural analysis of the bipartite DNA-binding domain of Tc3 transposase bound to transposon DNA. Nucleic Acids Res. 2004;32:4306–4312. doi: 10.1093/nar/gkh770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aggarwal AK, Rodgers DW, Drottar M, Ptashne M, Harrison SC. Recognition of a DNA operator by the repressor of phage 434: a view at high resolution. Science. 1988;242:899–907. doi: 10.1126/science.3187531. [DOI] [PubMed] [Google Scholar]
- 32.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 33.Segal E, Fondufe-Mittendorf Y, Chen L, Thåström A, Field Y, Moore IK, Wang JP, Widom J. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li S, Smerdon MJ. Nucleosome structure and repair of N-methylpurines in the GAL1-10 genes of Saccharomyces cerevisiae. J. Biol. Chem. 2006;277:44651–44659. doi: 10.1074/jbc.M206623200. [DOI] [PubMed] [Google Scholar]
- 35.Kaplan N, Moore I, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, Hughes TR, Lieb JD, Widom J, Segal E. Nucleosome sequence preferences influence in vivo nucleosome organization. Nat. Struct. Mol. Biol. 2010;17:918–920. doi: 10.1038/nsmb0810-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Moqtaderi Z, Rattner BP, Euskirchen G, Snyder M, Kadonaga JT, Liu XS, Struhl K. Reply to “Evidence against a genomic code for nucleosome positioning”. Nat. Struct. Mol. Biol. 2010;17:920–923. doi: 10.1038/nsmb0810-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee W, Tillo D, Bray N, Morse RH, Davis RH, Hughes RH, Nislow C. A high-resolution atlas of nucleosome occupancy in yeast. Nat. Genet. 2007;39:1235–1244. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- 38.Peckham HE, Thurman RE, Fu Y, Stamatoyannopoulos JA, Noble WS, Struhl K, Weng Z. Nucleosome positioning signals in genomic DNA. Genome Res. 2007;17:1170–1177. doi: 10.1101/gr.6101007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Møllegaard NE, Nielsen PE. Increased temperature and 2-methyl-2,4-pentanediol change the DNA structure of both curved and uncurved adenine/thymine-rich sequences. Biochemistry. 2003;42:8587–8593. doi: 10.1021/bi0343267. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.