

Abstract
Aplastic anemia (AA), a potentially fatal disease, may be cured with marrow transplantation. Survival in pediatric patients has been excellent early after transplantation, but only limited data are available regarding late effects. This study evaluates late effects among 152 patients followed 1-38 years (median, 21.8 years). Transplantation-preparative regimes were mostly cyclophosphamide with or without antithymocyte globulin. Survival at 30 years for the acquired AA patients is 82%, and for the Fanconi anemia patients it is 58% (P = .01). Multivariate analysis demonstrated that chronic GVHD (P = .02) and Fanconi anemia (P = .03) negatively impacted survival. Two Fanconi patients and 18 acquired AA patients developed a malignancy that was fatal for 4. There was an increased incidence of thyroid function test abnormalities among those who received total body irradiation. Cyclophosphamide recipients demonstrated normal growth, basically normal development, and pregnancies with mostly normal offspring. Quality-of-life studies in adult survivors of this pediatric transplantation cohort indicated that patients were comparable with control patients except for difficulty with health and life insurance. These data indicate that the majority of long-term survivors after transplantation for AA during childhood can have a normal productive life.
Introduction
Severe aplastic anemia (AA) is a potentially fatal disease characterized by acquired or genetic pancytopenia and significantly decreased or absent hematopoietic elements in the marrow without excess blasts.1 HSCT from an HLA-matched donor offers definitive curative treatment.2,3 Long-term survival of approximately 85%-100% has been observed for younger patients after BM transplantation from HLA-identical siblings after a preparative regimen with cyclophosphamide and antithymocyte globulin (CY + ATG) followed by posttransplantation immunosuppression with methotrexate and cyclosporine (MTX + CSP).2,4–6 As can be seen in Burroughs et al's report,7 5-year survival after 1981 is 95%-100% for children who underwent transplantation at Fred Hutchinson Cancer Research Center (FHCRC).
Late effects after transplantation have generally been reported for cohorts that included both pediatric and adult patients.8–10 The authors of a recent report demonstrated that thyroid function abnormalities occurred in < 20% long-term AA pediatric survivors.11 In a previous study, we showed that the patients who did not receive total body irradiation (TBI) had normal growth rates, and some appeared to be maturing normally.12 However, no comprehensive report of survival and late effects after transplantation exists for children who have undergone transplantation for AA.
Methods
Transplantation data and posttransplantation clinical and late effects data were retrospectively and prospectively collected for all patients who were < 18 years of age at the time of transplantation at FHCRC. All patients underwent transplantation and had follow-up data collected according to protocols approved by FHCRC's Institutional Review Board. All patients signed consent forms approved by the Institutional Review Board in accordance with the Declaration of Helsinki. Results are reported as of October 2010.
Patients
Between May 1971 and June 1, 2009, a total of 237 pediatric patients < 18 years of age with AA received a first marrow transplantation at FHCRC.2,4,13–15 The present analysis is focused on the 152 patients who survived for at least 1 year. Demographic data are summarized in Table 1. Patients were a median of 11.1 years of age at transplantation, and 79% were categorized as having an idiopathic etiology of AA. Table 1 also summarizes the transplantation characteristics: 86% received their transplantation from related donors and 94% from HLA-matched donors.
Table 1.
Patient and transplant characteristics
| Acquired | Fanconi | |
|---|---|---|
| Number | 137 | 15 |
| Gender F:M, n | 63:74 | 9:6 |
| Etiology, n | ||
| Idiopathic | 122 | – |
| Hepatitis associated | 10 | – |
| PNH | 2 | – |
| Insecticide | 3 | – |
| Age at diagnosis, median (range) | 10.7 (0.1-17.8) | 6.3 (3.0-13.3) |
| Time from diagnosis to transplantation, median (range) | 0.1 (0.1-9.1) | 2.0 (0.1-6.6) |
| Age at transplantation, median (range) | 11.1 (0.8-17.9) | 9.0 (5.1-15.9) |
| Length of follow-up, median (range) | 21.8 (1-38.1) | 20 (2-30) |
| First transplantation preparative regimen | ||
| Single-fraction TBI, n | 2 | – |
| Fractionated TBI | ||
| 12.0 Gy | 10 | – |
| 200-600 cGy | 12 | 4 |
| Cyclophosphamide ± ATG | 106 | 11 |
| Procarbazine ATG cyclophosphamide | 5 | – |
| None | 2 | – |
| Second/third preparative regimens, n | 15/4 | – |
| Donor, | ||
| Related | 118 | 12 |
| Unrelated | 16 | 3 |
| Syngenic | 3 | – |
| HLA-matched donor, yes:no | 128:19 | 15:0 |
| Source of stem cells | ||
| BM | 135 | 12 |
| Peripheral blood | – | 3 |
| Cord blood | 2 | – |
| Acute GVHD, grades 2-3, n (%) | 41 (30%) | 9 (60%) |
| Chronic GVHD, Clinical extensive, n | 36 | 5 |
ATG indicates antithymocyte globulin; PNH, paroxysmal nocturnal hemoglobinuria; and TBI, total body irradiation.
Conditioning regimen and posttransplantation care
The preparative regimens for the 15 Fanconi anemia patients were CY 50 mg/kg/d × 4 days (6 patients); CY 15-45 mg/kg/d × 4 days (5 patients); fludarabine plus 200 cGy of TBI (3 patients); and CY 20 mg/kg plus 450 cGy of TBI (1 patient; Table 1). The preparative regimen for first transplant for the idiopathic AA patients was intravenous CY 50 mg/kg/d given for 4 consecutive days alone (90 patients) or with horse ATG (ATGAM) 30 mg/kg/d (29 patients) given 12 hours after each of the first 3 CY doses. Five patients received procarbazine, ATG, and CY as previously described.16 Two survivors received 10 Gy of single-fraction TBI with CY 60 mg/kg/d × 2 consecutive days, and 10 patients received 60 mg/kg/d of CY × 2 days plus 200 cGy/d × 6 days of TBI. Sixteen patients received CY 50 mg/kg/d for 4 days and 200-600 cGy of TBI. Two syngeneic transplantation recipients received no preparative regimen. The majority of patients received BM from a related donor who was HLA matched (n = 129). Fifteen patients required second transplantations for graft rejection, and 4 patients received 3 transplantations. The transplantation-preparative regimens for the first transplantation are noted in Table 1. The second transplantation-preparative regimens were varied but included CY and ATG for 10 patients, with 6 patients receiving other immunosuppressive regimens. The third transplantation-preparative regimens consisted of CY 60 mg/kg/d for 2 days plus 200 cGy/d × 6 days' TBI (n = 2); CY 50 mg/kg/d for 4 days plus ATG (n = 1); and fludarabine 30 mg/m2 × 3 days plus 200 cGy of TBI (n = 1).
The majority of patients received posttransplantation prophylaxis for bacterial, fungal, and viral organisms. GVHD prophylaxis was MTX only (n = 71), CSP only (n = 1), and MTX + CSP (n = 75),4,13,14 with 5 patients receiving other regimens. Two syngeneic transplantation recipients did not receive GVHD prophylaxis. All patients who underwent transplantation after 1975 had chimerism evaluations performed at 1 year after transplantation. From 1976-1989 donor engraftment was confirmed with the use of red blood cell antigen and enzyme analysis, from 1989-1995 restriction fragments length polymorphism was used, from 1995-2004 the variable-number tandem repeats method was used, and after 2003 the method was changed to use short-tandem repeats.
Long-term follow-up
All patients had a departure work-up at approximately 80 days after transplantation, and all patients returned to FHCRC at 1 year and sporadically thereafter for a comprehensive medical evaluation with attention to late effects.17,18 Follow-up consisted initially of 6-monthly and later of annual contacts with the physician's office and the mailing of a questionnaire to the physician to obtain objective data. Patients were contacted once yearly on the anniversary of their transplantation, and beginning in August 1990 were mailed yearly questionnaires. The patients' responses were supplemented with results obtained from the physicians and provided a self-assessment of performance and well-being as well as results from a pulmonary function study for survivors > 5 years after transplantation and a quality-of-life study for adult survivors of this pediatric transplantation.19,20 In addition, since 1997, patients had dual-energy X-ray absorptiometry studies performed to measure bone mineral density at day 80 and 1 year and for patients who returned to FHCRC for subsequent annual evaluations.
Statistical analysis
Demographic data are reported with the use of range and median values for continuous variables. We estimated survival curves by using the Kaplan-Meier method, censoring at the time of last contact. More than 90% of patients had been contacted within the past 5 years. The time scale on the survival figures begins at 1 year after transplantation because patients had to survive that long to be included in the study. Late complications were determined from the data collected, and cumulative incidence of malignancy after transplantation was estimated by standard methods, with the treatment of death without malignancy as a competing risk. The SD growth curves were constructed from mean values of individual patient height measurements. Quality-of-life studies used were limited to those patients currently older than 18 years of age and surviving longer than 5 years after transplantation. The study used control subjects who were matched by sex with the survivor and within 5 years of the survivor's age. The measures used in this study have been previously reported.19
The impact of various factors on chronic GVHD at 1 year and mortality was determined by univariate or multivariate Cox regression analysis. Prognostic variables examined for their influence on survival or late complications included sex, previous acute GVHD, chronic GVHD, preparative regimen (without TBI or with low-dose (2-6 Gy) or high dose (10-12 Gy). For multivariate analysis, final models were determined on the basis of step-down regression methods with the use of P < .10 as inclusion criteria. Estimates of the expected 30-year survival of the acquired AA cohort were determined with 2001 population life tables from the National Center for Health Statistics. We used a weighted average of the sex-specific 30-year survival probabilities for female patients ages 10 years and male patients ages 12 years, which were the median ages at transplantation in the cohort.
Results
Survival
Patients have been followed for 1-38.1 years (median, 21.8 years) after transplantation and are currently 7.1-55.8 years (median, 35.2 years) of age. The survival of those with acquired AA is 82% at 30 years, but survival for the 15 Fanconi anemia patients is 58% at 30 years (P = .01; Figure 1A). Survival among those without chronic GVHD (n = 117) is 83%, and for those with any chronic GVHD (n = 35) is 62% (P = .02; Figure 1B). Currently, all syngeneic recipients, 15 of 16 with acquired AA and 2 of 3 Fanconi anemia patients who received unrelated transplantations, and 13 of the 15 patients who received second or third transplantations are alive. A multivariate analysis of the risk factors for mortality among the 152 patients surviving 1 year after transplantation is shown in Table 2. On the basis of the population survival rates, the estimated 30-year survival of a cohort with the same sex and age distribution as the acquired AA cohort would be approximately 97% when the median age at transplantation for male patients of 12 years and for female patients of 10 years is used.
Figure 1.
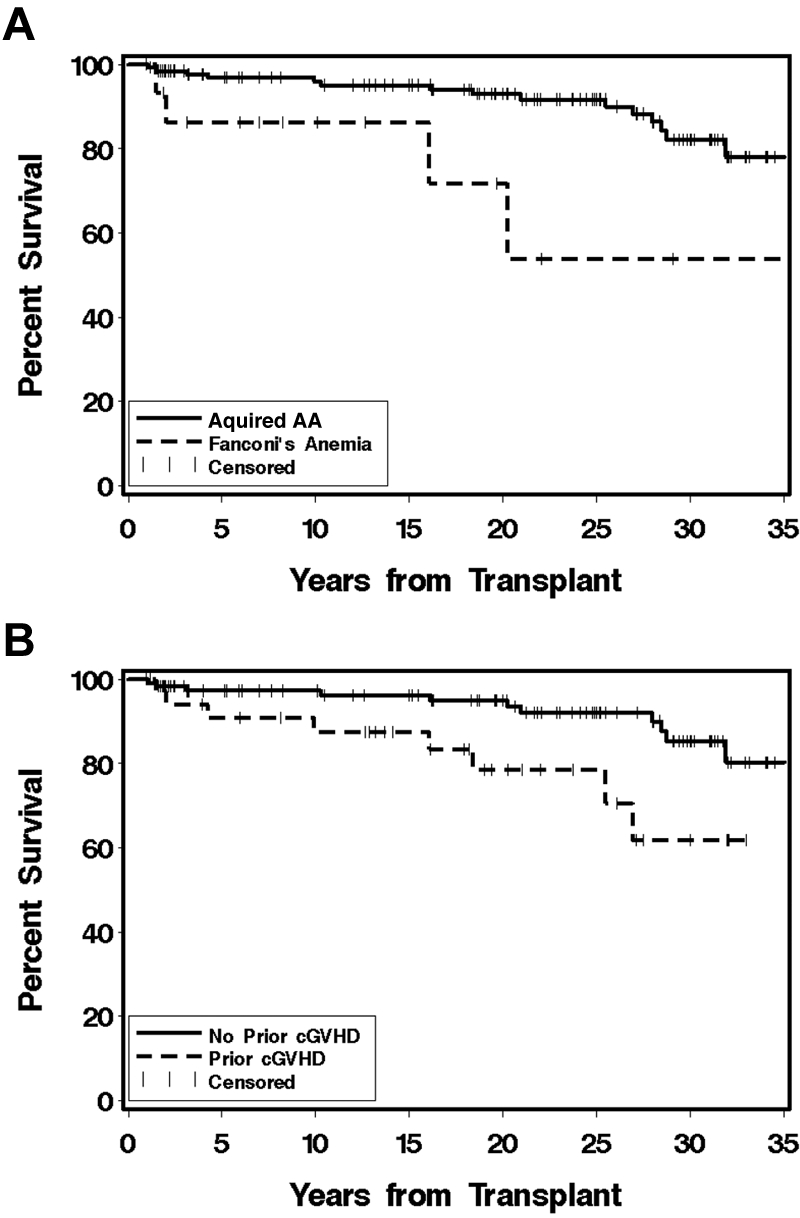
Survival 1 year after transplantation. (A) Percent survival among 152 children surviving at least 1 year after transplantation for severe AA. Survival of the 137 with acquired AA (solid line) is 82% and for the 15 Fanconi anemia patients (dashed line) is 58% at 30 years (P = .01). (B) Percent survival among 35 children who had chronic GVHD at 1 year after transplantation (dashed line) and 117 children without chronic GVHD (solid line) at 1 year after transplantation for severe AA (P = .02).
Table 2.
Mortality risk factors for 152 1-year survivors of HSCT for aplastic anemia
| Univariate |
Multivariate |
|||
|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | |
| Diagnosis | ||||
| Acquired AA | 1.0 | 1.0 | ||
| Fanconi anemia | 4.01 (1.3-12) | .03 | 4.43 (1.4-14) | .03 |
| Previous acute GVHD | ||||
| No | 1.0 | |||
| Yes | 2.04 (0.8-5.3) | .15 | ||
| Previous chronic GVHD | ||||
| No | 1.0 | 1.0 | ||
| Yes | 3.15 (1.3-7.9) | .02 | 3.73 (1.4-10) | .02 |
| Sex | ||||
| Female | 1.0 | |||
| Male | 1.56 (0.6-4.0) | .34 | ||
| Prophylaxis | ||||
| CSP | 1.0 | |||
| Combination | 1.63 (0.5-5.3) | .41 | ||
| Diagnosis to Tx | ||||
| 0-1 yr | 1.0 | |||
| 1 yr | 1.74 (0.5–6.1) | .41 | ||
| Regimen | ||||
| CY | 1.0 | |||
| TBI low | 5.62 (1.0-32) | |||
| TBI high | 0 (undefined) | .05 | ||
| Donor | ||||
| Matched related | 1.0 | |||
| Mismatch related | 0 (undefined) | |||
| Unrelated | 2.92 (0.6-14) | .28 | ||
| Age at HSCT, y | ||||
| 0-6 | 1.0 | |||
| 7-12 | 1.31 (0.3-5.3) | |||
| 13-17 | 1.95 (0.5-7.1) | .52 | ||
CI indicates confidence interval; CSP, cyclosporine; CY, cyclophosphamide; HR, hazard ratio; TBI, total body irradiation; and Tx, transplantation
Chimerism
Chimerism studies were not determined for the 22 patients who underwent transplantation before 1976. Among the 130 patients who underwent transplantation after that time, 2 were twins, 2 (1.5%) had documented autologous recovery, and 104 had donor chimerism documented from 1 to 6 years after transplantation. For these 104, there were 103 who had 100% donor chimerism and 1 who had stable mixed chimerism. Among the 22 who underwent transplantation before 1976, where chimerism studies are not documented, 5 had chronic GVHD, and 17 (77%) did not have chronic GVHD. In the 103 with 100% donor chimerism, 36 (35%) had chronic GVHD. Neither of the autologous recovery patients had chronic GVHD. Both of these autologous recovery patients rejected their allogeneic graft and have normal peripheral counts at 29 and 33 years after transplantation. The one patient with stable mixed chimerism did not have chronic GVHD.
Causes of death
Four of the 15 Fanconi anemia patients died (Figure 1A) between 2 and 20 years after transplantation. Two patients died of chronic GVHD complicated by poor marrow function < 5 years after transplantation. The other 2 Fanconi anemia patients deaths were between 5 and 20 years after transplantation because of metastatic squamous cell carcinoma (SCC) and because of liver failure secondary to hepatitis C (Table 3).
Table 3.
Late causes of death
| Number of patients |
||
|---|---|---|
| Acquired | Fanconi | |
| Total | 15 | 4 |
| 1-5 y after transplantation | 4 | 2 |
| Suicide | 1 | – |
| Chronic GVHD | 1 | 2 |
| Measles pneumonia | 1 | – |
| Hemolytic anemia | 1 | – |
| 6-32 y after transplantation | 11 | 2 |
| Pulmonary failure | 3 | – |
| Malignancy | 3 | 1 |
| Squamous cell cancer | 1 | 1 |
| Cervical cancer | 1 | – |
| Breast cancer | 1 | – |
| Suicide | 1 | – |
| HIV | 1 | – |
| Hepatitis C-related liver disease | 2 | 1 |
| Chronic GVHD | 1 | – |
Fifteen of the 137 acquired AA patients died; 4 between 1 and 5 years after transplantation and 11 between 6 and 32 years after transplantation (Table 3). The deaths < 5 years after transplantation were because of suicide, measles pneumonia, and refractory hemolytic anemia. The deaths beyond 5 years were because of pulmonary failure after lung transplantation for obstructive lung disease (1 patient), and for 2, severe obstructive lung disease associated with chronic GVHD. Three patients died of malignancy, including metastatic SCC (n = 1), metastatic cervical cancer (n = 1), and metastatic breast cancer (n = 1). Other late deaths were related to complications of hepatitis C (n = 2), HIV (n = 2), chronic GVHD (n = 1), and suicide (n = 1; Figure 1A).
Chronic GVHD
Chronic GVHD was present in 36 patients of the acquired AA patients and 5 of the Fanconi patients at 1 year after transplantation. These patients were treated with glucocorticoids, CSP, and other agents. Two patients received azathioprine for approximately 1 year, and both developed SCC. Risk factors for chronic GVHD are summarized in Table 4. The impact of having had chronic GVHD on survival is shown in Figure 1B. Here, patients with previous chronic GVHD had significantly poorer survival (P = .02) than those without chronic GVHD.
Table 4.
Risk factors for chronic GVHD among 1-year survivors
| Univariate |
Multivariate |
|||
|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | |
| Acute GVHD prophylaxis | ||||
| Single agent | 1.0 | |||
| Multiple agents | 1.47 (0.8-2.7) | .22 | ||
| Buffy coat | ||||
| No | 1.0 | 1.0 | ||
| Yes | 2.13 (1.1-4.2) | .04 | 2.79 (1.3-5.8) | .01 |
| Regimen | ||||
| Cyclophosphamide | 1.0 | |||
| TBI, low (2-6 Gy) | 1.18 (0.4-3.4) | |||
| TBI, high (10-12 Gy) | 3.68 (1.7-8.1) | .02 | ||
| Donor | ||||
| Matched related | 1.0 | 1.0 | ||
| Mismatched related | 5.36 (2.1-14) | .01 | 6.98 (2.6-19) | .004 |
| Unrelated | 1.53 (0.6-3.7) | 1.98 (0.8-4.9) | ||
CI indicates confidence interval; HR, hazard ratio; and TBI, total body irradiation.
Unrelated-donor transplantations
Of the 19 patients receiving transplants from unrelated donors, there were 16 with acquired AA and 3 had Fanconi anemia. Fifteen of the 16 (93%) of the acquired AA patients survived 1-24 years, and 5 had no chronic GVHD, 8 had mild chronic GVHD, and 3 had moderately severe chronic GVHD. All of these patients are no longer receiving immunosuppressive therapy after 1-4 years' treatment. One patient died at 1 year of refractory autoimmune hemolytic anemia. Two of the 3 Fanconi anemia patients survived for 5 and 10 years. One patient died at 2 years of severe chronic GVHD.
Malignancy
Twenty-one (13%) patients developed a posttransplantation malignancy that was the cause of death for 4. The proportion developing a malignancy was greater among the 28 patients who had TBI in their preparative regimen (37%) compared with the remainder who did not receive TBI (15%). The 5 malignancies among the 28 with TBI included 1 with SCC, 2 with thyroid malignancy, 1 with melanoma, and 1 with meningioma.
Among the 15 Fanconi anemia patients who received a transplant, 2 (13%) developed a malignancy at 9 years and 12 years after transplantation. One developed adenocarcinoma of the colon and is still alive. The second developed metastatic squamous carcinoma, which was the cause of death at 20 years after transplantation.
Among the remaining 137 patients with acquired AA, 19 (13%) developed a malignancy < 1-31 years after transplantation. Three patients developed a thyroid malignancy, all of whom successfully treated. One patient who underwent transplantation for paroxysmal nocturnal hemoglobinuria–associated AA subsequently developed myelodysplastic syndrome, for which he received a second transplant. After the successful second transplantation, he committed suicide. Two patients developed hepatocarcinoma associated with hepatitis C. This was the cause of death for one, and the other is alive after a liver transplantation. One patient developed a melanoma 2 months after receiving a transplant, which was treated with resection. The most common malignancy was SCC, which occurred in 7 patients and was the cause of death for 1. Five of the 7 also had severe skin chronic GVHD. Three additional patients developed breast cancer 31 years, 29 years, and 21 years after transplantation, respectively. This was the cause of death for one with invasive breast cancer, and the other 2 survivors had carcinoma in situ. A final patient died of metastatic cervical cancer.
Thirty-nine patients were treated with androgens for 3 weeks to months before 1995, and no patient was given androgen therapy pretransplantation after 1995. There were a total of 117 patients who received transplants before that time. In multivariate analysis (data not shown), androgens were not significantly associated with malignancy (hazard ratio 2.4, P = .07). Similarly, there was no association with TBI (hazard ratio = 1.2, P = .82).
Thyroid function
Thyroid function was evaluated in 137 (89%) of the patients tested on one or more occasion. Among these, normal thyroid function was found for 121 (88%). There were 9 of 106 (8.4%) evaluable patients who had not received TBI who had thyroid function abnormalities, and among these 5 were found to have compensated hypothyroidism, 1 had Hashimoto thyroiditis with autoantibodies, and 3 had hyperthyroidism. Among the 31 evaluable patients who had received TBI, 7 (22.6%) developed thyroid function abnormalities. Among these were 2 who developed compensated hypothyroidism, 2 had hypothyroidism of unknown type, 1 had central hypothyroidism, and 2 had hyperthyroidism.
Growth
Height growth was evaluated in the 67 boys and the 55 girls for whom at least 2 height data points were available. As can be seen in Figure 2, the height growth SD scores for these patients after transplantation was between 0 (50th percentile) and −1 SD below the mean. These children have basically normal height growth.
Figure 2.

Height SD growth curves for 67 boys (solid line) and the 55 girls (dashed line) for the patients whose preparative regimen was CY only.
Gonadal function
Among the 9 Fanconi girls, 7 were prepubertal at transplantation. Of these 7, 1 remains prepubertal, 5 had normal development through puberty, and 1 had delayed onset of puberty. Of the 2 postpubertal girls, both recovered gonadal function during the first year after transplantation. Among the 6 Fanconi anemia boys, 2 prepubertal boys had severe chronic GVHD, and neither boy had achieved pubertal development at time of death and are unevaluable. One of the other prepubertal boys had normal pubertal development, and 2 had delayed development. The 1 postpubertal boy had delayed development at the time of transplantation and has required hormonal therapy to promote puberty.
Development through puberty and gonadal function of postpubertal acquired AA patients at time of transplantation are shown in Table 5. Among the prepubertal girls, 11 remain prepubertal by virtue of age, and 30 of the 34 (88%) developed normally through puberty and had normal menarche and normal gonadotropins. Similarly for the prepubertal boys, 28 of 41 were evaluable by virtue of age, but data were only available for 21. Among these 21, 13 (62%) had normal development.
Table 5.
Gonadal function: acquired aplastic anemia
| Female patients |
Male patients |
|||
|---|---|---|---|---|
| Prepubertal | Postpubertal | Prepubertal | Postpubertal | |
| No. patients | 45 | 18 | 41 | 32 |
| Remains prepubertal | 11 | – | 13 | – |
| Evaluable | 34 | 18 | 28 | 32 |
| Normal menses | 30 | 15 | – | – |
| Normal development | 30 | – | 13 | – |
| Abnormal gonadotropin | 1 | 0 | 8 | 9 |
| NE | 0 | 1 | 5 | 4 |
| Early menopause | 0 | 1 | – | – |
| Low testosterone | NA | NA | 0 | 1 |
NE is not evaluable; and NA, not available.
Eighteen female patients were postpubertal at the time of transplantation, and of these 15 have normal gonadotropins and normal menses, 2 has abnormal gonadotropins, and 1 has early menopause. There were 32 postpubertal boys at time of transplantation, and data are available for 15, who seem to have normal function.
Pregnancies
After a CY-preparative regimen, there have been 73 evaluable pregnancies among women who were a median of 10 years (range 1.9-17.9 years) of age at the time of transplantation and a median of 24.1 years (range, 18.3-32.3 years) of age at time of first pregnancy (Table 6). The infants born to these women were normal, except for one with congenital aortic stenosis. There were 11 abortions; 7 spontaneous and 4 elective. In addition, there was one tubal pregnancy and one stillbirth. Among the men who received the CY-preparative regimen, their partners had 59 evaluable pregnancies, 47 (80%) of which resulted in live births. These infants were normal except for 2, who had hip dysplasia (n = 1) and cleft lip (n = 1). There were 2 women who received 12.0 Gy of TBI who also became pregnant, and both delivered normal infants. One of these women was age 3 years at transplantation, and the other was age 13 years.
Table 6.
Pregnancies after transplantation
| Preparative regimen | CY | TBI | |
|---|---|---|---|
| Sex | Female | Male* | Female |
| Prebirth | 1 | 2 | – |
| Total evaluable pregnancies | 73 | 59 | 2 |
| Live births, n (%) | 58 (82) | 47 (80) | 2 |
| Abortions | |||
| Spontaneous | 7 | 5 | – |
| Stillbirth | 1 | 0 | – |
| Elective | 4 | 6 | – |
| Tubal pregnancy | 1 | 1 | – |
| Birth defects | 1 | 2 | – |
CY indicates cyclophosphamide; and TBI, total body irradiation.
Partner of the male transplantation patient.
Bone mineral density
Bone mineral density studies at 100 days and 1 or more years after transplantation were initiated in 1997 and performed for 52 survivors. Among the 112 patients without chronic GVHD, 35 were evaluable, and of these 26 (74%) had normal bone mineral density. However, among the 42 patients with chronic GVHD, 17 (40%) were evaluable, and only 1 had normal bone mineral density. The 5 patients with Fanconi anemia who were studied all had normal bone mineral density.
Pulmonary function
Pulmonary function tests were performed for 108 patients when they were > 6 years of age. Among the 42 with chronic GVHD, 10 (24%) had evidence of restrictive lung disease as demonstrated by decreased lung volumes < 80% predicted, and 7 (17%) had obstructive lung disease as evidenced by forced expiratory volume in 1 second < 80% predicted and forced expired volume/forced vital capacity ratio < 70%. Among the 112 without chronic GVHD, 51 (46%) did not have pulmonary function testing performed, but of those who did, there were only 10 (9%) with restrictive lung disease and 10 with obstructive lung disease
Hepatitis C disease
Before 1992 none of the blood transfusion products patients received was screened for the presence of hepatitis C. One hundred four children received their transplants before 1992, and among these, 37 (35%) are known to be hepatitis C positive. An effort was made to have all of these patients tested, but the hepatitis C status of 22 survivors and 9 deceased patients is unknown. Of the 31 surviving who carry the hepatitis C virus, 1 developed liver failure and required a liver transplantation, and 12 were treated with ribavirin and/or interferon. Of these 12 treated patients, 5 no longer are hepatitis C positive, and 7 did not respond to treatment, with 1 receiving a liver transplantation. Eighteen remain untreated. Six patients died of hepatitis C disease complications, including one with hepatocellular carcinoma. One had failed treatment, 4 were untreated, and treatment status of the remaining patient is unknown. Among patients who were tested and underwent transplantation before 1995 (no hepatitis C-positive patient received a transplant after that), 2 of 47 (4%) hepatitis C-negative patients died, and 6 of 37 (16%) hepatitis C-positive patients died.
Quality of life
To understand how well these survivors are doing, 4 major complications were considered: chronic GVHD, pulmonary diseases, malignancy, and hepatitis C. Overall, 67 (44%) of the 152 patients had none of these complications, ie, 63 (46%) of the acquired AA patients and 4 (27%) of the Fanconi anemia patients. There were 49 (32%) who had one of these complications. There were 39 (26%) who had 2 or more of these complications, with 33 (24%) among the acquired AA survivors and 6 (40%) among the Fanconi anemia survivors.
Formal quality-of-life data were available for 49 adult survivors of a pediatric transplantation. These survivors were older than 18 years of age at the time the quality-of-life studies were performed. The Short Form-36 physical function and mental function studies among the AA survivors and the control patients (n = 197) were not significantly different from each other. Also not significantly different from controls was the educational status, work or school status, financial situation, or marital status of the patients. In contrast, insurance issues were significantly different for the transplant recipients compared with the control patients (P = .0001). Nine of 49 (18%) transplant recipients had been denied health insurance compared with 3 of 197 (2%) control patients. Similar data were found for life insurance. A total of 10 of 49 (20%) survivors had been denied life insurance compared with 2 of 197 (1%) of control patients.
Discussion
To determine a perspective of how the children survive long term after transplantation for severe AA, we evaluated the outcome and delayed effects in the patients who survived one or more years after transplantation and who were younger than 18 years of age when they underwent transplantation at FHCRC. With follow-up reaching 38 years, these findings are unique and should be instructive in guiding our counseling of future pediatric patients undergoing transplantation for severe AA.
Survival is excellent but continues to decrease even more than 20 years after transplantation, primarily from late pulmonary failure, malignancy, and liver failure secondary to hepatitis C disease. The advent of blood product screening for the hepatitis C virus has virtually eliminated hepatitis C as a cause of late transplantation-related death or complication. Among the deaths attributable to posttransplantation malignancy were deaths secondary to SCC, which occurred primarily in patients with significant skin chronic GVHD. However, as has been previously observed, chronic GVHD remains a significant factor for mortality, as does receiving 10-12.0 Gy of TBI.8 This study shows that future efforts must continue in the realm of controlling or preventing chronic GVHD as well as avoiding high-dose TBI whenever possible for children with AA.
Posttransplantation autologous recovery after allogeneic transplantation for AA has recently been documented by European Blood and Marrow Transplant group to occur in 4.2% of AA patients receiving transplants between 1973 and 2005. These 45 patients had survival for 10 years of 84%.21 In the present study the incidence of autologous recovery was similarly low at 1.5%. The European Blood and Marrow Transplant group reported that the chronic GVHD rate was significantly lower in the autologous recovery patients than in those with stable engraftment and that those with autologous recovery had improved rates of survival. The same observation cannot be made in the present study, where the overall incidence of chronic GVHD is 33%, survival of those without GVHD is 82%, and the number with autologous recovery is too small to be evaluable.
Chronic GVHD emerged as a risk factor for mortality, with survival among chronic GVHD patients significantly less than among those without chronic GVHD. This finding is similar to our previous observation among adult and pediatric long-term survivors who underwent transplantation for AA.8 Clearly improved methods for prevention, early diagnosis, and treatment of chronic GVHD are needed to improve survival. This is particularly important because the acceptable transplant donor pool continues to be expanded.
Malignancies were observed in 13% of the patients and were more common among those who received TBI. However, the most common malignancy, SCC, is thought not to be related to irradiation. Curtis et al22 reported that chronic GVHD and its therapy were strongly related to the risk of SCC, but no increased risk was found for non-SCC. In another report, the 20-year cumulative incidence of SCC was 3.4% among more than 4000 allogeneic transplantation patients evaluated.23 Excluding the 7 SCC patients among the 137 idiopathic AA patients leaves a malignancy incidence of 9.2%, which is greater than the 1.6% incidence rate reported by Surveillance, Epidemiology and End Results for any invasive malignancy (excluding SCC).24 In the current study, one SCC death was in a Fanconi anemia patient without chronic GVHD, and the remaining 7 instances of SCC occurred among patients with chronic GVHD and was the cause of death for one. Improved methods of preventing chronic GVHD are needed to decrease the incidence of SCC among survivors.
Although growth after marrow transplantation has been reported to be problematic, this has been noted among children who received high-dose TBI preparative regimens.25 Children who do not receive high-dose TBI-containing regimens have normal or near-normal growth. The evaluation of growth must always be tempered with consideration of the biology of parental growth. Parental height was not measured for the children in this report, but the observation that the standard deviation curves were normal for both boys and girls is gratifying. Concern should be raised if a child who received a CY-only preparative regimen has subnormal growth.
We have reported that the majority of young children progress normally through puberty when given CY-only preparative regimens.25 The present study confirms these previous observation with 85% of girls and 45% of boys developing normally. Not surprising is the observations of pregnancies after high-dose CY reported by us and others.8,26,27 The incidence of congenital heart disease in the present study was 1% (1/105), which is similar to the 0.8% expected. The present studies confirm these previous observations and notes that minimal number of minor birth defects was observed.
Decreased bone mineral density has been reported after marrow transplantation. This is a particularly important problem if it occurs in the adolescence and early adulthood because the attainment of sufficient bone mass during this time is an important determinant of long-term bone health.28 In the present study, nearly all of those with chronic GVHD and hence those most likely receiving glucocorticoid therapy developed osteopenia or osteoporosis. We were not able to evaluate the impact of therapy on subsequent fractures, but there were several documented fractures in the chronic GVHD group. The authors of previous studies have demonstrated the beneficial effect of bisphosphonate therapy versus standard calcium and vitamin D-replacement therapy for children with decreased bone mineral density.29
Overall the survival of this group of children is excellent at 82% for the acquired AA patients at 30 years after transplantation. No other study groups have evaluated these patients this long after transplantation. Survival is important, but the quality of survival is also very important. The previous report of the quality of life of these patients indicates that the majority of adult survivors of a childhood transplantation are functioning well.8,19 With the patients in the current report, some had insurance issues to overcome, but in general these former patients' physical and mental function was not different from healthy, normal patients who did not undergo transplantation. This finding is in contrast to the difficulties others have observed among adolescents or adults who received transplants as children.30,31 It is gratifying that these patients are not different from control patients in the overall quality of life.
In summary, this analysis confirms that marrow transplantation offers effective therapy for children with severe AA. The majority who survived beyond 1 year after transplantation grew up to become normal functioning adults. The likelihood was greater for this among those without chronic GVHD. Some patients who developed problems secondary to hepatitis C, which would not happen today because of testing for this virus. The children grow up and have normal children of their own. Overall, they are doing very well.
Acknowledgments
The authors thank all the patients and their parents, their physicians, and their nurses, who have continued to provide us with follow-up information.
This work was supported by grants HL 36444 and CA 15704.
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.E.S. participated in transplantation of these patients, designed the prospective collection of data, and wrote this study; A.E.W. and P.A.C. participated in the care and transplantation of these patients; B.E.S. performed the statistical data analysis; P.A.H. collected the data and managed the database; H.J.D. designed the unrelated donor transplantation-preparative regimens and participated in the manuscript preparation; M.E.D.F. designed the Fanconi anemia patient preparative regimens and participated in care of long-term patients; and R.F.S. designed the transplantation protocols, participated in the care of the patients, and in the preparation of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jean E. Sanders, MD, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave North, Suite D5-280, Seattle, WA 98109; e-mail: jsanders@fhcrc.org.
References
- 1.Camitta BM, Thomas ED, Nathan DG, et al. Severe aplastic anemia: a prospective study of the effect of early marrow transplantation on acute mortality. Blood. 1976;48(1):63–70. [PubMed] [Google Scholar]
- 2.Storb R, Etzioni R, Anasetti C, et al. Cyclophosphamide combined with antithymocyte globulin in preparation for allogeneic marrow transplants in patients with aplastic anemia. Blood. 1994;84(3):941–949. [PubMed] [Google Scholar]
- 3.Young NS. Acquired aplastic anemia [review]. Ann Intern Med. 2002;136(7):534–546. doi: 10.7326/0003-4819-136-7-200204020-00011. [DOI] [PubMed] [Google Scholar]
- 4.Storb R, Deeg HJ, Pepe M, et al. Graft-versus-host disease prevention by methotrexate combined with cyclosporin compared to methotrexate alone in patients given marrow grafts for severe aplastic anaemia: long-term follow-up of a controlled trial. Br J Haematol. 1989;72(4):567–572. doi: 10.1111/j.1365-2141.1989.tb04325.x. [DOI] [PubMed] [Google Scholar]
- 5.Kröger N, Zabelina T, Renges H, et al. Long-term follow-up of allogeneic stem cell transplantation in patients with severe aplastic anemia after conditioning with cyclophosphamide plus antithymocyte globulin. Ann Hematol. 2002;81(11):627–631. doi: 10.1007/s00277-002-0566-0. [DOI] [PubMed] [Google Scholar]
- 6.Fouladi M, Herman R, Rolland-Grinton M, et al. Improved survival in severe acquired aplastic anemia of childhood. Bone Marrow Transplant. 2000;26(11):1149–1156. doi: 10.1038/sj.bmt.1702699. [DOI] [PubMed] [Google Scholar]
- 7.Burroughs L, Woolfrey A, Storer B, et al. Improved survival following HLA-matched related marrow transplantation in pediatric patients with severe aplastic anemia: (a 39-year retrospective analysis) [abstract]. Biol Blood Marrow Transplant. 2011;17:S264. #302. [Google Scholar]
- 8.Deeg HJ, Leisenring W, Storb R, et al. Long-term outcome after marrow transplantation for severe aplastic anemia. Blood. 1998;91(10):3637–3645. [PubMed] [Google Scholar]
- 9.Ades L, Mary JY, Robin M, et al. Long-term outcome after bone marrow transplantation for severe aplastic anemia. Blood. 2004;103(7):2490–2497. doi: 10.1182/blood-2003-07-2546. [DOI] [PubMed] [Google Scholar]
- 10.Socié G, Salooja N, Cohen A, et al. Nonmalignant late effects after allogeneic stem cell transplantation [review]. Blood. 2003;101(9):3373–3385. doi: 10.1182/blood-2002-07-2231. [DOI] [PubMed] [Google Scholar]
- 11.Sanders JE, Hoffmeister PA, Woolfrey AE, et al. Thyroid function following hematopoeitic cell transplantation in children: 30 years experience. Blood. 2009;113(2):306–308. doi: 10.1182/blood-2008-08-173005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanders JE. Growth and development after hematopoietic cell transplant in children. Bone Marrow Transplant. 2008;41(2):223–227. doi: 10.1038/sj.bmt.1705875. [DOI] [PubMed] [Google Scholar]
- 13.Sanders JE, Whitehead J, Storb R, et al. Bone marrow transplantation experience for children with aplastic anemia. Pediatrics. 1986;77(2):179–186. [PubMed] [Google Scholar]
- 14.Storb R, Doney KC, Thomas ED, et al. Marrow transplantation with or without donor buffy coat cells for 65 transfused aplastic anemia patients. Blood. 1982;59(2):236–246. [PubMed] [Google Scholar]
- 15.Deeg HJ, Self S, Storb R, et al. Decreased incidence of marrow graft rejection in patients with severe aplastic anemia: changing impact of risk factors. Blood. 1986;68(6):1363–1368. [PubMed] [Google Scholar]
- 16.Storb R, Thomas ED, Weiden PL, et al. One-hundred-ten patients with aplastic anemia (AA) treated by marrow transplantation in Seattle. Transplant Proc. 1978;10(1):135–140. [PubMed] [Google Scholar]
- 17.Sullivan KM, Witherspoon RP, Storb R, et al. Chronic graft-versus-host disease: recent advances in diagnosis and treatment. In: Gale RP, Champlin R, editors. Bone Marrow Transplantation: Current Controversies. UCLA Symposia on Molecular and Cellular Biology, New Series. New York, NY: Alan R. Liss, Inc; 1989. pp. 511–522. [Google Scholar]
- 18.Loughran TP, Jr, Sullivan K, Morton T, et al. Value of day 100 screening studies for predicting the development of chronic graft-versus-host disease after allogeneic bone marrow transplantation. Blood. 1990;76(1):228–234. [PubMed] [Google Scholar]
- 19.Sanders JE, Hoffmeister PA, Storer BE, Appelbaum FR, Storb RF, Syrjala KL. The quality of life of adult survivors of childhood hematopoietic cell transplant. Bone Marrow Transplant. 2010;45(4):746–754. doi: 10.1038/bmt.2009.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffmeister PA, Madtes DK, Storer BE, Sanders JE. Pulmonary function in long-term survivors of pediatric hematopoietic cell transplantation. Pediatr Blood Cancer. 2006;47(5):594–606. doi: 10.1002/pbc.20531. [DOI] [PubMed] [Google Scholar]
- 21.Piccin A, McCann S, Socie G, et al. Survival of patients with documented autologous recovery after SCT for severe aplastic anemia: a study by the WPSAA of the EBMT. Bone Marrow Transplant. 2010;45(6):1008–1013. doi: 10.1038/bmt.2009.296. [DOI] [PubMed] [Google Scholar]
- 22.Curtis RE, Metayer C, Rizzo JD, et al. Impact of chronic GVHD therapy on the development of squamous-cell cancers after hematopoietic stem-cell transplantation: an international case-control study. Blood. 2005;105(10):3802–3811. doi: 10.1182/blood-2004-09-3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leisenring W, Friedman DL, Flowers MED, Schwartz JL, Deeg HJ. Nonmelanoma skin and mucosal cancers after hematopoietic cell transplantation. J Clin Oncol. 2006;24(7):1119–1126. doi: 10.1200/JCO.2005.02.7052. [DOI] [PubMed] [Google Scholar]
- 24.Bethesda, MD: National Cancer Institute; 2006. Cancer Epidemiology in Older Adolescents and Young Adults 15 to 29 Years of Age, Including SEER Incidence and Survival: 1975-2000. National Institutes of Health Pub. No. 06-5767. [Google Scholar]
- 25.Sanders JE. Growth and development after hematopoietic cell transplantation. In: Appelbaum FR, Forman SJ, Negrin RS, Blume KG, editors. Thomas' Hematopoietic Cell Transplantation. Oxford, UK: Wiley-Blackwell; 2009. pp. 1608–1619. [Google Scholar]
- 26.Sanders JE, Hawley J, Levy W, et al. Pregnancies following high-dose cyclophosphamide with or without high-dose busulfan or total-body irradiation and bone marrow transplantation. Blood. 1996;87(7):3045–3052. [PubMed] [Google Scholar]
- 27.Loren AW, Chow E, Jacobsohn DA, et al. Pregnancy after hematopoietic cell transplantation: a report from the late effects working committee of the Center for International Blood and Marrow Transplant Research (CIBMTR). Biol Blood Marrow Transplant. 2011;17(2):157–156. doi: 10.1016/j.bbmt.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshall D, Johnell O, Wedel H. Meta-analysis of how well measures of bone mineral density predict occurrence of osteoporotic fractures. BMJ. 1996;312(7041):1254–1259. doi: 10.1136/bmj.312.7041.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carpenter PA, Hoffmeister P, Chesnut CH, III, et al. Bisphosphonate therapy for reduced bone mineral density in children with chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2007;13(9):683–690. doi: 10.1016/j.bbmt.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Felder-Puig R, Peters C, Matthes-Martin S, et al. Psychosocial adjustment of pediatric patients after allogeneic stem cell transplantation. Bone Marrow Transplant. 1999;24(1):75–80. doi: 10.1038/sj.bmt.1701853. [DOI] [PubMed] [Google Scholar]
- 31.Nespoli L, Verri AP, Locatelli F, Bertuggia L, Taibi RM, Burgio GR. The impact of paediatric bone marrow transplantation on quality of life. Qual Life Res. 1995;4(3):233–240. doi: 10.1007/BF02260862. [DOI] [PubMed] [Google Scholar]