


Abstract
The vast majority of microbes are unculturable and thus cannot be sequenced by means of traditional methods. High-throughput sequencing techniques like 454 or Solexa-Illumina make it possible to explore those microbes by studying whole natural microbial communities and analysing their biological diversity as well as the underlying metabolic pathways. Over the past few years, different methods have been developed for the taxonomic and functional characterization of metagenomic shotgun sequences. However, the taxonomic classification of metagenomic sequences from novel species without close homologue in the biological sequence databases poses a challenge due to the high number of wrong taxonomic predictions on lower taxonomic ranks. Here we present CARMA3, a new method for the taxonomic classification of assembled and unassembled metagenomic sequences that has been adapted to work with both BLAST and HMMER3 homology searches. We show that our method makes fewer wrong taxonomic predictions (at the same sensitivity) than other BLAST-based methods. CARMA3 is freely accessible via the web application WebCARMA from http://webcarma.cebitec.uni-bielefeld.de.
INTRODUCTION
The vast majority of microbes cannot be cultivated in a monoculture and thus cannot be sequenced by means of traditional methods. To explore these microbes, they have to be analysed within their natural microbial communities. The new high-throughput sequencing (HTS) technologies like Roche's 454-sequencing, ABI's SOLiD or Illumina's Genome Analyzer make it possible to sequence microbial DNA samples of such communities. Due to the restricted read lengths currently produced by the different HTS technologies, reconstruction of complete genomic sequences is too difficult. Though, by comparing the metagenomic fragments with sequences of known function, it is possible to analyse the biological diversity and the underlying metabolic pathways in microbial communities.
To infer the taxonomic origin of metagenomic reads, two kinds of methods, composition-based and comparison-based, can be distinguished. The composition-based methods extract sequence features like GC content or k-mer frequencies, and compare them with features computed from reference sequences with known taxonomic origin (1–5). A disadvantage is that short reads are not suited for this method as rather long reads are required to obtain a reasonable classification accuracy. The comparison-based methods, in contrast, rely on homology information obtained by database searches. They can be further subdivided into methods that are based on hidden markov model (HMM) homology searches (6) and those that are based on BLAST homology searches (7,8). CARMA (9) as well as WebCARMA (10), a refined version of CARMA available as a web application for the taxonomic and functional classification of metagenomic reads belong to the HMM-based methods.
For the taxonomic classification of metagenomic reads based on BLAST, different methods have been developed. Probably the most basic method is to use BLAST to search for the best hit in a database of sequences with known origin, for example used in MG-RAST (11). Since the evolutionary distance between the source organisms of the metagenomic fragment and the database sequence is unknown, a classification result solely based on a best BLAST hit has to be interpreted carefully. In general, such a classification is more reliable on higher taxonomic levels (e.g. superkingdom or phylum) than on lower taxonomic levels (e.g. genus or species), but it is difficult to decide which taxonomic level is reliable enough, as this strongly varies for each metagenomic fragment.
The program MEGAN (12) is based on the lowest common ancestor (LCA) approach. A BLAST search is performed and all BLAST hits that have a bit score close to the bit score of the best hit are collected. The metagenomic fragment is then classified by computing the LCA of all species in this set. One of the reasons for the improved classification accuracy of this approach is that fragments with ambiguous hits are assigned at higher taxonomic levels.
The SOrt-ITEMS (13) method extends the LCA approach and uses additional techniques to reduce the number of false positive predictions. One approach is the reduction of the number of hits by using a reciprocal BLAST search step. Another technique used is the adaptation of the taxonomic assignment level for all hits, based on different alignment parameters like sequence similarity between the metagenomic fragment and the aligned database sequence.
Inspired by these techniques, in particular the reciprocal search step of SOrt-ITEMS, we have developed a new algorithm that further improves the accuracy of the taxonomic classification. Our method makes explicit use of the assumption of a model of evolution where different gene families have different rates of mutation, but within each family this rate does not change too much. We have adapted our method to work with both, BLAST and HMMER3. In the ‘Materials and Methods’ section, we first introduce the BLAST-based variant of our method and then we detail the adaptations necessary for the HMMER variant. In the ‘Results and Discussion’ section, we conduct four experiments. In the first experiment we compare our BLASTx and HMMER variants with each other, and in the second experiment we compare our BLAST-based variant with SOrt-ITEMS and MEGAN. In the last two experiments we evaluate CARMA3 on different real metagenomic data sets.
MATERIALS AND METHODS
Definitions
For a given BLAST hit h, let q(h) be the aligned query sequence without gap and frameshift characters. In case of BLASTx, q(h) is a translated substring of the DNA query sequence. Similarly, s(h) is the substring of the database sequence used in the alignment of h. Furthermore, score(h) is the bit score of the alignment of h and tax(h) is the taxonomic assignment of the database sequence of h. Given two taxa a and b, lca(a, b) is the LCA of a and b. Let RANKS be the set of the taxonomic levels {unknown, superkingdom, phylum, class, order, family, genus, species}, with the underlying taxonomic ordering relation unknown > superkingdom > … > species. For a given taxon a, rank(a) is the taxonomic rank of taxon a. The lineage of some taxon a denotes the set of taxa on the path from the root to a in the taxonomy tree. For a given rank k, ancestor(k, a) defines the taxon at rank k in the lineage of a. In the rest of this section, let query q be an unassembled metagenomic read with unknown taxonomic affiliation.
Reciprocal search
The basic idea of using a reciprocal search in the context of the taxonomic classification of metagenomic reads as described in the following goes back to (13). The first step of our method is to use BLASTx to search for homologs of q in the NCBI NR protein database. BLASTx hits with taxonomic assignment Other or Unclassified and hits without any taxonomic assignment are discarded. Furthermore, hits that have bit scores or alignment lengths that are below certain thresholds, are also discarded. Let B = {h1, …, h|B|} be the set of BLAST hits of q, such that score(h1) ≥ … ≥ score(h|B|). If B is empty, then q is classified as Unknown. Otherwise, the next step is the construction of a new BLAST database consisting of {q(h1), s(h1),…, s(h|B|)}. Then, BLASTp is used to search for hits of s(h1) in the new database. The result of this reciprocal search is (rquery, R), where rquery denotes the hit obtained by the alignment between s(h1) and q(h1), and R = {r1,…, r|R|} denotes the set of hits with known taxonomic affiliation, such that score(r1) ≥ … ≥ score(r|R|). In addition we require that in case of co-optimal results with the same highest score r1 ∈ R denotes the hit obtained by the alignment of s(h1) with itself. Let x = tax(rquery) and ti = tax(ri) for all ri ∈ R. Determining x, the species of the metagenomic fragment, is usually not possible if the species has not been sequenced before. The purpose of this method is to approximate y = lca(x, t1), which is the best possible classification, assuming t1 is the phylogenetically closest known homolog of x. For each r ∈ R, p(r) = rank(lca(tax(r), t1)) denotes the projection of r onto the lineage of t1. For each k ∈ RANKS, let Pk = {r ∈ R ∣ p(r) = k}. If Pk ≠ ∅, let Pmink = min{score(r) ∣ r ∈ Pk} and Pmaxk = max{score(r) ∣ r ∈ Pk}, otherwise Pmink = Pmaxk = 0. Pmink and Pmaxk define intervals for each taxonomic rank k.
Figure 1a depicts an example with projections of phylogenetic affiliations t2, … ,t8 of reciprocal BLAST hits r2, … ,r8 onto the lineage of t1. Note that this tree is not a phylogenetic tree. For example, the species t8, t7 and t6 share a common ancestor at taxonomic level order with t1, but this is not necessarily the last common ancestor of t8, t7 and t6. The dashed edges represent the projections of the hitherto unknown phylogenetic affiliations x and x′ of metagenomic sequences q and q′, respectively.
Figure 1.

(a) Projections of BLAST hits obtained from reciprocal search onto the lineage of t1. The dashed edges represent projections of unkown phylogenetic affiliations x and x′ of metagenomic sequences q and q′, respectively. (b) Intervals given by Pmink and Pmaxk for each taxonomic rank k and level assignments of x and x′ based on their score.
Figure 1b shows intervals defined by Pmink and Pmaxk that were obtained from the reciprocal scores in Figure 1a. For example the species t8, t7 and t6 define the interval (50,75) at taxonomic rank order and species t4 and t2 define the interval (95,120) at taxonomic rank genus.
Polishing
Under ideal conditions, one would expect that reciprocal hits that are phylogenetically further away from t1 should also have a lower bitscore. Thus, one would expect that for each taxonomic rank k ∈ RANKS ∖ {unknown}, Pmaxk ≥ Pmaxk+1 holds. As this is not always the case for real data, Pmaxk is set to zero for all ranks k with Pmaxk < Pmaxk+1.
Values of Pmaxk that are zero, because there was no hit at this taxonomic rank or because they have been set to zero in the previous step, can be approximated by a linearly interpolated score if there exists at least one higher and one lower taxonomic rank for which Pmax is non-zero. Note that there always exists some lower taxonomic rank with Pmax ≠ 0, since r1 provides a lower bound at taxonomic rank species. Thus, if a higher taxonomic rank with Pmax ≠ 0 exists, the smallest rank kh > k with Pmaxkh ≠ 0 and the largest rank kl < k with Pmaxkl ≠ 0 are taken and used as anchors for the linear interpolation. If Pmink = 0, Pmink is set to Pmaxk. If no kh exists, an interpolation is not possible.
Classification
Another formulation of the best possible classification y = lca(x,t1) is y = ancestor(k,t1), assuming that rank k = rank(y) is given. Similarly, yapprox, an approximation of the best possible classification, can be obtained by ancestor(kapprox, t1) if rank kapprox is given. Therefore, the goal of our method is to find such an approximation kapprox. This step requires that there exists some reciprocal BLAST hit r ∈ R with score(r) ≤ score(rquery). If this is not the case, a fall-back method, which is described below, will be used. Otherwise, we obtain kapprox by min{k ∈ RANKS ∣ Pmink ≤ score(rquery) and for all l > k : Pmaxl < score(rquery)}. The algorithm for this works as follows: Starting at taxonomic rank k = unknown, k is decreased until Pmaxk−1 ≥ score(rquery). If k is above the taxonomic rank species and score(rquery) ≥ Pmink−1, then k will be decreased once again. The rank kapprox is then given by k.
Two examples for the taxonomic classification are given in Figure 1b. The metagenomic read q with unknown phylogenetic affiliation x has a reciprocal score of 90 and k is decreased until Pmaxk−1 ≥ 90. Since the interval at taxonomic rank genus contains a reciprocal hit (t2) with a score of 120 which is higher than that of q, k is set to rank family. Because the score of q is also smaller than the lowest score Pmink−1 of any reciprocal hit in the interval at rank genus, k remains at its last rank and kapprox is set to family. For the metagenomic read q′ with reciprocal score of 105, k is similarly placed at taxonomic rank family in the first phase, but in contrast to q its score is higher than the lowest score in the interval at taxonomic rank genus. Therefore, kapprox is set to genus for metagenomic read q′.
Fall-back
As mentioned before, the previous step will only work if there exists some reciprocal BLAST hit r ∈ R with score(r) ≤ score(rquery). If there is no such r, the highest taxonomic rank klow with Pklow ≠ ∅ will only provide a lower bound for the approximation of y. As a fall-back method for this case, the lower bound prediction klow will be combined with a technique introduced in (13) that is based on the assumption of a uniform rate of evolution. Different BLASTx alignment parameters, e.g. percent identity, are used to estimate the taxonomic rank of the LCA of the metagenomic sequence and the database sequence. A high similarity between both sequences will result in the estimation of a lower taxonomic rank and a lower similarity will result in a higher taxonomic rank, respectively. For example a metagenomic read with a BLAST hit h to some database sequence, with length(q(h)) = 200 bp and percent identity = 60, is assigned at the taxonomic rank family of the database sequence. In contrast, the same metagenomic read with an alignment with a percent identity of only 55 will be assigned at the higher taxonomic rank order, as it is assumed to be evolutionarily further away from the database sequence. The thresholds for the alignment parameters used in this method are the same as in SOrt-ITEMS (13). Let kuni be the taxonomic rank obtained by this technique using the alignment parameters of the best BLAST hit h1 from the initial BLAST search. Both predictions are combined by taking the maximum, i.e. kmax = max(klow, kuni). The final classification yapprox is then given by ancestor(kmax, t1).
HMMER variant
It is also possible to apply the same classification technique within the context of HMMER3-based homology searches against the Pfam database (14).
For convenience, some of the previous notations are reused. Let h be a pairwise alignment, q(h), s(h) and tax(h) are defined analogously. The value score(h) is given by computing a similarity score over the pairwise alignment with the BLOSUM62 score matrix (15). The first step is to translate all six reading frames of the metagenomic sequence into protein sequences and to search them against Pfam-A using hmmscan. If there is no significant match, the metagenomic sequence is classified as Unknown. Otherwise, let 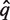 be the aligned sequence of the match with the lowest Pfam-HMM E-value. Then,
be the aligned sequence of the match with the lowest Pfam-HMM E-value. Then, 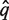 is aligned against the full multiple alignment of the Pfam family using hmmalign. Let q* be the alignment row corresponding to
is aligned against the full multiple alignment of the Pfam family using hmmalign. Let q* be the alignment row corresponding to 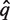 and let F = {f1,… f|F|} be the set of alignment rows of the Pfam family members of the full multiple alignment.
and let F = {f1,… f|F|} be the set of alignment rows of the Pfam family members of the full multiple alignment.
The next step is similar to the BLAST approach, where the closest homologue of the (translated) metagenomic sequence 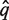 is searched for: For each pair in {(q*, f) ∣ f ∈ F}, a pairwise alignment is obtained where columns that correspond to leading and trailing gaps of q* as well as columns where both sequences have a gap are discarded. Pairwise alignments that are too short or have too low a score will not be considered for further processing.
is searched for: For each pair in {(q*, f) ∣ f ∈ F}, a pairwise alignment is obtained where columns that correspond to leading and trailing gaps of q* as well as columns where both sequences have a gap are discarded. Pairwise alignments that are too short or have too low a score will not be considered for further processing.
Let B = {h1,…, h|B|} be the set of all these pairwise alignments, such that score(h1) ≥ … ≥ score(h|B|). The reciprocal search is performed by computing the pairwise similarity between s(h1) and all other Pfam family members. The following steps, the creation of intervals and the classification are performed in the same way as for the BLAST variant. The alignment parameters that are needed for the fall-back method can easily be computed by counting the number of identities, positives and gaps in the alignment.
Since HMMER3 does not support DNA to Protein alignments yet, frameshifts cannot be detected directly. This decreases both, the sensitivity of homology detection and the classification accuracy. In order to incorporate frameshifts, it is possible to add to the default six reading frame translations the BLASTx-based translation q(h1) if available. In this case, seven translations, instead of six, are searched against Pfam-A.
Parameter p
Except for the homology search thresholds and the fall-back method, our classification algorithm is parameter-free. For evaluation and comparison purposes, we introduced a parameter p to trade off sensitivity against specificity of the taxonomic classification. It is used to artificially increase or decrease the score of the metagenomic sequence in the reciprocal phase, i.e. scorenew(rquery) = min(p·score(rquery), score(r1)). For example, values of p > 1 will increase sensitivity and decrease specificity of the classifications. The parameter is suited only for small changes in the sensitivity-specificity trade-off because the fall-back method is not effected by the parameter.
Taxonomic classification of amino acid sequences
Both, the BLAST and the HMMER variant of CARMA3 can also be used for the taxonomic classification of amino acid sequences. In the case of the BLAST variant of CARMA3, BLASTx is replaced by BLASTp. In the HMMER variant the amino acid sequences are now passed directly to HMMER3, in contrast to DNA that first requires translation into six reading frames.
Functional classification
An important feature of the HMMER variant is the functional classification of metagenomic reads based on Gene Ontology Identifiers (GO-Ids) (16). The Gene Ontology provides a controlled vocabulary for gene products, distinguishing between their associated biological processes, cellular components and molecular functions. A metagenomic sequence that has a significant match to some Pfam family can then be classified by the set of GO-Ids that are assigned to this Pfam family.
RESULTS AND DISCUSSION
Both, the BLAST and the HMMER variant of the method described above have been implemented in C/C++ as version 3 of our CARMA/WebCARMA pipeline. In the following, CARMA3BLASTx denotes the BLASTx variant and CARMA3HMMER3 denotes the HMMER3 variant. The pipeline takes metagenomic reads as input and sequentially starts BLASTx, CARMA3BLASTx, HMMER3 and CARMA3HMMER3. The resulting taxonomic and functional classifications are further processed to create taxonomic and functional profiles. The pipeline runs on the compute cluster of the Bielefeld University Bioinformatics Resource Facility at the Center for Biotechnology (CeBiTec) and is freely accessible at http://webcarma.cebitec.uni-bielefeld.de (10). The complete source code (C/C++) has been released under the GPL and is available for download at the WebCARMA homepage.
Compared methods
In the first experiment CARMA3BLASTx, CARMA3HMMER3 and their predecessor CARMA2.1HMMER2 have been compared to each other, in the second experiment CARMA3BLASTx, SOrt-ITEMS (13) and MEGAN (12).
The taxonomic classification methods assign to a metagenomic read one taxon and therefore also one taxonomic rank. This taxon implicitly provides a taxonomic classification also for the higher taxonomic ranks. For example, the taxon Gammaproteobacteria at the taxonomic rank class, implicitly provides the taxonomic classification Bacteria at the taxonomic rank superkingdom. The taxonomic ranks below the predicted taxon can be considered to be classified as ‘unknown’. Therefore, for each taxonomic rank a metagenomic read can either be correctly classified and counts as a true positive (TP), can be wrongly classified and counts as a false positive (FP), or it is not classified and counts as unknown (U). As for each taxonomic rank the numbers TP, FP and U sum up to the total number N of reads used in the evaluation and U equals N − TP − FP, U will not explicitly be given in the results.
Metagenomes
For the evaluation of CARMA3 a synthetic and two real data sets were used. The synthetic metagenome (Supplementary Table S1) was constructed consisting of 25 randomly chosen bacterial genomes from the NCBI ftp site (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/). N = 25 000 metagenomic reads were simulated using MetaSim (17) with the default 454 sequencing error model resulting in an average read length of 265 bp. The real data set used in Experiment 3 consists of over 600 000 unassembled reads from a biogas plant microbial community (18). The reads were obtained by 454 sequencing and have an average length of 230 bp. The real data set used in Experiment 4 consists of 3.3 million non-redundant microbial genes of the gene catalogue of the human gut microbiome (19). Faecal samples from different individuals were sequenced with the Illumina Genome Analyser (GA) which yielded in 576.7 Gb of sequence. The reads were assembled into longer contigs and a gene finder was used to detect open reading frames (ORFs). Similar ORFs were clustered to obtain the final non-redundant gene set. We downloaded this gene set and translated the ORFs into protein sequences using the NCBI Genetic Code 11. The simulated metagenomes and the results of the CARMA3 analyses of the real metagenomes used in the evaluation are available for download at the WebCARMA homepage.
Databases
To evaluate the different BLAST-based methods regarding their ability to classify sequences of unknown source organism, three BLAST NR protein databases were created: ‘order-filtered’, without sequences from species that share the same order as any of the species from the synthetic metagenome, ‘species-filtered’, without sequences from species in the synthetic metagenome, and ‘All’, the complete NR database.
Similarly, for CARMA3HMMER3, the curated Pfam-A database from Pfam 24.0 was used to create the three databases, ‘order-filtered’, ‘species-filtered’ and ‘All’, by removing corresponding sequences from the full multiple alignments.
Parameters used
The BLASTx runs for CARMA3BLASTx, SOrt-ITEMS and MEGAN were performed with default E-value threshold (-e 10), soft sequence masking (-F “m S”), and frameshift penalty 15 (-w 15). To ensure comparability, CARMA3BLASTx used the same thresholds as SOrt-ITEMS regarding the BLASTx hits, a minimal bit score of 35 and a minimal alignment length of 25. For our first experiment, the CARMA3 parameter p was set to 1. For the second experiment, p was set differently for each of the three databases, since for p = 1, CARMA3BLASTx has fewer TPs and fewer FPs than SOrt-ITEMS (except for taxonomic rank superkingdom). In order to be comparable, p was chosen for the order and the species-filtered databases such that CARMA3BLASTx had about the same number of TPs as SOrt-ITEMS on the lowest taxonomic rank that had not been filtered. For the unfiltered database (all), SOrt-ITEMS gave no classifications on the taxonomic rank species. Therefore p was chosen with respect to the taxonomic rank genus. The values of p were 1.024 for order-filtered, 1.033 for species-filtered and 1.15 for the unfiltered database.
The parameter for the minimal number of reads that are required to report a taxon in SOrt-ITEMS and MEGAN was set to 1 in all experiments. To ensure comparability of MEGAN with the other two BLAST-based methods, the top percent parameter was increased from 10 (default) to 15 resulting in more conservative predictions.
CARMA3HMMER3 was run with an E-value of 0.1 for hmmscan, a minimal alignment length of 25 and a minimal score of 30 for the pairwise alignments. CARMA2.1HMMER2 was run with an E-value of 0.0001 for hmmpfam.
Experiment 1
In the first experiment CARMA3BLASTx and CARMA3HMMER3 were compared with each other in order to see which of both variants provides better taxonomic classification results (Table 1). As a third variant the older version CARMA2.1HMMER2 was also included in the comparison.
Table 1.
Comparison of the taxonomic classification accuracy of the different CARMA variants CARMA3BLASTx, CARMA3HMMER3 and CARMA2.1HMMER2
| Order-filtered |
Species-filtered |
All |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C3BLASTx |
C3HMMER3 |
C3BLASTx |
C3HMMER3 |
C3BLASTx |
C3HMMER3 |
C2.1HMMER2 |
||||||||
| TP | FP | TP | FP | TP | FP | TP | FP | TP | FP | TP | FP | TP | FP | |
| Superkingdom | 12282 | 799 | 6668 | 660 | 20059 | 113 | 9563 | 516 | 22725 | 31 | 11276 | 544 | 6099 | 140 |
| Phylum | 8532 | 1094 | 4194 | 657 | 18968 | 183 | 8065 | 377 | 22626 | 17 | 10255 | 345 | 5724 | 238 |
| Class | 3700 | 1257 | 1983 | 721 | 15793 | 274 | 6329 | 322 | 20584 | 25 | 8822 | 223 | 4969 | 278 |
| Order | – | 2019 | – | 1158 | 14829 | 275 | 5084 | 367 | 20869 | 30 | 8066 | 220 | 4756 | 385 |
| Family | – | 926 | – | 531 | 11126 | 239 | 3400 | 324 | 18301 | 25 | 6485 | 223 | 4149 | 346 |
| Genus | – | 144 | – | 175 | 6897 | 427 | 1852 | 517 | 16025 | 107 | 5366 | 303 | 3487 | 746 |
| Species | – | 9 | – | 25 | – | 142 | – | 214 | 1142 | 31 | 809 | 176 | 2092 | 1135 |
Table entries ‘–’ indicate taxonomic ranks where the corresponding species have been filtered away and therefore true positives are not possible.
For the order-filtered database, CARMA3BLASTx has more TPs but also more FPs than CARMA3HMMER3 at all taxonomic ranks. In the species-filtered database, CARMA3BLASTx has more TPs than CARMA3HMMER3 as before, but this time it also has fewer FPs than CARMA3HMMER3. Similar results are provided for the Unfiltered database: CARMA3BLASTx has significantly more TPs and at the same time considerably fewer FPs than CARMA3HMMER3 at all taxonomic ranks. While for the order-filtered database it is not obvious which variant should be preferred over the other, for the species-filtered and unfiltered databases CARMA3BLASTx clearly outperforms CARMA3HMMER3.
The comparison of CARMA3HMMER3 and CARMA2.1HMMER2 using the unfiltered database shows that CARMA3HMMER3 is superior to CARMA2.1HMMER2 on all taxonomic ranks from class to genus.
Fraction of fall-back method on the overall classification
About 10–20% of all metagenomic reads that have been classified with CARMA3BLASTx were classified using the fall-back method (see Table 2). Of these, about one half of the reads were classified with the fall-back method because they had only one BLAST hit in the corresponding database.
Table 2.
The total number of reads (‘total’) classified with CARMA3BLASTx and the number of reads classified with the fall-back method (‘fall-back’)
| Total | Fall-back | Fall-back |
||
|---|---|---|---|---|
| Single | Multiple | |||
| Order-filtered | 13081 | 2668 | 1397 | 1271 |
| Species-filtered | 20172 | 1907 | 878 | 1029 |
| All | 22756 | 2203 | 1159 | 1044 |
‘Single’ represents the number of metagenomic reads that had only one BLAST hit and ‘Multiple’ represents the number of reads with two or more BLAST hits.
Performance on different read lengths
The performance of CARMA3BLASTx was also evaluated for other read lengths and different error models. To simulate a metagenome sequenced with 454 GS FLX Titanium, reads were created with the default 454 error model of MetaSim with an average read length of 400 bp. For the 454-GS20 and Illumina sequencing technology, reads of length 80 bp were simulated. The error model for the Illumina reads (errormodel-80bp.mconf) was downloaded separately from the MetaSim homepage. As no error model for Illumina reads longer than 80 bp was available, the 454-GS20 reads were adapted to this length. Each of the three simulated metagenomes (454-400 bp, 454-80 bp and Illumina-80 bp) was analysed using the order-, species- and unfiltered protein databases. The results are given in Supplementary Tables S2–4.
In general, the 400 bp reads provide more classifications than the 265 bp. In addition, in many cases the 400 bp reads account for more TPs and fewer FPs than the 265 bp reads. This is the case in the species-filtered database at taxonomic ranks class to family, but also for the unfiltered database at taxonomic ranks superkingdom, family and genus. As expected, the shorter 454-80 bp reads perform worse than the 454-265 bp reads. This is clearly shown for the species-filtered database at taxonomic rank family and the unfiltered database at taxonomic ranks phylum to family.
The comparison of the 454-80 bp and Illumina-80 bp reads shows that Illumina reads are about twice as often classified as the 454 reads for all databases. For the species-filtered database at taxonomic rank superkingdom and the unfiltered database at taxonomic ranks superkingdom to genus the Illumina error model clearly outperfoms the 454 error model in terms of accuracy. A comparison of the simulated reads revealed that the 454 error model has produced many more base substitutions than the Illumina error model. In addition, the 454 error model accounts for insertions and deletions, which the Illumina error model does not. It is unclear to the authors how representative the MetaSim default error models are for the currently available sequencers by 454 and Illumina. Therefore, rather than as a comparison of two different sequencing technologies, the comparison of both error models should be understood as a demonstration of the influence of sequencing errors on the accuracy of the taxonomic classification.
Experiment 2
In the second experiment our new method CARMA3BLASTx was compared to the two other BLASTx-based methods, SOrt-ITEMS and MEGAN (Table 3).
Table 3.
Comparison of the taxonomic classification accuracy of the different BLASTx-based methods CARMA3BLASTx, SOrt-ITEMS and MEGAN
| Order-filtered |
Species-filtered |
All |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CARMA3 |
SOrt-ITEMS |
MEGAN |
CARMA3 |
SOrt-ITEMS |
MEGAN |
CARMA3 |
SOrt-ITEMS |
MEGAN |
||||||||||
| TP | FP | TP | FP | TP | FP | TP | FP | TP | FP | TP | FP | TP | FP | TP | FP | TP | FP | |
| Superkingdom | 12696 | 861 | 12576 | 786 | 12626 | 1849 | 20266 | 118 | 20345 | 128 | 20840 | 453 | 22890 | 36 | 23979 | 30 | 23900 | 105 |
| Phylum | 8989 | 1224 | 9254 | 1736 | 8079 | 1985 | 19268 | 227 | 19466 | 356 | 19010 | 535 | 22832 | 30 | 23909 | 43 | 23607 | 91 |
| Class | 4066 | 1495 | 4062 | 1937 | 3649 | 2479 | 16206 | 349 | 16259 | 401 | 15921 | 735 | 20932 | 38 | 21912 | 41 | 21418 | 107 |
| Order | – | 2507 | – | 4011 | – | 4975 | 15671 | 366 | 15684 | 535 | 15105 | 954 | 21994 | 65 | 22871 | 58 | 21543 | 155 |
| Family | – | 1186 | – | 2565 | – | 4087 | 12117 | 345 | 13104 | 606 | 11625 | 1101 | 20089 | 62 | 20864 | 59 | 18937 | 143 |
| Genus | – | 210 | – | 798 | – | 4041 | 8328 | 752 | 8299 | 1112 | 8031 | 1889 | 20430 | 314 | 21124 | 483 | 17758 | 263 |
| Species | – | 23 | – | 0 | – | 3544 | – | 995 | – | 0 | – | 4346 | 15232 | 685 | 0 | 0 | 11786 | 550 |
In the table CARMA3 refers to the BLAST variant CARMA3BLASTx.
While for the order-filtered database CARMA3 performs better than SOrt-ITEMS at rank class, for the ranks superkingdom and phylum it is not clear which method is better. At the taxonomic ranks order to genus, where the metagenomic sequences have been filtered away, CARMA3 has much fewer (∼37–74%) false positives than SOrt-ITEMS. CARMA3 has better results than MEGAN at all taxonomic ranks, while SOrt-ITEMS has better results than MEGAN at all taxonomic ranks below superkingdom. For the species-filtered database CARMA3 has better results than SOrt-ITEMS and MEGAN at taxonomic rank genus. For the other taxonomic ranks the results of CARMA3 and SOrt-ITEMS are not comparable, since SOrt-ITEMS has more TPs and more FPs. Only at taxonomic rank species CARMA3 has FPs which SOrt-ITEMS does not have. The reason for this is that SOrt-ITEMS requires a minimal alignment length of 550 bp in order to make classifications at the taxonomic rank species, but the simulated metagenome contains only reads with an average length of 265 bp. The advantage of avoiding FPs at rank species in the order and species-filtered databases is traded off against the disadvantage of not detecting species in the unfiltered database. CARMA3 performs better than MEGAN at all taxonomic ranks, except superkingdom, where the results are not comparable. To provide comparability between the methods also for the unfiltered database we tried to increase the number of TPs of CARMA3 at the taxonomic rank genus. We were able to increase the number of TPs by 4 405 from 16 025 (Table 1) to 20 430, but not higher. The reason for this is that classifications of reads from the fall-back method cannot be changed with the parameter p. Although CARMA3 performs worse than SOrt-ITEMS at three taxonomic ranks (superkingdom, order and family) in the unfiltered data set, the corresponding TP and FP numbers at each taxonomic rank except species are quite similar. CARMA3 is able to detect many species where SOrt-ITEMS does not detect any. On all taxonomic ranks, except ranks genus and species, CARMA3 and SOrt-ITEMS perform better than MEGAN.
Assuming that about 10%–20% of microbial genomes consist of non-protein coding sequences (20), it is clear that many of the metagenomic reads can not be classified using protein homology information. But because many of these reads do overlap at least partly with a coding region, it can be observed that 92–96% of the reads are correctly assigned to bacteria by the BLASTx based methods using the unfiltered database.
Overlap
Figure 2 shows Venn diagrams for the overlap of (a) correct and (b) wrong classifications for the order-filtered data set at taxonomic rank class. Although each method has about 3 600–4 000 correct classifications, only about 2 100 reads have been correctly classified by every method. In this particular case each of the three compared methods correctly classifies a significant proportion of the reads, which the other methods do not. However, for higher taxonomic ranks and the species- and unfiltered data set the overlap of correct classifications is much higher and therefore the differences between the methods are smaller. Figure 2(b) shows that the overlap of wrong classifications is relatively smaller than that of the correct classifications. As expected, a high number of wrong classifications are unique to MEGAN. For the Venn diagrams of the other taxonomic ranks and data sets see Supplementary Figures S1–6.
Figure 2.

Overlap of 25 000 simulated metagenomic reads classified by CARMA3, SOrt-ITEMS and MEGAN for the order-filtered data set at taxonomic rank class.
Experiment 3
For the evaluation on a real data set of unassembled 454 reads, the metagenome of a biogas plant microbial community was analysed with CARMA3BLASTx, SOrt-ITEMS and MEGAN. Figure 3 shows Venn diagrams for the number of reads being classified at taxonomic ranks superkingdom and class (see Supplementary Figure S7 for the other ranks). Reads that are classified by two or all methods are not necessarily assigned to the same taxon. The Venn diagrams show that the fraction of reads that are classified by all methods is bigger at higher taxonomic ranks than on lower taxonomic ranks. For a qualitative comparison of the taxonomic classifications of the three methods, comparative taxonomic profiles for each taxonomic rank have been created. Figure 4 shows the profile for taxonomic rank class, Supplementary Figures S8–14 contain the full set of profiles. In order to restrict the number of taxa in the taxonomic profiles to the most abundant ones, all taxa with a relative abundance <0.01 were discarded. Taxa for which any of the classification methods predicted an abundance of 0.01 or higher were not discarded. After this threshold was applied, the remaining taxa were normalized such that the relative abundances sum up to one for each of the methods ensuring comparability between the methods. In contrast to the profiles of the other taxonomic ranks, the profile of taxonomic rank superkingdom includes the relative abundance of reads that have been classified as ‘unknown’.
Figure 3.

Venn diagrams for a biogas plant metagenome with over 600 000 reads. The subset sizes depict the numbers of reads being classified with CARMA3, SOrt-ITEMS and MEGAN at taxonomic ranks superkingdom and class.
Figure 4.

Comparative taxonomic profile of a biogas plant metagenome analysed with CARMA3, SOrt-ITEMS and MEGAN at taxonomic rank class.
The comparative taxonomic profiles reveal a strong consistency between the compared methods regarding the relative abundances of the most abundant taxa. Only at taxonomic ranks genus and species, bigger differences can be found: CARMA3 predicts more Clostridia, SOrt-ITEMS more Methanocullei and MEGAN predicts more Cloacamonas. The reason for the high consistency between the three methods above taxonomic rank genus is that low abundant species have been filtered away. Filtering of low abundant taxa provides a trade-off between filtering noise produced by FPs and the detection of low abundant true positive taxa. Supplementary Table S5 shows how many reads of each method have been filtered away. For example at taxonomic rank order, about 7% of all reads classified by CARMA3, 11% of all reads classified by SOrt-ITEMS and about 28% of all reads classified by MEGAN have been filtered away. This effect and the differences between the methods are even stronger at lower taxonomic ranks. The results of the evaluation in Experiment 2, showing that SOrt-ITEMS and in particular MEGAN produce more FPs than CARMA3, are an indication that most of the filtered taxa in this data set are actually wrong predictions rather than truly low abundant taxa.
This biogas plant metagenome has formerly been analysed using two different approaches, (a) construction of bacterial and archaeal 16S-rDNA amplicon libraries and (b) screening for reads in the 454 data set that encode for 16S-rDNAs (21). Both 16S-rDNA approaches and our results coincide in the identification of the main abundant taxa. For example at taxonomic rank order, the archaea Methanomicrobiales and the bacteria Clostridiales and Bacteroidales have by all approaches been predicted as the main abundant taxa. Apart from these consistent predictions the differences in the relative abundances of the other taxa might also be explained by various biases that are inherent to the compared methods. For example, the database reference sequences come mainly from culturable species and therefore are biased towards certain bacterial phyla (22). On the other side, the oligonucleotide primers that are used to amplify the 16S-rDNA can exhibit substantial variations in their specificity towards different clades (23). Considering these potential biases, the taxonomic classifications of the BLASTx-based methods show a high consistency with the results of the 16S-rDNA analyses.
Running times
To determine the running time of our method 10 000 metagenomic reads from the biogas plant metagenome with the complete CARMA3 pipeline were analysed. For comparative purposes, the running times of SOrt-ITEMS and MEGAN were also measured. The computation was conducted on a 2.5 GHz Intel Core 2 Duo processor with 8 GB RAM, running Linux (64-bit Ubuntu 10.04, kernel version 2.6.35.23). The observed running times, measured with the GNU time command (user+sys), are given in Table 4.
Table 4.
Running times for the homology searches (BLASTx and HMMER3) and the taxonomic classifications with CARMA3, SOrt-ITEMS and MEGAN
| CARMA3 | SOrt-ITEMS | MEGAN | |
|---|---|---|---|
| BLASTx | 54 h 15 m | 54 h 15 m | 54 h 15 m |
| -classification | 52 m 22 s | 12 m 36 s | 3 m 4 s |
| HMMER3 | 6 h 20 m | – | – |
| -classification | 41 m 8 s | – | – |
The results show that for the BLAST-based classifications, the BLAST homology search accounts for more than 98% of the total running time. Among the three BLAST-based classification methods, MEGAN is the fastest method, more than 4 times faster than SOrt-ITEMS. SOrt-ITEMS in turn is about 4 times faster than CARMA3BLASTx. In contrast to MEGAN, CARMA3BLASTx and SOrt-ITEMS spend additional time on performing reciprocal BLAST searches and therefore are slower. CARMA3BLASTx is slower than SOrt-ITEMS because it does not use a top-percent filter and therefore creates bigger BLAST databases in the reciprocal search step.
To measure the time needed to run BLASTx on shorter Illumina reads, 10 000 75 bp-reads sequenced with Illumina Genome Analyser (GA) from a human gut microbial community (24) were searched against the full NR protein database. The running time of about 14.5 h for the BLASTx run is in terms of bases per second quite similar to the running time of the BLASTx analysis of the 454 data. While a BLASTx analysis of a complete 454 run is feasible on a compute cluster in the order of hours or a few days, this approach seems to be less practical for the analysis of all unassembled reads produced by a complete run of an Illumina sequencing machine that produces one to two orders of magnitudes more bases in total than a 454 sequencing machine in a single run. The usage of data reduction techniques, as shown in Experiment 4, can be a way to overcome this limitation.
Experiment 4
Data reduction techniques are a common method to handle the amount of data produced by Illumina sequencing machines (24,25). Typical steps involve the assembly of reads into longer fragments, gene detection with a gene finder to detect open reading frames (ORFs), clustering of highly similar ORFs, and translation of the non-redundant ORFs into protein sequences. Such a metaproteome has, in contrast to the full set of unassembled Illumina reads, a size that makes the analysis with the BLASTp variant of CARMA3 possible on a compute cluster in the order of hours or a few days.
To evaluate the applicability of CARMA3 on amino acid sequences derived from assembled Illumina reads, the BLASTp variant of CARMA3 was used to analyse the gene catalogue of the human gut microbiome (24) (Supplementary Figures S15–21). The results were compared to the taxonomic classification of another study of the human intestinal microbial flora based on 13 355 prokaryotic 16S ribosomal RNA gene sequences (19).
Both methods, the 16S-rDNA analysis and CARMA3, identify Firmicutes and Bacteroidetes as the most abundant phyla, followed by Proteobacteria, Actinobacteria, Verrucomicrobia and Fusobacteria. Also, in both analyses the phylum Firmicutes consists mainly of the class Clostrida. Nearly all genera of the Clostridia that have been predicted by the 16S-rDNA analysis, like Eubacterium, Ruminococcus, Dorea, Butyrivibrio and Coprococcus, have also been predicted by CARMA3 (Supplementary Figure S22). Also most of the species of Clostridia like E. rectale, E. hallii, R. torques, R. gnavus, F. prausnitzii, D. formicigenerans and D. longicatena that are found by the 16S-rDNA analysis could be confirmed by CARMA3 (Supplementary Figure S23). However, the species E. hadrum and R. callidus that have been found by 16S-rDNA were not found by CARMA3. The genus Clostridium which is the taxon found by CARMA3 to have the highest abundance in the class Clostrida is not reported by the 16S-rDNA analysis. The reason for this might be that the 16S-rDNA sequence of Clostridium bartlettii, which mostly contributes to the genus Clostridium and is known to be found in human faeces, might not have been available at the time of the 16S-rDNA analysis (26). Also the species R. inulinivorans and R. intestinalis of the genus Roseburia, which are found by CARMA3 but not by the 16S-rDNA analysis, are known to occur in human faeces (27,28). For the second most abundant phylum, the Bacteroidetes, the authors of the 16S-rDNA analysis report a high variability in the distribution of phylotypes in samples from different subjects. Nevertheless, all phylotypes reported by the authors of the 16S-rDNA analysis, B. vulgatus, Prevotellaceae, B. thetaiotaomicron, B. caccae and B. fragilis, were among the 11 or, in case of B. putredinis, among the 22 most abundant taxa predicted by CARMA3 (Supplementary Figures S24 and 25).
The comparison of the taxonomic predictions of the 16S-rDNA analysis and CARMA3 has revealed a high consistency in the results of both methods. This shows that CARMA3 can also be used for the taxonomic classification of amino acid sequences obtained from assembled Illumina reads.
CONCLUSION
We have introduced a new method for the taxonomic classification of assembled and unassembled metagenomic sequences that can be used in combination with BLAST- and HMMER-based homology searches. Except for the homology search and the fall-back scenario, our method is parameter-free. In addition, for the HMMER-based variant, our method also provides a functional classification of the metagenomic sequence. Typically, a metagenomic sample contains many novel species that have not been sequenced before. We have simulated such a scenario with the order-filtered database and have shown that in most cases CARMA3 not only performs better than existing BLAST-based methods, but most strikingly, it is better at avoiding FP predictions on lower taxonomic ranks when only remote homologues are available for the classification of novel species.
We think that our method works because reciprocal hits provide a reasonable estimation of the last common ancestor of the metagenomic sequence and its best hit in the sequence database. In contrast to the other BLAST-based methods our method is not based on the LCA and therefore does not discard reciprocal hits that can provide valuable information for the taxonomic classification.
CARMA3 uses both BLAST and HMMER3 for the taxonomic classification of metagenomic reads. One of the reasons we developed the HMMER3 variant was the idea that we could improve the speed of the reciprocal search by first finding the corresponding protein family with HMMER3 and then restrict the search of reciprocal hits to this smaller set of sequences from the same family. Indeed, for the future, we plan to further increase the speed of CARMA3HMMER3 by using BLASTp to search for the reciprocal hits within the protein family instead of computing the pairwise alignments for every Pfam family member. However, in nearly all cases the BLASTx-based variant classified significantly more reads than the HMMER3-based variant. In many cases it also had fewer FPs. Therefore, we think that the BLASTx-based variant is in our current setting the preferable method for the taxonomic classification. A drawback of using BLASTx is its running time. The computational bottleneck of the CARMA3 pipeline is the homology search, in particular the BLAST search. In our evaluation the initial BLAST search accounted for over 98% of the total running time. However, this is a problem shared with all BLAST-based approaches. Furthermore, we have shown in our evaluation that this problem can be dealt with by the use of data reduction strategies which include assembly and gene detection steps. One of the reasons that the HMMER3-based variant does not perform as well as the BLASTx-based variant might be that the Pfam-A database contains less sequence information than the NR protein database. In our evaluation the NR protein database contained 3.55 billion amino acids while Pfam-A contained only 0.77 billion amino acids. The Pfam database also provides multiple alignments that have been created by aligning NCBI GenPept sequences (29) against Pfam-A. As this additional sequence information might increase the classification accuracy we are planning to incorporate these alignments into the HMMER-based variant of CARMA3. Also, we are considering including the Pfam-B database in the homology search as this should increase the fraction of metagenomic reads being classified.
Currently available biological sequence databases are known to be biased because they mainly contain sequences of species that are culturable. Although we have tried to minimize the effect of this bias on the results of our evaluation by creating the order-filtered database, this bias has to be kept in mind when generalizing our evaluation results to metagenomic reads from unculturable species.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
DFG Graduiertenkolleg 635 Bioinformatik (to W.G.). Funding for open access charge: Bielefeld University.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Abe T, Sugawara H, Kinouchi M, Kanaya S, Ikemura T. Novel phylogenetic studies of genomic sequence fragments derived from uncultured microbe mixtures in environmental and clinical samples. DNA Res. 2005;12:281–290. doi: 10.1093/dnares/dsi015. [DOI] [PubMed] [Google Scholar]
- 2.Diaz NN, Krause L, Goesmann A, Niehaus K, Nattkemper TW. TACOA: taxonomic classification of environmental genomic fragments using a kernelized nearest neighbor approach. BMC Bioinformatics. 2009;10:56. doi: 10.1186/1471-2105-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karlin S, Mrázek J, Campbell AM. Compositional biases of bacterial genomes and evolutionary implications. J. Bacteriol. 1997;179:3899–3913. doi: 10.1128/jb.179.12.3899-3913.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McHardy AC, Martín HG, Tsirigos A, Hugenholtz P, Rigoutsos I. Accurate phylogenetic classification of variable-length dna fragments. Nat. Methods. 2007;4:63–72. doi: 10.1038/nmeth976. [DOI] [PubMed] [Google Scholar]
- 5.Teeling H, Waldmann J, Lombardot T, Bauer M, Glöckner FO. TETRA: a web-service and a stand-alone program for the analysis and comparison of tetranucleotide usage patterns in dna sequences. BMC Bioinformatics. 2004;5:163. doi: 10.1186/1471-2105-5-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eddy SR. Profile hidden markov models (review) Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 7.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 8.Gish W, States DJ. Identification of protein coding regions by database similarity search. Nat. Genet. 1993;3:266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- 9.Krause L, Diaz NN, Goesmann A, Kelley S, Nattkemper TW, Rohwer F, Edwards RA, Stoye J. Phylogenetic classification of short environmental dna fragments. Nucleic Acids Res. 2008;36:2230–2239. doi: 10.1093/nar/gkn038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerlach W, Jünemann S, Tille F, Goesmann A, Stoye J. WebCARMA: a web application for the functional and taxonomic classification of unassembled metagenomic reads. BMC Bioinformatics. 2009;10:430. doi: 10.1186/1471-2105-10-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haque MM, Ghosh TS, Komanduri D, Mande SS. SOrt-ITEMS: Sequence orthology based approach for improved taxonomic estimation of metagenomic sequences. Bioinformatics. 2009;25:1722–1730. doi: 10.1093/bioinformatics/btp317. [DOI] [PubMed] [Google Scholar]
- 14.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, et al. The pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology the gene ontology consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richter DC, Ott F, Auch AF, Schmid R, Huson DH. Metasim: a sequencing simulator for genomics and metagenomics. PLoS ONE. 2008;3:e3373. doi: 10.1371/journal.pone.0003373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlüter A, Bekel T, Diaz NN, Dondrup M, Eichenlaub R, Gartemann K-H, Krahn I, Krause L, Krömeke H, Kruse O, et al. The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. J. Biotechnol. 2008;136:77–90. doi: 10.1016/j.jbiotec.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 19.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rogozin IB, Makarova KS, Natale DA, Spiridonov AN, Tatusov RL, Wolf YI, Yin J, Koonin EV. Congruent evolution of different classes of non-coding dna in prokaryotic genomes. Nucleic Acids Res. 2002;30:4264–4271. doi: 10.1093/nar/gkf549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kröber M, Bekel T, Diaz NN, Goesmann A, Jaenicke S, Krause L, Miller D, Runte KJ, Viehöver P, Pühler A, et al. Phylogenetic characterization of a biogas plant microbial community integrating clone library 16S-rDNA sequences and metagenome sequence data obtained by 454-pyrosequencing. J. Biotechnol. 2009;142:38–49. doi: 10.1016/j.jbiotec.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 22.Hugenholtz P. Exploring prokaryotic diversity in the genomic era. Genome Biol. 2002;3:REVIEWS0003. doi: 10.1186/gb-2002-3-2-reviews0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baker GC, Cowan DA. 16S rDNA primers and the unbiased assessment of thermophile diversity. Biochem. Soc. Trans. 2004;32(Pt 2):218–221. doi: 10.1042/bst0320218. [DOI] [PubMed] [Google Scholar]
- 24.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hess M, Sczyrba A, Egan R, Kim T-WW, Chokhawala H, Schroth G, Luo S, Clark DS, Chen F, Zhang T, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–467. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- 26.Song YL, Liu CX, McTeague M, Summanen P, Finegold SM. Clostridium bartlettii sp. nov., isolated from human faeces. Anaerobe. 2004;10:179–184. doi: 10.1016/j.anaerobe.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Duncan SH, Hold GL, Barcenilla A, Stewart CS, Flint HJ. Roseburia intestinalis sp. nov., a novel saccharolytic, butyrate-producing bacterium from human faeces. Int. J. Syst. Evol. Microbiol. 2002;52(Pt 5):1615–1620. doi: 10.1099/00207713-52-5-1615. [DOI] [PubMed] [Google Scholar]
- 28.Scott KP, Martin JC, Chassard C, Clerget M, Potrykus J, Campbell G, Mayer C-D, Young P, Rucklidge G, Ramsay AG, et al. Microbes and health sackler colloquium: substrate-driven gene expression in roseburia inulinivorans: Importance of inducible enzymes in the utilization of inulin and starch. Proc. Natl Acad. Sci. USA. 2010;108:4672–4679. doi: 10.1073/pnas.1000091107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2010;38:D5–D16. doi: 10.1093/nar/gkp967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.