


Abstract
Taxonomy-independent analysis plays an essential role in microbial community analysis. Hierarchical clustering is one of the most widely employed approaches to finding operational taxonomic units, the basis for many downstream analyses. Most existing algorithms have quadratic space and computational complexities, and thus can be used only for small or medium-scale problems. We propose a new online learning-based algorithm that simultaneously addresses the space and computational issues of prior work. The basic idea is to partition a sequence space into a set of subspaces using a partition tree constructed using a pseudometric, then recursively refine a clustering structure in these subspaces. The technique relies on new methods for fast closest-pair searching and efficient dynamic insertion and deletion of tree nodes. To avoid exhaustive computation of pairwise distances between clusters, we represent each cluster of sequences as a probabilistic sequence, and define a set of operations to align these probabilistic sequences and compute genetic distances between them. We present analyses of space and computational complexity, and demonstrate the effectiveness of our new algorithm using a human gut microbiota data set with over one million sequences. The new algorithm exhibits a quasilinear time and space complexity comparable to greedy heuristic clustering algorithms, while achieving a similar accuracy to the standard hierarchical clustering algorithm.
INTRODUCTION
Microbes play an essential role in processes as diverse as human health and biogeochemical activities critical to life in all environments on earth. The descriptions of complex microbial communities, however, remain poorly characterized. Currently available pyrosequencing technologies easily and inexpensively determine millions of signature sequences in a matter of hours. However, analyzing such massive nucleotide sequence collections can overwhelm existing computational resources and analytic methods, and consequently new computational algorithms are urgently needed (1).
Providing a detailed description of microbial populations, including high, medium and low abundance components, is typically the first step in microbial community analysis (2,3). PCR amplification of the 16S rRNA gene, followed by DNA sequencing, is now a standard approach to studying microbial community dynamics at high resolution (4–8). Existing algorithms for microbial classification using 16S rRNA sequences can be generally categorized into taxonomy-dependent or -independent analyses (9). In the former methods, query sequences are first compared against a database and then assigned to the organism of the best-matched reference sequences [e.g. BLAST (10)]. Since most microbes have not been formally described yet, these methods are inherently limited by the completeness of reference databases (9). In contrast, taxonomy-independent analysis compares query sequences against each other to form a distance matrix followed by clustering analysis to group sequences into operational taxonomic units (OTUs) at a specified level of sequence similarity (e.g. sequences grouped at 97% identity are often used as proxies for bacterial species). Various ecological metrics can then be estimated from the clustered sequences to characterize a microbial community. This analysis does not rely on any reference database, and can thus enumerate novel pathogenic and uncultured microbes as well as known organisms. In addition to microbial diversity estimation, there is currently increased interest in applying taxonomy-independent analysis to analyze millions of sequences for comparative microbial community analysis (11,12).
The key step in taxonomy-independent analysis is to group sequences into OTUs based on pairwise sequence differences, where hierarchical clustering is one of the most widely employed approaches (13,14). Hierarchical clustering is a classic unsupervised learning technique (15), and has been used in numerous biomedical applications [e.g. (12,16,17)]. The main drawback of hierarchical clustering is its high computational and space complexities. In computer science, this computational complexity is represented in so-called ‘Big-O’ notation, where the number given indicates how the time or space scales for large problem sizes: for example, an (N) algorithm takes time proportional to the size of the input, and an (N2) algorithm takes time proportional to the square of the size of the input (e.g. computing all pairwise distances between sequences takes time proportional to the square of the number of sequences, because each sequence must be compared to each other sequence). Given N objects, a brute-force algorithm takes (N2 log N) time, and improved methods take (N2) time (18). The memory needed for conventional methods also grows quadratically with respect to the data size. In the last decade, researchers developed several approximate hierarchical clustering algorithms with sub-quadratic time complexity (19,20). The basic idea is to employ a space-partitioning technique [e.g. dynamic closest-pair tree (21)] to organize objects hierarchically into cells so that the nearest neighbor of each object can be found within its adjacent cells by using a divide-and-conquer strategy. These algorithms can perform the analysis in (N log N) time and are good approximations. However, they can only handle low-dimensional data in a numerical space where hyper-planes can be defined to partition the space as well as the samples. Mathematically, in order to partition a space with a hyper-plane, an inner-product operator has to be defined so that a direction in the space can be specified to indicate the inner and outer sides of the plane. For nucleotide sequence data, there does not exist such an inner-product operator, and thus no hyper-planes can be defined to efficiently partition the data. Moreover, unlike numeric-valued vectors, the distances between pairs of sequences can only be computed through sequence alignment, because sequences vary in length and can have deletions and insertions. Hence, a sequence can be considered as a data point in a nucleotide space of undefined dimensions, which poses additional mathematical challenges.
Several algorithms have been developed in the past decade for taxonomy-independent analysis. DOTUR is probably the first published hierarchical clustering algorithm for pyrosequencing data analysis and widely used by the microbiology community (14). It requires users to provide a distance matrix and load it into the main memory. Due to its quadratic complexity, it can process only several ten thousand sequences (13). We recently developed a new algorithm, referred to as ESPRIT, that enables researchers to handle up to one million sequences by using a computer cluster (12,13). An online learning-based hierarchical clustering algorithm called hcluster was developed within the ESPRIT framework that addressed the memory issue associated with cluster merging. Although ESPRIT uses k-mer statistics to remove a large amount of unnecessary sequence comparisons, it is still an (N2) algorithm. Hcluster is incorporated in the well-known mothur pipeline (22) that replaced DOTUR. Unlike ESPRIT, mothur computes pairwise distances by aligning input sequences against a pre-aligned reference database. Since a reference database can be maintained off-line, the computational complexity of the sequence-alignment step grows only linearly with respect to the number of input sequences. However, the algorithm suffers the same problem as those used for taxonomy-dependent analysis. Since most bacterial genomes have not been sequenced yet, a large proportion of input sequences from unknown microorganisms may not be able to find significant hits and can only be aligned to distantly related reference sequences, leading to inaccurate estimates of pairwise distances. Moreover, the overall space and computational complexities remain (N2). Another line of research is to develop greedy heuristic-clustering methods. Two well-known methods are CD-HIT (23) and UCLUST (24). Both methods use pairwise sequence alignment and process input sequences sequentially. Given a predefined threshold, an input sequence is either assigned to an existing cluster if the distance between the sequence and a seed is smaller than the threshold, or becomes a seed otherwise. The computational complexity of greedy heuristic clustering is on the order of (NM), where M is the number of seeds and usually M ≪ N. CD-HIT and UCLUST are the two only methods that we are aware of that are capable of handling millions of sequences using a desktop computer. Although, CD-HIT and UCLUST organize data in a hierarchical structure, they are not hierarchical clustering algorithms and there is no guarantee that the true data structure can be recovered. In a numerical study presented below, we show that although CD-HIT and UCLUST run several orders of magnitude faster than a hierarchical clustering algorithm, their accuracy is much worse. Interested readers may refer to a companion paper (25) for a comprehensive review of existing algorithms for taxonomy-independent analysis.
In this article, we propose a new algorithm, referred to as ESPRIT-Tree, for hierarchical clustering analysis of massive sequence data. To avoid confusion, we note that ESPRIT-Tree is not a program for determining phylogenetic trees, but rather for producing hierarchical clusters of sequences based on sequence similarity, using a tree-like data structure. We extended the concept of space partition used by previous methods for handling sequence data of varying lengths. By assuming that sequence data lives in a pseudometric space, we created a distance-based partition of the data without explicitly defining an inner-product operator to divide the space, and organized the partition results in a pseudometric based partition tree. By repeatedly applying the triangular inequality, a fast closest-pair searching algorithm was developed within the ESPRIT-Tree framework. An efficient method for dynamic insertion and deletion of tree nodes were also developed. In order to avoid exhaustive computation of pairwise distances between clusters, we represented a cluster of sequences as a probabilistic sequence, and defined a set of operations to align probabilistic sequences and to compute genetic distances and k-mer distances between probabilistic sequences. The analyses of space and computational complexities of the algorithm are presented. A large-scale test was conducted on a human gut microbiota data set consisting of over one million sequences that demonstrated the effectiveness of the newly proposed algorithm.
METHODS
This section presents a detailed description of the newly proposed ESPRIT-Tree algorithm. Throughout the article, we use a boldfaced lowercase letter (e.g. a) to represent a vector or a sequence string, and a boldfaced uppercase letter (e.g. M) to represent a matrix, the ij-th element of which is written as Mij.
Prerequisites
Pseudometric space
We assume that sequence data lives in a pseudometric space. Precisely, given a data set and a scoring function d(·) used to measure the similarity between two sequences, for x, y, z ∈ , the following properties hold: (1) d(x, y) = d(y, x), (2) d(x, x) = 0 and (3) d(x, y) ≤ d(x, z) + d(z, y) (this is the triangular inequality, which states that there cannot be a shorter path from A to B that goes through a third point C than the direct path from A to B). The first two properties trivially hold. Although sequence data does not strictly follow the triangular inequality, the above assumption is very weak. A Monte Carlo experiment was performed where only 7 out of 100K trials were observed that violated the inequality (See ‘Experimental results’ section).
Pseudometric based partition tree
A pseudometric based partition (PBP) tree is a height-balanced tree consisting of multiple layers of nodes at pre-designated distance levels. Figure 1 depicts a toy tree with four layers. A similar technique was first used in the well-known BIRCH algorithm for clustering large-scale numerical-valued data (26). In this article, we extend the concept to handle sequence data of varying lengths. Each node in the tree represents a hypersphere region in the space and includes all sub-nodes and sequences that are positioned in the region, except for those that have been included by a preceding node. A non-leaf node is characterized by 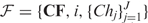 , where J is the total number of children of the node,
, where J is the total number of children of the node, 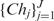 is an ordered list of pointers to its child nodes, and i is the order of the node in the child list of its parent. CF = {N, r, c} is a triple summarizing the sequences absorbed in the node, where N is the total number of the sequences, c is a sequence or a probabilistic sequence (described in the next section) defining the center of the node, and r is the distance level used to determine whether to absorb a newly arrived sequence into the node or to create a new node. A leaf node contains only a single sequence or a single cluster, and for ease of presentation, a root node is created with no center and level defined that includes all descendent nodes (Figure 1b). We call one node a sibling of another node if both share the same parent. For two sibling nodes A and B, assuming the order of A is smaller than B, A is then called the predecessor of B. A detailed description of how to build a PBP tree given a sequence data set is given in the ‘Constructing a PBP tree’ section.
is an ordered list of pointers to its child nodes, and i is the order of the node in the child list of its parent. CF = {N, r, c} is a triple summarizing the sequences absorbed in the node, where N is the total number of the sequences, c is a sequence or a probabilistic sequence (described in the next section) defining the center of the node, and r is the distance level used to determine whether to absorb a newly arrived sequence into the node or to create a new node. A leaf node contains only a single sequence or a single cluster, and for ease of presentation, a root node is created with no center and level defined that includes all descendent nodes (Figure 1b). We call one node a sibling of another node if both share the same parent. For two sibling nodes A and B, assuming the order of A is smaller than B, A is then called the predecessor of B. A detailed description of how to build a PBP tree given a sequence data set is given in the ‘Constructing a PBP tree’ section.
Figure 1.

PBP tree. A PBP tree partitions an input space into a set of non-overlapped hyperspherical regions at various distance levels indicated by circles with different colors (a), and organizes input sequences in a tree-like hierarchical structure (b). Each point in (a) represents a sequence, and the color of a node in (b) corresponds to the color of a circle in (a). For ease of presentation, the leaf nodes are omitted, and a root node is created that includes all descendent nodes. The partition, though not necessarily reflecting the true structure of a data set as shown in (a), can significantly accelerate a clustering process by removing most unnecessary sequence alignment operations and distance computation.
Probabilistic sequences
A probabilistic sequence is a statistical model used to describe a group of similar sequences. Suppose we have two sequences a and b, the optimal global alignment of which is given by:
| (1) |
We create a 5 × 11 matrix P, where each row from the top to the bottom represents a nucleotide A, T, C, G and a gap, respectively, and each column represents a nucleotide base of the aligned sequences (The matrix P is presented in Supplementary Table S1). For notational convenience, we associate with matrix P a virtual sequence x of length 11, and collectively call {x, P} a probabilistic sequence. Each element of x can take one of the four nucleotides or a gap, the probability (or occurrence frequency) of which is specified in P. For example, the first column of P in the above example reads [0.5, 0, 0, 0.5, 0]T, where T is the matrix transpose. By using the probabilistic Needleman–Wunsch algorithm, which will be detailed in the following section, the update of P when given a newly arrived sequence and the computation of the genetic distance between two probabilistic sequences only involve the application of simple linear algebra.
Probabilistic Needleman–Wunsch algorithm
The newly proposed probabilistic Needleman–Wunsch algorithm is a generalization of the Needleman–Wunsch algorithm (27) and used to optimally align two virtual sequences. Suppose we have two probabilistic sequences {x, P} and {y, Q}. Denote x = [x1, …, xJ] and y = [y1, …, yL]. Given a scoring matrix, the best alignment score between {x, P} and {y, Q} can be computed by using the following recursive equation:
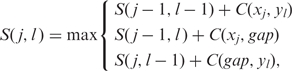 |
(2) |
where C(xj, yl), C(xj, gap) and C(gap, yl) are the costs of aligning xj to yl, xj to a gap, and yl to a gap, respectively. However, since both xj and yl can take a nucleotide base or a gap with a certain probability, by denoting = {A, T, C, G, gap}, C(xj, yl) and C(xj, gap) can be computed as:
| (3) |
| (4) |
where Pij and Qnl are the ij-th and nl-th elements of matrices P and Q, respectively, and i is the i-th element of . C(xj, yl) and C(xj, gap) can be interpreted as the expected cost of aligning xj with yl or a gap, respectively. The alignment score for each position is stored in an array with a pointer that records the current optimal operation and provides an effective path to backtrack the optimal alignment. In the above descriptions, we use a linear gap penalty for simplicity. Extension to an affine gap penalty is straightforward.
The proposed probabilistic Needleman–Wunsch algorithm shares the same idea as the profile–profile alignment (PPA) used in the well-known MUSCLE algorithm for multiple sequence alignment (28). However, PPA works on two groups of sequences, rather than two probabilistic sequences. With the concept of probabilistic sequence, we can go beyond PPA and compute genetic distances directly based on alignment results, which is described below.
Genetic distances between probabilistic sequences
The genetic distance between two globally aligned sequences is computed as the number of mismatches divided by the total length of the sequences. The distance between two virtual sequences can be calculated analogously. Let {x, P} and {y, Q} be two aligned probabilistic sequences of length L. Suppose that we randomly select two sequences s = [s1, …, sL] and t = [t1, …, tL] following the probabilities specified by P and Q, respectively. The probability that the two sequences differ at a given position, say l, can be computed as:
| (5) |
and the genetic distance between virtual sequences x and y can be computed as
| (6) |
By construction, the matrices P and Q record the distributions of the sequences of two clusters. We call d(x, y) a probabilistic average distance of two clusters, and use it as an approximation of average inter-cluster distances. A simulation study was performed that showed that the two distances have a nearly perfect correlation, and using probabilistic distances only has a negligible impact on clustering outcomes (see Figures 2 and 3). A highly desirable property is that the complexity of computing probabilistic distances is a constant ‘independent’ of the sizes of clusters.
Figure 2.

(a) NMI scores of four methods evaluated at 10 distance levels. (b) Box-plot of the maximum NMI scores of four methods. The species assignments of input sequences were used as ground truth.
Figure 3.

(a) NMI scores of four methods evaluated at 14 distance levels. (b) Box-plot of the maximum NMI scores of four methods. The genus assignments of input sequences were used as ground truth.
We next define the operation of merging two aligned probabilistic sequences. Suppose we have two probabilistic sequences {x, P} and {y, Q}, representing N and M sequences, respectively. After alignment, P and Q are updated as 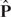 and
and  . The probabilistic matrix T of the merged sequence can be readily computed as
. The probabilistic matrix T of the merged sequence can be readily computed as 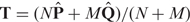 .
.
K-mer distances between probabilistic sequences
The data structure imposed by a PBP tree and using a probabilistic sequence to represent a group of similar sequences enable us to remove a large number of unnecessary sequence comparisons. Yet, it is still computationally expensive to align millions of sequence pairs. One commonly used strategy to alleviate the above issue is to use k-mer distances to identify a short list of candidate sequences for exact sequence comparison. This technique has been used by various bioinformatics algorithms, including MUSCLE (28), ESPRIT (13), CD-HIT (23), UCLUST (24) and RDP classifier (29). It has been shown that k-mer distances are highly correlated with genetic distances and can be computed thousands of times faster than sequence alignment. In this section, we extend the concept of k-mer distances to handle probabilistic sequences.
A k-mer is a sequence string consisting of k nucleotides. By specifying the value of k, a complete alphabet of k-mers can be constructed. Given a sequence x, a k-mer statistics vector v is computed, where the i-th element of v records the occurrence frequency of the i-th k-mer of in sequence x. Then, the k-mer distance between two sequences x and y can be computed as:
| (7) |
where || is the number of elements in , and L1 and L2 are the lengths of x and y, respectively.
In order to compute k-mer distances for probabilistic sequences, we need to re-define k-mer statistics vector v. Let = {A, T, C, G} and τ = [τ1,τ2, …, τK] be the i-th k-mer of . We further assume that τ1, …, τK take the j1, …, jK-th nucleotides of , respectively. The occurrence frequency of τ in probabilistic sequence {x, P} of length L can then be computed as:
| (8) |
 can be interpreted as the probability of observing k-mer τ at the i-th base of x.
can be interpreted as the probability of observing k-mer τ at the i-th base of x.
The k-mer statistics are used in two places in ESPRIT-Tree. First, when constructing a PBP tree, we use k-mer distances as a filter to determine whether the distances between a newly arrived sequence and the centers of the nodes are within a predefined threshold before performing exact sequence comparison. Secondly, when searching for the nearest neighbor of a query sequence in a set of candidate sequences, we calculate k-mer distances to optimize the order of comparison and perform a branch-and-bound search. Specifically, the candidate sequence with the smallest k-mer distance to the query sequence is considered first, and the obtained genetic distance is then used to compute an upper bound based on the correlation relationship between k-mer and genetic distances. Sequences with k-mer distances larger than the bound are removed from the candidate set, and the remaining sequences are sorted and compared to the query sequence. The procedure repeats until the candidate set becomes empty. The sequence with the smallest genetic distance is identified as the nearest neighbor of the query sequence.
ESPRIT-Tree
With the above prerequisites, we are ready to present the proposed algorithm. ESPRIT-Tree consists of two steps. First, a PBP tree is constructed that roughly partitions an input space into a set of hyperspherical cells. Second, a refinement procedure is carried out that iteratively finds the closest pairs of sequences or clusters and merges them into a new cluster. The two steps are detailed below.
Constructing a PBP Tree
A PBP tree contains multiple levels, uniformly spaced on a logarithmic scale starting from the top at 0.1 and to the bottom at 0.01, and grows incrementally to include all input sequences. The number of levels created is a parameter of the algorithm. Initially, the tree comprises only one branch of nodes, the centers of which are all assigned to the first arrived sequence. Given a new sequence, we first compare it with the center of the child nodes of the top node in a fixed order. If the resulting pairwise distance is larger than the threshold, a new node is created; otherwise, the sequence travels down toward the leaf nodes through the first branch that yields a pairwise distance smaller than the threshold. The reason that we do not assign the sequence to the nearest center is that since the tree is constructed dynamically, the hyperspherical cells can overlap (see Figure 1a), which makes the search of the closest pairs in the second step difficult. After a sequence is absorbed into a node, the parameter of that node is updated using the operations defined in the previous section. The procedure is repeated until the sequence reaches a leaf node at the bottom. If we are not interested in microbial diversities at distance levels larger than 0.1, there is no need to grow the tree upwards.
A PBP tree provides a coarse representation of the entire data, and the representation is most accurate at the bottom level. Since only the sequence index information and cluster feature vector 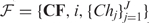 of each node are saved in a PBP tree, the memory required is very small. We also see that when a sequence travels from the top to the bottom, it visits only a very small fraction of the tree and aligns only with the center sequences of the visited nodes. For example, for the tree presented in Figure 1b, if we decide to use the right branch, the nodes on the left branch will never be touched. The computational complexity of constructing a PBP tree is on the order of (N), instead of (N2). For the problems we are most interested in, N is on the order of 106. Hence, a PBP tree can be built very efficiently. In our simulation study, it took only ∼0.5 h to build a PBP tree for one million sequences using a desktop computer.
of each node are saved in a PBP tree, the memory required is very small. We also see that when a sequence travels from the top to the bottom, it visits only a very small fraction of the tree and aligns only with the center sequences of the visited nodes. For example, for the tree presented in Figure 1b, if we decide to use the right branch, the nodes on the left branch will never be touched. The computational complexity of constructing a PBP tree is on the order of (N), instead of (N2). For the problems we are most interested in, N is on the order of 106. Hence, a PBP tree can be built very efficiently. In our simulation study, it took only ∼0.5 h to build a PBP tree for one million sequences using a desktop computer.
Clustering refinement
Clustering refinement consists of two steps: finding the closest pair among existing clusters and dynamically updating a PBP tree. The first step is computationally the most expensive module. A naive implementation of finding distances between all pairs of clusters and selecting the minimum requires (N2) operations. This problem has been intensively studied in the field of computational geometry (30). Approximation algorithms can solve the problem in (N log N) time. However, they work only for numerical-valued data of fixed dimension, and little work has been done to handle sequence data of varying lengths. We below show how to utilize the data structure imposed by a PBP tree to design a fast and accurate closest-pair searching algorithm. A PBP tree partitions an input sequence space into a set of non-overlapped hyperspherical regions at various distance levels and organizes them in a hierarchical structure (Figure 1). Similar sequences are grouped into the same or adjacent cells, which suggests that the nearest neighbor of a sequence can be found locally, avoiding the need to explore the entire space.
Suppose that we have a data set consisting of N sequences. In order to find the nearest neighbor of sequence x, we first compare it with its successor sibling sequences that share the same parent as x and have an order larger than that of x, and find a sequence y that yields the minimal pairwise distance among the sequences compared. We then move upwards to compare x with the center of the parent node T to validate the identified pair. The distance between x and the boundary of the parent node is computed as db = r1 − d(x, c1), where r1 and c1 are the distance level and the center of T, respectively. There are two possibilities.
Case 1: If d(x, y) > db, it can be inferred that there may exist a sequence outside the region covered by T that is closer to x than y. We thus move up one level and explore sequences belonging to the successor sibling nodes of T, and use the parent of T, K, as the new reference node. Note that we ignore all of the sequences in the predecessor nodes of T, which greatly speeds up the searching process. For each successor node with center c2 and level r2, we first compute the distance between sequence x and its center. If d(x, c2) > d(x, y) + r2, it can be proved by using the triangular inequality that there does not exist a sequence in the node that can be closer to x than y, and hence all sequences within that node can be ignored; if d(x, c2) ≤ d(x, y) + r2, we check the child nodes of the successor node by using the triangular inequality, and repeat the above process until we reach a leaf node. After all successor nodes are explored, a sequence z is identified that yields the minimal distance among all sequences checked. If d(x, z) < d(x, y), y is replaced by z and compared to the center of K to determine the next move.
Case 2: If d(x, y) ≤ db, we bypass all of the sibling nodes of T and go on to check if the sibling nodes of K need to be explored.
The above described searching process continues until the root node is reached. Sequences x, y and the corresponding pairwise distance is recorded. It should be noted that y is not necessarily the nearest neighbor of x since only successor nodes are searched. We thus call y the successor nearest neighbor (SNN) of x. Given N sequences, a list of N sequence pairs is generated. By using a heap structure (31), the minimal distance among the N sequence pairs can be found in (log N) time, and the algorithm requires only (N) space. For ease of presentation, we above consider only how to find the closet pairs of sequences. The derivation can be easily generalized to find the closest pairs of nodes containing a group of sequences by using the probabilistic Needleman–Wunsch algorithm described in the ‘Probabilistic Needleman–Wunsch algorithm’ section. The following lemma proves that although we do not explore the entire data set, the sequence pair found by the proposed algorithm is the closest pair among the input sequences.
Lemma 1 —
Let be a set of input sequences in a pseudometric space. For the algorithm described as above, the sequence pair found by the algorithm is the closest pair of the input sequences.
Proof —
It is trivial to prove that if all sequences in successor nodes are searched, a SNN list contains the closest pair. We then prove that if d(x, y) ≤ r1 − d(x, c1), no sequence in the sibling successor nodes of T is closer to x than y and thus can be ignored. Suppose there exists a sequence z in the sibling successor nodes so that d(x, z) < d(x, y). By using the triangular inequality, d(z, c1) ≤ d(z, x) + d(x, c1) < d(x, y) + d(x, c1) < r1, which contradicts the assumption that z is a sequence in the sibling successor nodes of T. The same strategy can be used to prove that if d(x, c2) > d(x, y) + r2, there does not exist a sequence in the node that can be closer to x than y. ▪
After the closest pair is found, we then merge the identified sequence pair into one cluster, remove the two sequences from the tree, and insert the newly formed cluster into the PBP tree by using the same procedure we used to construct the tree in the first step. More specifically, a probabilistic sequence is generated from the two merged sequences, and then compared to the center of the children of the root node by using the probabilistic Needleman–Wunsch algorithm. If the resulting pairwise distance is larger than the threshold, a new node is created; otherwise, the sequence travels down toward the leaf nodes through the first branch that yields a pairwise distance smaller than the threshold. The SNN table is then updated. First, the two identified sequences are removed from the table, the nearest neighbor of the newly formed probabilistic sequence is then identified and added to the table, and finally the sequences that previously set either of the two identified sequences as the nearest neighbor are reassigned new nearest neighbors. It should be noted that since the new formed cluster or probabilistic sequence is added to the tree dynamically, in order to maintain a correct SNN list, the new cluster is compared with all existing sequences, except for those that are bypassed according to the triangular inequality. Nevertheless, due to the hierarchical partitioning of the data set by the PBP tree, only a small fraction of sequences are actually compared. The iteration of finding the closest pair, creating new clusters and updating the SNN table continues until only one cluster is left or the distance between the closest pair is larger than a predefined threshold.
Unlike conventional hierarchical clustering algorithms, the proposed algorithm does not require the generation of a distance matrix. All of the operations are executed on the fly, and the distances are computed only when they are needed. The memory required to store a SSN table is on the order of (N). The algorithm thus addresses the space-complexity issue of conventional methods. It is difficult to conduct a computational complexity analysis. However, since our algorithm uses a divide-and-conquer-based strategy to recursively partition a sequence space and refine clustering results, it is well known that this type of algorithm has a quasilinear complexity (19–21), which is empirically verified in a simulation study.
NUMERICAL EXPERIMENTS
We present a numerical experiment to compare ESPRIT-Tree with three other methods, namely, CD-HIT (23), UCLUST (24) and ESPRT (13). CD-HIT and UCLUST are two greedy heuristic-clustering methods widely used by the microbiology community. ESPRIT is a standard implementation of hierarchical clustering and used to benchmark the performance of ESPRIT-Tree. We demonstrate that the proposed algorithm achieves a similar accuracy to the standard hierarchical clustering algorithm but with a computational complexity comparable to CD-HIT and UCLUST. All experiments were performed on a desktop computer with Intel E5462 2.8GHz and 16GB RAM.
Experimental setup
A real-world sequence data set was used to benchmark the performance of the four methods. The data set was originally used to study the connection between obesity and altered compositions of the human gut microbial community (8). It consists of ∼1.1 M sequences with an average length of 232 nucleotides, covering the V2 hypervariable region of 16S rRNAs collected from the stool samples of 154 individuals. This is one of the most comprehensive 16S rRNA based surveys of the human gut microbiota available to date.
One of the major obstacles of a benchmark study is that for complex microbial communities there is no ground-truth information about what species are actually in the community. To overcome this difficulty, we first constructed a reference database from the RDP-II database (9), where each reference sequence was fully annotated. We then ran a MegaBlast (32) search of the gut data against the reference database, and used a stringent criterion to retain the annotated sequences if the identity percentage >97% and the length of the aligned region >97% of the total length. This resulted in a total of ∼750K reads classified into 671 species and 283 genera. We then applied the four methods to the annotated sequences and used the commonly used normalized mutual information (NMI) criterion (33) to evaluate how the outcome of a clustering algorithm agrees with the ground truth. NMI penalizes both assigning sequences with the same label into different clusters and assigning sequences with different labels into the same clusters. NMI = 1 means that a clustering result completely agrees with the ground-truth partition, and NMI = 0 means that each sequence is randomly assigned. A mathematical description of NMI can be found in the Supplementary section S2. In order to remove statistical variations, the experiment was repeated 20 times. In each iteration, 30K sequences were randomly extracted from the annotated data set, the four methods were used to group the sequences into clusters at various distance levels ranging from 0.01 to 0.15, a NMI score was computed at each distance level by using either the species or genus assignments of input sequences as the ground truth, and the maximum NMI score was recorded. We observed high, medium and low abundance components (i.e. a long tail) in the test data sets, which is similar to what observed in a real microbial community and much more complicated than the previously used mock community generated from 43 known 16S rRNA sequences (13,34). We acknowledge that this benchmark is limited to known taxa and is subject to a certain degree of inaccuracy because not all taxa evolve at equal rates and so OTUs are not expected to map perfectly onto species; however, these factors should not bias the evaluation in favor of any specific method since they were all applied to the same data set, and, all else being equal, a method that gives clusters consistent with existing taxonomic knowledge should generally be preferred over one that is less consistent.
For UCLUST, the clustering outcomes may depend on the order of sequences presented to the algorithms. The default setting is to sort input sequences based on their lengths, while another possibility is based on their abundances (i.e. a sequence and its subsequences are considered as one sequence). We found that abundance sort yielded better results, which are reported in the article. For ESPRIT, we used a loose k-mer threshold of 0.8 to remove unnecessary sequence alignments. At the distance levels <0.2, the results of ESPRIT are exactly the same as those generated by the standard method. We considered both average and complete linkage functions in ESPRIT, and found that both performed similarly in terms of clustering performance. Only the result of average linkage was reported.
Experimental results
A key assumption of the proposed method is that sequence data lives in a pseudometric space. We performed a simulation study to justify the above assumption. We first randomly selected 30 K sequences from the gut data set and applied ESPRIT with the average linkage function (ESPRIT-AL) to group the selected sequences into clusters at various distance levels ranging from 0.01 to 0.10. We then randomly selected three clusters, and applied the probabilistic Needleman–Wunsch algorithm to generate three probabilistic sequences, represented by x, y and z, respectively, from the sequences within the three selected clusters. The ratio of the pairwise distances of the three sequences d(y, z)/(d(x, y) + d(x, z)) was computed. A ratio of less than or equal to 1 means that the triangular inequality is satisfied. We repeated the experiment 100 K times, and observed only 7 cases where the inequality was violated. This experiment suggests that it is generally true that sequence data follows the triangular inequality.
We next conducted a benchmark study to compare the clustering performance of the four methods. Figure 2a depicts the NMI scores as a function of distance levels, averaged over the 20 runs, by using the species assignments of input sequences as ground truth. We observe that all curves have a bell shape. This can be explained by the fact that when a distance level is small, sequences belonging to the same species are partitioned into different clusters, and when a distance level is large, sequences belonging to different species are grouped into the same clusters, and by definition both result in suboptimal NMI scores. We also see that the NMI scores of the four methods may peak at different positions, due to the different formulations used to define the distance between two clusters. Hence, the NMI scores obtained at the same distance level are not directly comparable. We thus compared the maximum NMI score of each method, which by definition corresponds to the best clustering result that a method can achieve. From Figure 2b, we observe that ESPRIT-Tree performed similarly to ESPRIT-AL, and significantly better than CD-HIT and UCLUST (P-value ≤ 10−5 based on a Student's t-test). We repeated the analysis by using the genus assignments as ground truth, and observed similar results (Figure 3).
One of the main purposes of performing taxonomy-independent analysis is to estimate the biodiversity of a microbial community. In the microbiology literature, 3% and 5% are the two most commonly used criteria to define species and genus-level OTUs, respectively, although these definitions are controversial (14,35,36). Table 1 reports the numbers of species and genera estimated at the 0.03 and 0.05 distance levels, and those at the positions where the NMI scores peak. We see that the numbers of OTUs observed at the 0.03 and 0.05 distance levels are significantly larger than the ground truths, and vary significantly for different methods although they were all applied to the same data sets. It was previously thought that sequencing errors are the main reason for severe overestimation of microbial diversity and several sequencing-error-correction algorithms have been developed to address the above issue [e.g. (34,37)]. However, we observe from Table 1 that the numbers of OTUs for each method obtained at the peak positions are always much closer to the ground truths than those obtained at the 0.03 and 0.05 distance levels. This suggests that the overestimation to some extent is due to the incorrect use of distance levels. The commonly used 3% and 5% are not proper for defining species and genus-level OTUs, which was also observed in (38), and researchers should be careful when interpreting their diversity estimates. ESPRIT-Tree and ESPRIT-AL yielded the most accurate estimates of microbial diversity among the four methods.
Table 1.
The numbers of OTUs observed at the 0.03 and 0.05 distance levels and at the peak positions for the four methods
| ESPRIT-AL | ESPRIT-Tree | UCLUST | CD-HIT | |
|---|---|---|---|---|
| 0.03 level | 1045 (19) | 1137 (30) | 1193 (26) | 920 (23) |
| 0.05 level | 241 (7) | 268 (6) | 362 (11) | 314 (9) |
| peak NMI-species | 402 (9) | 400 (9) | 590 (13) | 314 (9) |
| peak NMI-genus | 190 (5) | 176 (7) | 216 (6) | 243 (7) |
The ground truths of the numbers of species and genera are 371±7 and 170±5, respectively. The number in the parenthesis is one SD. ESPRIT-Tree and ESPRIT with the average linkage function (ESPRIT-AL) yielded the most accurate estimates of microbial diversity among the four methods.
The massive amount of data generated by high-throughput pyrosequencing technologies poses serious challenges to existing algorithms. In addition to accuracy, computational complexity is another important issue that needs to be considered. To demonstrate the scaling property of the new method, we compared ESPRIT-Tree with CD-HIT and UCLUST using a human gut data set with a varying number of sequences, ranging from 1K to 1.1M. It is computationally intractable to run ESPRIT on 1.1M sequences using a desktop computer. Figure 4 reports the CPU times of the three methods as a function of the numbers of sequences. The empirical complexity and confidence interval are also reported. In terms of computational efficiency, UCLUST performs the best, ESPRIT-Tree the second and CD-HIT the third. However, all three methods have a quasilinear computational complexity of (N1.2). It took ESPRIT-Tree ∼11 h to process 1.1 M reads to generate OTUs at ten distance levels (0.01–0.1). We have previously applied ESPRIT to the same gut data set using a computer cluster of 100 processors (12). It took ESPRIT ∼4 days to finish the analysis, which is about 800 times slower than ESPRIT-Tree.
Figure 4.

Scalability of ESPRIT-Tree, CDHIT and UCLUST performed on a human gut microbiota data set with a varying number of sequences ranging from 1K to 1.1M. The empirical complexity and confidence interval (CI) are also reported.
We performed additional experiments using other hypervariable region and near full-length 16S rRNA sequences and observed similar results. Due to space limitations, the results are presented in the Supplementary Data.
CONCLUSION
With the advent of the massively parallel pyrosequencing technology, researchers can now obtain millions of signature sequences easily and inexpensively for diverse applications ranging from human epidemiological studies to global ocean surveys. The molecular studies of microbial communities are recently entering an era of quantization, where not only species richness but also detailed compositions of microbial communities are required in order to query multiple biological and ecological questions. Considering that bacterial populations usually contain a significant amount of unknown species, taxonomy-independent analysis is a widely accepted and powerful tool for studying microbial community dynamics at high resolutions, and hierarchical clustering is an essential step to explore the taxonomical information of bacterial populations. In this article, we have proposed a novel computational algorithm that enables researchers to perform clustering analysis of millions of 16S rRNA tag sequences on a desktop computer, while maintaining a clustering accuracy comparable to the standard hierarchical clustering algorithm. The new algorithm can be extended for parallel computing. While parallel computing is generally not a viable solution to scaling up (N2) algorithms, the quasilinear space and computational complexities of the proposed algorithm make it computationally tractable to process tens of millions of sequences by using a small computer cluster. Taxonomy-independent analysis plays a key role in several recently developed pipelines (e.g. QIIME, mothur and PANGEA), and our algorithm can significantly improve the utility of these pipelines, as each includes an OTU picking step that is distinct from the chimera checking step (we note that ESPRIT-Tree can either be applied to chimera-checked data, or that the representative or consensus sequences from clusters of nearly identical sequences generated from ESPRIT-Tree output can be chimera-checked, to obtain improved OTU counts from real data in which chimeras are frequent). Although in this article we mainly focused on 16S rRNA based studies, the new algorithm can be used for other large-scale sequence based studies that require large-scale clustering analyses. The ESPRIT-Tree software is available at http://plaza.ufl.edu/sunyijun/ES-Tree.htm.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: NSF Award No. 1062362.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Rob Knight for his thoughtful comments on the draft of this paper. The authors also would like to thank the editor Alan Kimmel and four anonymous reviewers for their valuable comments that significantly improved the quality of this paper.
REFERENCES
- 1.Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, et al. The NIH human Microbiome Project. Genome Res. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huse SM, Dethlefsen L, Huber JA, Welch DM, Relman DA, Sogin ML. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008;4:e1000255. doi: 10.1371/journal.pgen.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fabrice A, Didier R. Exploring microbial diversity using 16S rRNA high-throughput methods. J. Comput. Sci. Syst. Biol. 2009;2:74–92. [Google Scholar]
- 5.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLOS Biol. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huber JA, Welch DBM, Morrison HG, Huse SM, Neal PR, Butterfield DA, Sogin ML. Microbial population structures in the deep marine biosphere. Science. 2007;318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- 7.Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl Acad. Sci. USA. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–485. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, et al. The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 11.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun Y, Cai Y, Mai V, Farmerie W, Yu F, Li J, Goodison S. Advanced computational algorithms for microbial community analysis using massive 16S rRNA sequence data. Nucleic Acids Res. 2010;38:e205. doi: 10.1093/nar/gkq872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun Y, Cai Y, Liu L, Yu F, Farrell ML, McKendree W, Farmerie W. ESPRIT: estimating species richness using large collections of 16S rRNA pyrosequences. Nucleic Acids Res. 2009;37:e76. doi: 10.1093/nar/gkp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 2005;71:1501–1506. doi: 10.1128/AEM.71.3.1501-1506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward JH. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 1963;58:236–244. [Google Scholar]
- 16.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl Acad. Sci. USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yanagisawa K, Shyr Y, Xu B, Massion PP, Larsenc PH, White BC, Roberts JR, Edgerton M, Gonzalez A, Nadafa S, et al. Proteomic patterns of tumour subsets in non-small-cell lung cancer. Lancet. 2003;362:433–439. doi: 10.1016/S0140-6736(03)14068-8. [DOI] [PubMed] [Google Scholar]
- 18.Murtagh F. Complexities of hierarchic clustering algorithms: state of the art. Comput. Stat. Quart. 1984;1:101–113. [Google Scholar]
- 19.Krznaric D, Levcopoulos C. Optimal algorithms for complete linkage clustering in D dimensions. Theor. Comput. Sci. 2002;286:139–149. [Google Scholar]
- 20.Franti P, Virmajoki O, Hautamaki V. Fast agglomerative clustering using a k-nearest neighbor graph. IEEE Trans. Patt. Anal. Mach. Intell. 2006;28:1875–1881. doi: 10.1109/TPAMI.2006.227. [DOI] [PubMed] [Google Scholar]
- 21.Bespamyatnikh SN. An optimal algorithm for closest-pair maintenance. Discrete Comput. Geom. 1998;19:175–195. [Google Scholar]
- 22.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: opensource, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 24.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 25.Sun Y, Cai Y, Huse S, Knight R, Farmerie W, Wang X, Mai V. A large-scale benchmark study of existing algorithms for taxonomy-independent microbial community analysis. Brief. Bioinformatics. 2011 doi: 10.1093/bib/bbr009. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang T, Ramakrishnan R, Livny M. BIRCH: a new data clustering algorithm and its applications. Data Min. Knowl. Disc. 1997;12:141–182. [Google Scholar]
- 27.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 28.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleinberg J, Tardos E. Algorithm Design. Addison Wesley; 2006. [Google Scholar]
- 31.Cormen TH, Leiserson CE, Rivest RL. Introduction to Algorithms. MIT Press/McGraw-Hill; 1990. [Google Scholar]
- 32.Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 33.Fred ALN, Jain AK. Proceedings IEEE Conference on Computer Vision and Pattern Recognition. 2003. Robust data clustering. 3 pp. 128–136. [Google Scholar]
- 34.Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borneman J, Triplett EW. Molecular microbial diversity in soils from eastern Amazonia: evidence for unusual microorganisms and microbial population shifts associated with deforestation. Appl. Environ. Microbiol. 1997;63:2647–2653. doi: 10.1128/aem.63.7.2647-2653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sait M, Hugenholtz P, Janssen PH. Cultivation of globally distributed soil bacteria from phylogenetic lineages previously only detected in cultivation-independent surveys. Environ. Microbiol. 2002;4:654–666. doi: 10.1046/j.1462-2920.2002.00352.x. [DOI] [PubMed] [Google Scholar]
- 37.Quince C, Lanzen A, Curtis TP, Davenport RJ, Hall N, Head IM, Read LF, Sloan WT. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods. 2009;6:639–641. doi: 10.1038/nmeth.1361. [DOI] [PubMed] [Google Scholar]
- 38.White JR, Navlakha S, Nagarajan N, Ghodsi MR, Kingsford C, Pop M. Alignment and clustering of pylogenetic markers—implications for microbial diversity studies. BMC Bioinfomatics. 2010;11:152. doi: 10.1186/1471-2105-11-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.