

Abstract
Background
Globally, gastric cancer is the second most common cause of cancer-related death, with the majority of the health burden borne by economically less-developed countries.
Methods
Here, we report a genetic characterization of 50 gastric adenocarcinoma samples, using affymetrix SNP arrays and Illumina mRNA expression arrays as well as Illumina sequencing of the coding regions of 384 genes belonging to various pathways known to be altered in other cancers.
Results
Genetic alterations were observed in the WNT, Hedgehog, cell cycle, DNA damage and epithelial-to-mesenchymal-transition pathways.
Conclusions
The data suggests targeted therapies approved or in clinical development for gastric carcinoma would be of benefit to ~22% of the patients studied. In addition, the novel mutations detected here, are likely to influence clinical response and suggest new targets for drug discovery.
Background
Despite recent decline of mortality rates from gastric cancer in North America and in most of Northern and Western Europe, stomach cancer remains one of the major causes of death worldwide and is common in Japan, Korea, Chile, Costa Rica, Russian Federation and other countries of the former soviet union [1]. Despite improvements in treatment modalities and screening, the prognosis of patients with gastric adenocarcinoma remains poor [2]. To understand the pathogenesis and to develop new therapeutic strategies, it is essential to dissect the molecular mechanisms that regulate the progression of gastric cancer. In particular, the oncogenic mechanisms which can be targeted by personalized medicine.
The term "oncogene addiction" to describe cancer cells highly dependent on a given oncogene or oncogenic pathway was introduced by Weinstein [3,4]. The concept underscores the development of targeted therapies which attempt to inactivate an oncogene, critical to survival of cancer cells whilst sparing normal cells which are not similarly addicted.
Several oncogenes activated at high frequency in other cancers have also been shown to be mutated in gastric cancer. It follows that marketed therapeutics targeting these oncogenes would effectively treat a proportion of gastric carcinomas, either as single agents or in combination. In January 2010, trastuzumab was approved in combination with chemotherapy for the first-line treatment of ERBB2-positive advanced and metastatic gastric cancer. Trastuzumab is the first targeted agent to be approved for the treatment of gastric carcinoma and an increase of 12.8% in response rate was seen with addition of Trastuzumab to chemotherapy in ERBB2 positive gastric adenocarcinoma [5,6]. It has been estimated that 2-27% of gastric cancers harbour ERBB2 amplifications and may be treated with ERBB2 inhibitors [7,8]. Similarly, overexpression of another receptor tyrosine kinase (RTK) EGFR, has been noted in gastric cancer and multiple trials of EGFR inhibitors in this cancer type are ongoing (reviewed in [9,10]). Furthermore some gastric cancers harbour DNA amplification or overexpression of the RTK MET [11,12] and its paralogue MST1R [13] and may be treated with MET or MST1R inhibitors [14-20]. Finally, FGFR2 over expression and amplification has been observed in a small proportion of gastric cancers (scirrhous) [21] and inhibitors have shown some efficacy in clinic [22].
Downstream of the RTKs, KRAS wildtype amplification and mutation has also been found in about 9-15% of gastric cancers [23,24] and may be effectively treated with MEK inhibitors [25,26]. Activation of the Pi3K/AKT/mTOR pathway has also been seen in 4-16% of gastric cancer [27-30] and so may be sensitive to PI3K inhibitors [31-34]. Similarly, cell cycle kinase AURKA has been shown to be activated in gastric cancer [35,36] and AURKA inhibitors in clinical development [37] may have clinical benefit.
Reports of the frequency of different types of oncogenic activation and their co-occurrence are limited. In contrast to gastrointestinonal stromal tumours (GIST) which are characterized by a high frequency of KIT and PDGFRA activation [38] and hence effectively treated in the majority by imitanib and sunitinib [39,40], gastric adenocarcinoma appears to be a molecularly heterogeneous disease with no high-frequency oncogenic perturbation discovered thus far. This is illustrated by a recent survey of somatic mutation in kinase coding genes across 14 gastric cancer cell lines and three gastric cancer tissues which discovered more than 300 novel kinase single nucleotide variations and kinase-related structural variants. However, no very frequently recurrent mutation or mutated kinase was uncovered [41].
With the aim of elucidating the potential for treatment of gastric carcinoma with targeted therapies either on the market, in development or to be discovered, we have characterized clinical gastric carcinoma samples to detect oncogene activation.
We took a global approach by assaying the samples on affymetrix SNP arrays and Illumina mRNA expression arrays. These technologies are well validated for detection of genotype, DNA copy number variation and mRNA expression profile. They are amenable to heterogeneous clinical samples. The samples were also interrogated by second generation (Illumina) sequencing. Relatively novel second generation sequencing technologies offer both increased throughput and deep sequencing capacity. The latter is especially important for characterizing cancer samples which tend to include a mixture of cell types including infiltrating normal cells, vasculature and tumour cell of different genotypes. In this study we utilized target enrichment and Illumina sequencing technology to sequence the coding regions of 384 genes. We decided to favour depth of coverage over wider coverage in order to capture mutations present in subpopulations within the tumours. Recent studies have shown cancers tend to harbour many mutations in a smaller number of signalling pathways [42,43] therefore we concentrated on genes in these pathways. We also included genes coding for proteins previously shown to affect response to targeted therapies and more likely to be successfully targeted by small molecule intervention, as our aim is to find more effective and novel ways of treating gastric carcinoma.
Methods
Tissue samples
DNA and RNA samples were obtained from hospitals in Russia and Vietnam according to IRB approved Protocols and with IRB approved Consent forms for molecular and genetic analysis. The medical centres themselves also have internal ethical committees with reviewed the protocol and ICFs. The samples were sourced through Tissue Solutions Ltd http://www.tissue-solutions.com/. For sample characteristics see additional file 1 table S1
Arrays
Genotypes and copy number profiles were generated for each samples using 1 μg of DNA run on Affymetrix SNP V6 arrays using Affymetrix protocols. Copy number variation data was analysed within the ArrayStudio software http://www.Omicsoft.com. Data was normalized using Affymetrix algorithm and segmented using CBS. A transcript profile was generated for each sample using 1 μg of total RNA run on Illumnia HG-12 RNA expression arrays following the Illumina protocols. Data was analysed within the Illumina GenomeStudio software http://www.illumina.com/software/genomestudio_software.ilmn. As a data pre-processing procedure, a probe set was only retained if it has a "present" (i.e. two standard deviations above background) call in at least one of the samples. Signal values of the remaining probe sets were transformed to 2-based logarithm scale and quantile normalization was performed. DNA copy and RNA expression levels were integrated at the gene level within the ArrayStudio software http://www.Omicsoft.com. Pathway enrichment analysis was performed within the GeneGO metacore analysis suite http://www.genego.com/. All array data from this study is available in GEO http://www.ncbi.nlm.nih.gov/geo/ under series accession number GSE29999.
Targeted deep DNA sequencing
5 μg of DNA was PCR-enriched for the coding exons of any known transcript of 384 genes of interest (additional file 2 table S2) using the Raindance platform http://www.raindancetechnologies.com/.
The resulting target libraries were sequenced using Illumnia GAII at a read-length of 54 nt. Sequence reads were mapped to the reference genome (hg18) using the BWA program [44]. Bases outside the targeted regions were ignored when summarizing coverage statistics and variant calls. SAMtools was used to parse the alignments and make genotype calls [45], and any call that deviates from reference base was regarded as a potential variant. The SAMtools package generates consensus quality and variant quality estimates to characterize the genotype calls. Accuracy of genotype calls was estimated by concordance to genotype calls from the Affymetrix 6.0 SNP microarray. Concordance matrices of samples based on both SNP and sequence data were generated to check for sample mislabelling (additional file 3 figure S1). Concordance and quantity of genotype calls were tabulated for thresholds of consensus quality, variant quality, and depth. The final set of variant calls were identified using consensus quality greater than or equal to 50 and variant quality greater than 0. To exclusively identify somatic changes, only those mutations present in the cancer sample and not detected in any of the normal samples were retained. As an additional filter for germline variants, all variants present in dbSNP and 1000 genome polymorphism datasets were removed.
Q-PCR
Q-PCR was performed via standard protocol using Fluidigm 48*48 dynamic array. Firstly, a validation run was conducted using pooled control RNA from three specimens. Four input RNA amounts were tested (125 ng, 250 ng, 375 ng and 500 ng). Triplicate data points were obtained for the subsequently 10-point serial dilution per each condition per assay. The best overall results were at 250 or 500 ng, which yielded efficiency values ~85%. Therefore 250 ng input amount for the experimental samples. Data was produced in triplicate and mean combined. CT values were converted to abundance using standard formula abundance = 10(40-CT/3.5). Test data was normalised to housekeepers using the analysis of covariance method whereby the two housekeepers (GAPDH and beta-actin) were used to compute a robust score and the score was used as a covariate to adjust the other genes. Data analysis was performed in the Arraystudio software.
Sanger Sequencing
Genomic DNA PCR primers were ordered from IDT (Integrated DNA Technologies Inc, Coralville, Iowa). PCR reactions were carried out using Invitrogen Platnium polymerase (Invitrogen, Carlsbad, CA). 50 ng of genomic DNA was amplified for 35 cycles at 94°C for 30 seconds, 58°C for 30 seconds and 68°C for 45 seconds. PCR products were purified using Agencourt AmPure (Agencourt Bioscience Corporation, Beverly, MA). Direct sequencing of purified PCR products with sequencing primers were performed with AB v3.1 BigDye-terminator cycle sequencing kit (Applied Biosystems, Foster City, CA) and sequencing reactions were purified using Agencourt CleanSeq (Agencourt Bioscience Corporation, Beverly, MA). The sequencing reactions were analyzed using a Genetic Analyzer 3730XL (Applied Biosystems, Foster City, CA). All sequence results data were assembled and analyzed using Codon Code Aligner (CodonCode Corporation, Dedham, MA).
Results
DNA and RNA amplification patterns across samples are consistent with previous studies
Consistent with most other human cancers, copy number changes occurred across the genomes of the 50 gastric cancer samples compared to matched normal samples (Figure 1). Large regions of frequent amplification were found at chromosomal regions 8q, 13q, 20q, and 20p. Known oncogenes MYC and CCNE1 are located in the 8q and 20p amplicons, respectively and likely contribute to a growth advantage conferred by the amplification. These amplifications have been seen in prior studies in gastric cancer along with amplification of 20p for which ZNF217 and TNFRSF6B have been suggested as candidate driver genes [46].
Figure 1.
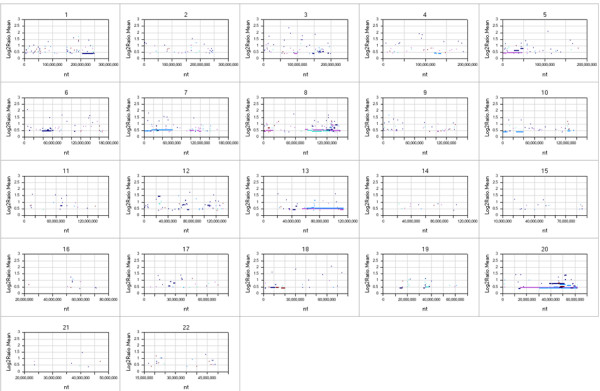
View of CNV aberrations across all 50 gastric carcinoma samples, for each autosome. The y-axis corresponds to the sum of the number of positive or negative changes for a particular segment with the log2 ratio of those change. Areas with increased or decreased copy number consistent throughout all the samples analysed or very large changes in few samples will show large positive and negative change sizes. Each dot or segment in figure is colored by sample. The colour code is arbitrary with each of the 50 cancer samples being assigned a colour. Amplified segments include chromosome 8q, 20q, 20p, 3q, 7p, and 1q.
Concordance between DNA copy number gain and RNA expression among the cancer samples was evaluated and the top 200 genes contained within a region of frequent high DNA copy in cancer samples and which had high mRNA levels (compared to matched normal tissue) are tabulated in additional file 4 table S3. Most of the genes on this list are from chromosomal regions 20q and 8q, suggesting that these amplifications have the most effect on mRNA levels, in the minority are genes for 20p, 3q, 7p, and 1q. Figure 2 shows the RNA profiles measured by Q-PCR of an exemplar gene from each region showing general overexpression in gastric cancer, particularly in certain samples. Besides MYC and CCNE1, there are multiple genes in these regions, which could contribute to a growth advantage for the cancer cell. The biological pathways most significantly enriched for amplified and overexpressed genes are involved in regulation of translation (p = 0.000015) and DNA damage repair (p = 0.003). Samples with amplifications in these genomic regions are annotated in Figure 3. There is no discernible tendency for amplifications in these regions to co-occur or to be exclusive. In agreement with a previous study [47], the PERLD1 locus was amplified (within the ERBB2 amplicon) in sample 08280 and MMP9 was overexpressed but not discernibly amplified. Also in Figure 3 focal DNA amplifications with concordant RNA expression of genes likely to affect the response to targeted therapies are denoted, for example underlying data see additional file 5 figure S2.
Figure 2.

Expression of example genes from each amplified chromosomal region across study samples confirmed by Q-PCR. Red dots denote cancer samples and white dots denote normal samples. The y-axis denotes the mRNA abundance.
Figure 3.

Mutational profile of samples. Tissue samples are displayed across the top and annotations relevant to them are in columns below. Red boxes denote DNA amplification and concordant mRNA overexpression, orange boxes denote RNA overexpression with no evidence of DNA amplification, red dots denote DNA loss. Blue boxes denote somatic nonsynonymous mutation validated by Sanger sequencing and purple boxes denote nonsynonymous somatic mutations, observed in the Illumina data with no attempt to confirm by Sanger sequencing. Amino changes are noted in the boxes and changes leading to loss or gain of a stop codon are in red text.
Sequencing data shows high concordance with genotyping
Sequencing library preparation failed for six of the original 50 cancer samples and fourteen of the original matched normal samples. Therefore two more matched pairs were added to the analysis, resulting in a dataset of 44 cancer samples, 36 with matched normal pairs (additional file 1 table S1). The targeted region included 3.28 MB across 6,547 unique exons in 384 genes (additional file 2 table S2). Median coverage of across all samples was 88.3% and dropped to 74% when requiring minimum coverage of 20. All sequencing was carried out to a minimum of 110x average read coverage across the enriched genomic regions for each sample. The reads were aligned against the human genome and variants from the reference genome were called. As a control, an analysis to compare genotyping calls from the Affymetrix V6 SNP arrays and the Illumina sequencing was performed. The regions targeted for sequencing contained 1005 loci covered by the Affymetrix V6 SNP arrays. With no filtering of the sequencing variant calls for quality metrics, the median agreement between the genotyping and sequencing results was 97.8% with a range of 65-99% (additional file 6a, Figure S3a). The raw overall genotype call concordance was 96.8%. Quality metrics were chosen to maximize the agreement between the genotyping and the sequencing calls while minimizing false negatives. The most informative metric was consensus quality and a cut-off of ≥50 resulted in loss of about 10% of the shared genotypes but an overall 2% increase in concordance to 98.7% (additional file 6b, Figure S3b). Variant genotype calls were isolated for further concordance analysis. In this set, a variant quality threshold of > 0 increased accuracy of variant genotype calls to 98.9% (additional file 6c, Figure S3c). When both quality thresholds were applied the median sample concordance is 99.5% (additional file 6d, Figure S3d) which is within the region of genotyping array error. Six samples (08362T1, 08373T2, 336MHAXA, 08337T1, 89362T2, DV41BNOH) had a concordance of < 98% and two of these (08393T2 and DV41BNOH) had a concordance of 82% and 88% respectively. Therefore with a consensus quality ≥ 50 and a variant quality > 0, the false positive rate was 0.5% and 1.6% for reference genotypes and variant genotypes, respectively (additional file 6e Figure S3e).
From all single nucleotide changes passing the above thresholds, all variants present in any of the normal samples or in the polymorphism databases of dbSNP (v130) or 1000 genomes were assumed to be germline variants and discarded. Variants present only in the exons of cancer samples were assumed to be somatic and retained. 18,549 somatic variants were detected in total across all 44 samples (additional file 7 Table S4), 3357 were predicted to be exonic and nonsynonymous. To prioritise for mutations with functional impact we concentrate all further analyses on nonsynonymous mutations and highlighted mutations leading to loss or gain of stop codons. We have applied the SIFT algorithm [48] to predict amino acid changes that are not tolerated in evolution and so are more likely to affect the function of the protein, 1509 somatic nonsynonymous mutations have a SIFT score of < 0.05. The rate of mutations with SIFT score < 0.05 per gene, corrected for CDS length was calculated (4). Figure 4 shows, the genes with the highest concentration of low SIFT scoring mutations were S1PR2, LPAR2, SSTR1, TP53, GPR78 and RET, with S1PR2 being most extreme. There are fifteen mutations with SIFT score <0.05 across the 353aa CDS of S1PR2, concentrated in nine samples. S1PR2 also known as EDG5 codes for a G-protein coupled receptor of S1P and activates RhoGEF, LARG [49]. Little is known of its role in cancer and somatic mutations have not been observed in the 44 tissues sequenced for S1PR2 in the COSMIC database [50].
Figure 4.
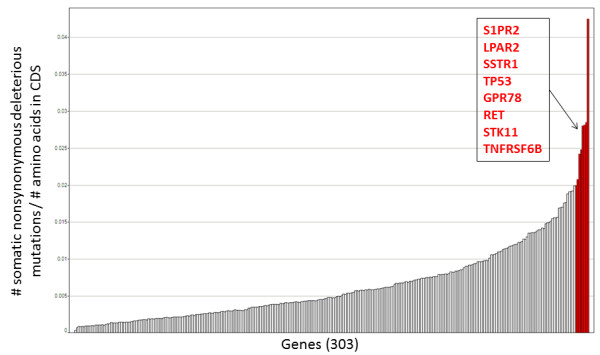
Bar chart of rate of deleterious mutations across gene sequenced. Genes sequenced are shown on the x-axis. The number of deleterious somatic nonsynonymous mutations observed in each gene/number of amino acids in each CDS in plotted.
Sequencing data is confirmed by Sanger sequencing
Some nonsynonymous somatic mutations were selected to be confirmed by Sanger sequencing. All mutations reported in blue in Figure 3 were confirmed by Sanger sequencing and were also confirmed to be somatic by sequencing of the wildtype sequence in the matched normal tissue (see additional file 8 Figure S4 for example sequencing traces). Although 74% were confirmed, some mutations detected in the Illumnia sequencing were not confirmed as somatic mutations by Sanger sequencing. Sixteen of the 68 (24%) mutations we attempted to confirm were present in the normal and cancer sample, these are germline mutations but not detected in any of the normal samples by Illumina sequencing and also not represented in dbSNP or 1000 genomes data. Five of the sixteen germline mutations were from cancer samples with no matched normal tissue included in the dataset, the other eleven came from cancer samples with matched normal tissue sequence included in the dataset. This evidences a rate of germline contamination not eliminated by the matched normal controls or the comparison to known polymorphism databases. It may be that the coverage of the substitutions in the normal tissue happens to be lower than in the cancer sample and so some germline mutations remain despite the somatic filters. Two of the 68 (3%) mutations we attempted to confirm were not present in the normal or cancer sample by Sanger sequencing. One cause could be false positives in the Illumnia data due to artefact; however additional file 6 Figure S3 shows the false positive rate to be low at least for those variants represented on the Affymetrix V6 arrays. Another possibility is that these are present in a subset of the sample below the sensitivity of the Sanger methodology but detected by the Illumina sequencing. Therefore, mutations reported in the Illumina sequencing are also reported in purple in Figure 3, some caution is warranted when interpreting these results as they may be germline polymorphisms or present only in a subset of the tumour sample.
Alterations in the RAS/RAF/MEK/ERK pathway
Three tumour samples had KRAS genetic alterations (Figure 3) suggesting therapeutic opportunity for treatment with MEK inhibitors. One of these alterations is a G12D mutation. KRAS G12D mutations have been shown to initiate carcinogenesis and tumour survival [51]. Amplification and overexpression of wildtype KRAS was seen in the other 2 samples. KRAS amplification has been observed before in 5% of primary gastric cancers. Gastric cancer cell lines with wildtype KRAS amplification show constitutive KRAS activation and sensitivity to KRAS RNAi knockdown [24]. A novel mutation in KRAS was also observed; (in sample 08393) the functional consequence is unknown.
The PIK3CA mutation co-occurring with KRAS G12D, is known to affect sensitivity to MEK inhibitors [25]; in addition, novel mutations observed in this study may also have consequences for the same class of therapeutics. For instance: KSR2 functions as a molecular scaffold to promote ERK signalling [52,53]. Therefore, mutations in KSR2 such as seen in seven samples may affect sensitivity to MEK inhibitors. A second example is ULK1, which positively controls autophagy downstream of mTOR [54] and is mutated in fourteen samples. Autophagy is increased along with ERK phosphorylation when gastric cancer cells are treated with a proteasome inhibitor [55], therefore mutations in ULK1 may affect sensitivity to proteasomal inhibitor treatments such as bortezomib as a single agent or in combination with MEK inhibitors.
Alterations in the PI3K/AKT pathway
There was substantial sequence disruption of the phosphoinositide-3-kinase (Pi3K) pathway genes in the sample set. There are a number of PI3K/AKT/mTOR inhibitors in clinical development and patients with activating mutations in the pathway are candidates for treatment [56]. PIK3CA mutations of known oncogenicity were found in four samples. This results in a frequency of PIK3CA hotspot mutation of 9%, slightly higher than previous estimates of 6% (12/185) [27] and 4.3% (4/94) [57]. The common PIK3CA hotspot mutations of known oncogenicity (E545K and H1047R) [58] were observed twice each. Another mutation in PIK3CA K111E, which has also been observed before in four samples in COSMIC, was observed once and potentially novel somatic mutations were observed in two more samples.
Five nonsynonymous AKT1 mutations were observed. Although AKT1 mutations are found in about 2% of all cancers, they mainly occur at amino acid 15 and the functional importance of mutation at other sites is unknown. Another nonsynonymous mutation in AKT2 was observed in sample 08407. AKT2 mutations are much rarer than AKT1 mutations, although an AKT2 mutation has been observed before in gastric carcinoma, at a 2% frequency [59]. Finally mutation of PTEN or MTOR may affect response to pathway inhibitors. Several PTEN mutations are noted and MTOR mutations are frequent.
Alterations in Receptor Tyrosine Kinases
The receptor tyrosine kinases (RTKs) and drug targets EGFR, ERBB2 and MET were each amplified (log2 > 0.6) and overexpressed at the RNA level in one cancer sample. It follows that the tumours may be sensitive to the inhibitors of the amplified RTKs. In addition, multiple nonsynonymous mutations are observed in their coding regions. Downstream mutations would be expected to influence response. For instance, in the MET amplified sample a truncating mutation in AKT3 may affect sensitivity to MET inhibitors.
FGFR2 is amplified and RNA overexpressed in two samples, there are also multiple mutations in FGFR1-4. Broad range RTK inhibitors, which target FGFRs among other kinases, may be efficacious in these patients [60,61].
Alterations in Cell Cycle Proteins
The viral oncogene homolog SRC is mutated in four of the tumour samples, two of the mutations are predicted to have a deleterious effect including introduction of a stop codon. This may counter-indicate SRC inhibitors. MET amplification is also a known resistance marker for anti-SRC therapeutics such as dasatanib [62,63]. The cell cycle related kinase, AURKA was amplified and overexpressed in one sample. AURKA inhibitors are in development for solid tumours [37] and may be indicated in this case. CCNE1 was amplified in two samples (08390 and 08357). High levels of CCNE1 have been shown to be frequently associated with early gastric cancer and metastasis but expression levels do not correlate with survival [64,65]. High CCNE1 levels have been suggested as a sensitivity marker for the gene-directed pro-drug enzyme-activated therapies [66]
Activation of wnt pathway is common in the carcinoma samples
Mutations were observed in the APC gene in 22 samples. APC is a tumour suppressor known to activate CTNNB1 and wnt pathway signalling, amongst other effects [67]. The wnt pathway has been previously found to be frequently activated in gastric cancer [68]. We used a transcriptional signature, generated from previous studies [69,70] and available at the Broad Institute MSigDB database to classify the study samples by their wnt transcriptional signatures. Figure 5A shows a heat map of the transcriptional levels of the WNT signature genes in the datasets. Activation of this pathway is higher in nearly all the cancer samples compared to the normal samples. Wnt inhibitors are the subject of intense investigation in pharmaceutical and academic research [71-73]. These results suggest they will have an indication in gastric cancer as well as many other cancers.
Figure 5.

Transcriptional signatures across samples. Clustered heatmap showing expression of A wnt signature genes and B hedgehog signature genes, across samples in the study. All expression values are Zscore normalized. Zscore <-1 are blue, Z-score > 1 are red with a graded coloring through white at 0. Sample names are on the x-axis, they are clustered by expression pattern and samples with high signature scores are to the right. Samples with somatic nonsynonymous APC mutations (A) or PTCH1 mutations (B) and denoted by an asterisk above the heatmaps. WNT signature genes (top to bottom): FSTL1, DACT1, CD99, LMNA, SERPINE1, TNFAIP3, GNAI2, ID2, MVP, ACTN4, CAPN1, LUZP1, MTA1, RPS19, PTPRE, AXIN2, NKD2, SFRS6, CCND1, SCAP, CPSF4, SENP2, DKK1, PRKCSH, SLC1A5, HDGF, CBX3, SCML1, PCNA, RPS11, SNRPA1, TGM2, LY6E, IFITM1, NSMAF, TCF20, BCAP31, AXIN1, AGRN, PLEKHA1, SLC2A1, CTNNB1, EIF5A, IMPDH2, GSK3B, PFN1, UBE, MAP3K11, ARHGDIA, HNRPUL1, FLOT2, GYPC, NCOA3, CENTB1, SYK, POLR2A, KRT5, DHX36, ELF1, SMG2, FGD6, MAPKAP1, LOC389435, RPL27A, SRP19, RPL39L, SFRS2IP, FUSIP1; Hedgehog signature genes (top to bottom): LRFN4, JAG2, RPL29, WNT5A, SNAI2, FST, MYCN, BMP4, CCND1, BMI1, CFLAR, PRDM1, GREM1, FOXF1, CCND2, CD44.
Activation of the hedgehog pathway is also common in the carcinoma samples
PTCH1 is a tumour suppressor and acts as a receptor for the hedgehog ligands and inhibits the function of smoothened. When smoothened is freed, it signals intracellularly leading to the activation of the GLI transcription factors [74]. Multiple somatic mutations of PTCH1 are recorded in COSMIC, consistent with its tumour suppressor role. The D362Y mutation seen in this study in sample FICJG, is in the fourth transmembrane domain of PTCH1 and has been previously seen as a loss-of-function germline mutation in a patient with Gorlin syndrome, predisposing to neoplasms (numbered D513Y due to different transcript) [75]. Therefore, sample FICJG is very likely to have deregulated hedgehog signalling and does indeed have high levels of GLI target genes (as defined by [74] (Figure 5B)). Other samples also contain PTCH1 mutations in the Illumina sequence data, including a truncating stop codon (Y140X) in sample 08379 and have high levels of hedgehog signature genes. Hedgehog signalling has previously been shown be frequently activated in gastric cancer [76] though no genetic cause has been previously implicated. Inhibitors of the hedgehog pathway are in clinical development [77,78].
Loss of Epithelial phenotype
Epithelial or mesenchymal status has been shown to affect response to multiple drugs [79] and samples may be more resistant due to loss of an epithelial phenotype. Both hedgehog and wnt signalling upregulate mesenchymal precursors such as BMP4 and mutations can lead directly to loss of epithelial phenotype. CDH1 is a marker of an epithelial phenotype and is often lost in gastric tumours due to the process of epithelial to mesenchymal transformation (EMT) and is a negative prognostic marker [80]. Mutations in CDH1 were observed in nine samples, including a D254G mutation in CDH1 was detected in sample 08359. A mutation at the same site (D254Y) has been recorded in COSMIC in a breast tumour and 211 somatic mutations have been observed in the 2732 samples sequenced for CDH1 in COSMIC. Mutation in SMAD4 is also likely to affect epithelial phenotype. Loss of SMAD4 function facilitates EMT and its re-expression reverses the process in cancer cell lines [81]. Mutations in tumour suppressor SMAD4 were observed in ten samples.
Sensitivity to chemotherapy
Multiple substitutions in BRCA1 were observed in ten samples, including three cases of substitution of a stop codon. Germline mutations in BRCA1 predispose patients to breast and ovarian cancer, multiple somatic mutations have been found in tumours [82]. BRCA1 expression levels and polymorphic status has been shown to correlate with sensitivity to chemotherapeutics in gastric cancer [83,84]. Therefore, the observed mutations of BRCA1 may affect sensitivity to chemotherapy.
Another commonly mutated gene which is linked to sensitivity to chemotherapy in gastric cancer is TP53 [85]. Eight examples of TP53 mutation including two stop codons are seen in the dataset.
Mutations in TRAPP were found in 22 samples, including one mutation to a stop codon. TRRAP is a component of histone acetyltransferase complexes and is implicated in oncogenic transformation and cell fate decisions through chromatin regulation [86]. Loss of function mutations of the Sacchromyces pombe orthologue of TRRAP, cause defects in G2/M cell cycle control and resistance to CHK1 overexpression [87]. Mutations in TRAPP are likely to affect response to HDAC and CHK1 inhibitors currently approved and in trials for use as anticancer agents [88-92].
Novel targets for therapies in gastric cancer
An additional aim of our study was to uncover novel drug targets for gastric cancer. Many novel perturbations were observed in tractable target genes, following are three examples which warrant further investigation.
Thyrotropin receptor (TSHR) is mutant in four samples. The A553T mutation of TSHR found in sample 08360, has been previously been observed in two siblings with congenital hypothyroidism and was found to be inactivating [93]. Both loss and gain of function TSHR mutations are often found in thyroid cancer [94]. However, a role for TSHR in other cancers has not been elucidated, although infrequent mutations in lung cancer are recorded in COSMIC and TSHR has been shown to be lost at the DNA level, in some gastric cancers [95]. Three of the four TSHR mutations found have very low SIFT scores and may suggest deregulation of this growth hormone pathway.
We used the COPA algorithm [96] to identify mRNAs with outlier expression in the cancer samples. The top gene identified was KLK6. KLK6 is not detected or detected at very low levels in the normal samples, whilst its expression is very high in eleven of the cancer samples. Figure 6 shows the expression profile of KLK6 across the samples, confirmed by Q-PCR. KLK6 has previously been shown to be over expressed in gastric cancer and RNAi mediated knockdown of KLK6 in gastric cancer cell lines has been shown to be anti-proliferative and anti-invasive [97,98].
Figure 6.
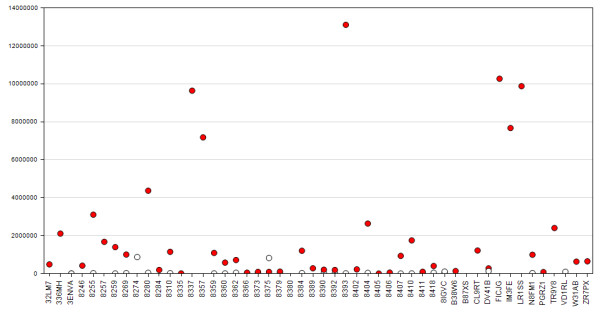
Expression of KLK6 across study samples confirmed by q-PCR. Red dots denote cancer samples and white dots denote normal samples. Patient IDs are arranged on the x-axis. The y-axis is the mRNA abundance.
Finally, mutations in the Rho associated coiled-coil containing protein kinases (ROCK1 and ROCK2) are interesting in view of their role as effectors of RhoA GTPase and the recent finding that truncating mutations in ROCK1 (similar to the confirmed ROCK2 mutation in this study) are activating and lead to increased motility and adhesion in cancer cells [99].
Discussion
Gastric adenocarcinoma rates vary widely across geographical regions, gender, ethnicity and time [100]. Diet has been shown to significantly influence gastric cancer risk as have tobacco smoking and obesity [101]. The infectious agent Helicobacter pylori is intimately associated with the most common types of gastric adenocarcinoma development [102]. H. pylori colonizes the stomach of at least half the world's population, virtually all persons infected with H. pylori develop gastric inflammation, which confers an increased risk for developing gastric cancer; however, only a fraction of infected individuals develop the clinical disease [103]. H. pylori induces generalized mutation and genomic instability in host DNA [104], which along with the complex risk profile suggests diverse routes to oncogenesis in gastric adenocarcinoma.
Therefore, an individualized personal medicine approach, measuring molecular targets in tumours and suggesting treatment regimens based on the results, is attractive. A recent study using this approach across tumour types has reported improved outcomes [105]. The trial used IHC, FISH and microarray technologies to assay levels of molecular targets in tumours, as the authors mention, second generation sequencing techniques offers a more complete picture of tumour mutagenic profile and will be even more informative in identifying sensitivity and resistance biomarkers.
Conclusions
This study evidences previously observed perturbations of the KRAS, ERBB2, EGFR, MET, PIK3CA, FGFR2 and AURKA genes in gastric cancer and suggests some of the targeted therapies approved or in clinical development would be of benefit to 11 of the 50 patients studied. The data, also suggests that agents targeting the wnt and hedgehog pathways would be of benefit to a majority of patients. The previously undocumented DNA mutations discovered are likely to affect clinical response to marked therapeutics and may be good drug targets. Detection of these mutations was enabled by Illumina sequencing and the concordance with genotyping arrays shows its suitability for heterogeneous cancer samples. These "nextgen sequencing" techniques are just at the beginning of expanding our abilities to detect genome wide DNA mutation, DNA copy number, RNA levels and epigenetic changes, in each patient's genome. However, it remains a challenge to filter germline from somatic mutations and sort driver mutations with functional import from passenger mutations.
Whole genome studies using both Sanger and nextgen sequencing have revealed mutagenic profiles of other cancers in unprecedented completeness and detail [41,106-112]. Similar studies with large numbers of samples will be critical to fully appreciate the mutagenic diversity in gastric cancer and identify the important driver mutations. Bodies such as the ICGC (International Cancer Genomics Consortium) are currently collecting gastric adenocarcinoma samples.
Translation of these findings to clinic will require pinpointing of important mutations as well as easier access to broad diagnostic assays and clinical development of agents targeting low-frequency events [113]. Data such as that presented here, is a necessary preliminary step in delivering the maximum benefit from the major advances of targeted therapies and personalized medicine to gastric cancer patients.
Competing interests
The authors declare that they have no competing interests.
JDH, KTG, WSH, AMH, PFL and PK are, or were employees of Glaxosmithkline plc and hold stock.
JSP and VJW are an employees of Expression Analysis Inc., who were financially compensated for some of the work in this manuscript by Glaxosmithkline.
Authors' contributions
JDH, PFL and RK: Developed the initial idea and design of the study
JDH: managed data acquisition, analysed the array, qPCR and sequence data, interpreted the findings and drafted the manuscript.
RK: contributed to the manuscript
JSP and VJW: Analysed Illumina sequence data
KTG: Managed samples and performed translocation discovery
WSH and AMH: Carried out Sanger sequencing
All authors revised and commented on drafts of the manuscript
Supplementary Material
Table S1: Sample characteristics.
Table S2: List of genes sequenced.
Figure S1: Concordance matrices of samples based on array and sequence data.
Table S3: Top 200 genes with amplification at the DNA levels and concordant overexpression at the mRNA level.
Figure S2: Array data evidencing focal amplifications. Top panels show mRNA expression data from arrays, bottom panels show log2 value for DNA abundance in genomic context as derived from SNP arrays.
Figure S3: Comparison of genotyping calls with sequencing data. A total of 1005 common loci were mapped between the Affymetrix 6.0 SNP microarray and the targeted regions. Concordance of genotype calls between affymetrix 6.0 SNP and SAMtools with no filters applied (top left). Application of a consensus quality filters (threshold values plotted as points) improves concordance (y-axis) but reduces the total number of calls (x-axis)(top right). A similar trend is observed for the variant quality thresholds, but at different threshold values (plotted points)(middle left). Sample concordance of genotype calls is improved with consensus quality filter >= 50 and variant quality > 0 (middle right). The total number of genotype calls stratified by reference or variant genotype, and concordance (bottom left).
{kind=link}
Table S4: All somatic variants detected.
Figure S4: Sanger sequencing traces. Sanger sequencing traces for variants denoted by blue boxes in Figure 3 (i.e. confirmed in Illumnia and Sanger) are provided.
Contributor Information
Joanna D Holbrook, Email: joanna_holbrook@sics.a-star.edu.sg.
Joel S Parker, Email: jparker@expressionanalysis.com.
Kathleen T Gallagher, Email: Kathleen.T.Gallagher@gsk.com.
Wendy S Halsey, Email: Wendy.S.Halsey@gsk.com.
Ashley M Hughes, Email: Ashley.M.Hughes@gsk.com.
Victor J Weigman, Email: vweigman@expressionanalysis.com.
Peter F Lebowitz, Email: Peter.F.Lebowitz@gmail.com.
Rakesh Kumar, Email: Rakesh.2.Kumar@gsk.com.
Acknowledgements
We would like to thank Don Gregory of GenomeQuest, for help in data management and processing.
References
- Bertuccio P, Chatenoud L, Levi F, Praud D, Ferlay J, Negri E, Malvezzi M, La Vecchia C. Recent patterns in gastric cancer: a global overview. Int J Cancer. 2009;125:666–673. doi: 10.1002/ijc.24290. [DOI] [PubMed] [Google Scholar]
- Khosravi Shahi P, Diaz Munoz de la Espada VM, Garcia Alfonso P, Encina Garcia S, Izarzugaza Peron Y, Arranz Cozar JL, Hernandez Marin B, Perez Manga G. Management of gastric adenocarcinoma. Clin Transl Oncol. 2007;9:438–442. doi: 10.1007/s12094-007-0082-8. [DOI] [PubMed] [Google Scholar]
- Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077–3080. doi: 10.1158/0008-5472.CAN-07-3293. discussion 3080. [DOI] [PubMed] [Google Scholar]
- Okines AF, Cunningham D. Trastuzumab in gastric cancer. Eur J Cancer. 2010;46:1949–1959. doi: 10.1016/j.ejca.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T. et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- Grabsch H, Sivakumar S, Gray S, Gabbert HE, Muller W. HER2 expression in gastric cancer: Rare, heterogeneous and of no prognostic value - conclusions from 924 cases of two independent series. Cell Oncol. 2010;32:57–65. doi: 10.3233/CLO-2009-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainberg ZA, Anghel A, Desai AJ, Ayala R, Luo T, Safran B, Fejzo MS, Hecht JR, Slamon DJ, Finn RS. Lapatinib, a dual EGFR and HER2 kinase inhibitor, selectively inhibits HER2-amplified human gastric cancer cells and is synergistic with trastuzumab in vitro and in vivo. Clin Cancer Res. 2010;16:1509–1519. doi: 10.1158/1078-0432.CCR-09-1112. [DOI] [PubMed] [Google Scholar]
- Arkenau HT. Gastric cancer in the era of molecularly targeted agents: current drug development strategies. J Cancer Res Clin Oncol. 2009;135:855–866. doi: 10.1007/s00432-009-0583-7. [DOI] [PubMed] [Google Scholar]
- Ku GY, Ilson DH. Esophagogastric cancer: targeted agents. Cancer Treat Rev. 2010;36:235–248. doi: 10.1016/j.ctrv.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Huang TJ, Wang JY, Lin SR, Lian ST, Hsieh JS. Overexpression of the c-met protooncogene in human gastric carcinoma--correlation to clinical features. Acta Oncol. 2001;40:638–643. doi: 10.1080/028418601750444204. [DOI] [PubMed] [Google Scholar]
- Kuniyasu H, Yasui W, Kitadai Y, Yokozaki H, Ito H, Tahara E. Frequent amplification of the c-met gene in scirrhous type stomach cancer. Biochem Biophys Res Commun. 1992;189:227–232. doi: 10.1016/0006-291X(92)91548-5. [DOI] [PubMed] [Google Scholar]
- Zhou D, Pan G, Zheng C, Zheng J, Yian L, Teng X. Expression of the RON receptor tyrosine kinase and its association with gastric carcinoma versus normal gastric tissues. BMC Cancer. 2008;8:353. doi: 10.1186/1471-2407-8-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan SG, Hendle J, Lee PS, Smith CR, Bounaud PY, Jessen KA, Tang CM, Huser NH, Felce JD, Froning KJ. et al. SGX523 is an exquisitely selective, ATP-competitive inhibitor of the MET receptor tyrosine kinase with antitumor activity in vivo. Mol Cancer Ther. 2009;8:3181–3190. doi: 10.1158/1535-7163.MCT-09-0477. [DOI] [PubMed] [Google Scholar]
- Dai Y, Siemann DW. BMS-777607, a small-molecule met kinase inhibitor, suppresses hepatocyte growth factor-stimulated prostate cancer metastatic phenotype in vitro. Mol Cancer Ther. 2010;9:1554–1561. doi: 10.1158/1535-7163.MCT-10-0359. [DOI] [PubMed] [Google Scholar]
- Munshi N, Jeay S, Li Y, Chen CR, France DS, Ashwell MA, Hill J, Moussa MM, Leggett DS, Li CJ. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther. 2010;9:1544–1553. doi: 10.1158/1535-7163.MCT-09-1173. [DOI] [PubMed] [Google Scholar]
- Pan BS, Chan GK, Chenard M, Chi A, Davis LJ, Deshmukh SV, Gibbs JB, Gil S, Hang G, Hatch H. et al. MK-2461, a novel multitargeted kinase inhibitor, preferentially inhibits the activated c-Met receptor. Cancer Res. 2010;70:1524–1533. doi: 10.1158/0008-5472.CAN-09-2541. [DOI] [PubMed] [Google Scholar]
- Qian F, Engst S, Yamaguchi K, Yu P, Won KA, Mock L, Lou T, Tan J, Li C, Tam D. et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009;69:8009–8016. doi: 10.1158/0008-5472.CAN-08-4889. [DOI] [PubMed] [Google Scholar]
- Smolen GA, Sordella R, Muir B, Mohapatra G, Barmettler A, Archibald H, Kim WJ, Okimoto RA, Bell DW, Sgroi DC. et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci USA. 2006;103:2316–2321. doi: 10.1073/pnas.0508776103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MH, Padhye SS, Guin S, Ma Q, Zhou YQ. Potential therapeutics specific to c-MET/RON receptor tyrosine kinases for molecular targeting in cancer therapy. Acta Pharmacol Sin. 2010;31:1181–1188. doi: 10.1038/aps.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori Y, Itoh H, Uchino S, Hosokawa K, Ochiai A, Ino Y, Ishii H, Sakamoto H, Yamaguchi N, Yanagihara K. et al. Immunohistochemical detection of K-sam protein in stomach cancer. Clin Cancer Res. 1996;2:1373–1381. [PubMed] [Google Scholar]
- Yashiro M, Shinto O, Nakamura K, Tendo M, Matsuoka T, Matsuzaki T, Kaizaki R, Miwa A, Hirakawa K. Synergistic antitumor effects of FGFR2 inhibitor with 5-fluorouracil on scirrhous gastric carcinoma. Int J Cancer. 2010;126:1004–1016. doi: 10.1002/ijc.24763. [DOI] [PubMed] [Google Scholar]
- Liu ZM, Liu LN, Li M, Zhang QP, Cheng SH, Lu S. Mutation detection of KRAS by high-resolution melting analysis in Chinese with gastric cancer. Oncol Rep. 2009;22:515–520. doi: 10.3892/or_00000465. [DOI] [PubMed] [Google Scholar]
- Mita H, Toyota M, Aoki F, Akashi H, Maruyama R, Sasaki Y, Suzuki H, Idogawa M, Kashima L, Yanagihara K. et al. A novel method, digital genome scanning detects KRAS gene amplification in gastric cancers: involvement of overexpressed wild-type KRAS in downstream signaling and cancer cell growth. BMC Cancer. 2009;9:198. doi: 10.1186/1471-2407-9-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greshock J, Bachman KE, Degenhardt YY, Jing J, Wen YH, Eastman S, McNeil E, Moy C, Wegrzyn R, Auger K. et al. Molecular target class is predictive of in vitro response profile. Cancer Res. 2010;70:3677–3686. doi: 10.1158/0008-5472.CAN-09-3788. [DOI] [PubMed] [Google Scholar]
- Yoon YK, Kim HP, Han SW, Hur HS, Oh do Y, Im SA, Bang YJ, Kim TY. Combination of EGFR and MEK1/2 inhibitor shows synergistic effects by suppressing EGFR/HER3-dependent AKT activation in human gastric cancer cells. Mol Cancer Ther. 2009;8:2526–2536. doi: 10.1158/1535-7163.MCT-09-0300. [DOI] [PubMed] [Google Scholar]
- Lee JW, Soung YH, Kim SY, Lee HW, Park WS, Nam SW, Kim SH, Lee JY, Yoo NJ, Lee SH. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005;24:1477–1480. doi: 10.1038/sj.onc.1208304. [DOI] [PubMed] [Google Scholar]
- Oki E, Baba H, Tokunaga E, Nakamura T, Ueda N, Futatsugi M, Mashino K, Yamamoto M, Ikebe M, Kakeji Y, Maehara Y. Akt phosphorylation associates with LOH of PTEN and leads to chemoresistance for gastric cancer. Int J Cancer. 2005;117:376–380. doi: 10.1002/ijc.21170. [DOI] [PubMed] [Google Scholar]
- Velho S, Oliveira C, Ferreira A, Ferreira AC, Suriano G, Schwartz S Jr, Duval A, Carneiro F, Machado JC, Hamelin R, Seruca R. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer. 2005;41:1649–1654. doi: 10.1016/j.ejca.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang J, Chen Y, Wang X, Pan J, Li G, Jia Z, Li Q, Yao JC, Xie K. Overexpression of phosphorylated mammalian target of rapamycin predicts lymph node metastasis and prognosis of chinese patients with gastric cancer. Clin Cancer Res. 2009;15:1821–1829. doi: 10.1158/1078-0432.CCR-08-2138. [DOI] [PubMed] [Google Scholar]
- Cejka D, Preusser M, Fuereder T, Sieghart W, Werzowa J, Strommer S, Wacheck V. mTOR inhibition sensitizes gastric cancer to alkylating chemotherapy in vivo. Anticancer Res. 2008;28:3801–3808. [PubMed] [Google Scholar]
- Cejka D, Preusser M, Woehrer A, Sieghart W, Strommer S, Werzowa J, Fuereder T, Wacheck V. Everolimus (RAD001) and anti-angiogenic cyclophosphamide show long-term control of gastric cancer growth in vivo. Cancer Biol Ther. 2008;7:1377–1385. doi: 10.4161/cbt.7.9.6416. [DOI] [PubMed] [Google Scholar]
- Doi T, Muro K, Boku N, Yamada Y, Nishina T, Takiuchi H, Komatsu Y, Hamamoto Y, Ohno N, Fujita Y. et al. Multicenter phase II study of everolimus in patients with previously treated metastatic gastric cancer. J Clin Oncol. 2010;28:1904–1910. doi: 10.1200/JCO.2009.26.2923. [DOI] [PubMed] [Google Scholar]
- Fuereder T, Jaeger-Lansky A, Hoeflmayer D, Preusser M, Strommer S, Cejka D, Koehrer S, Crevenna R, Wacheck V. mTOR inhibition by everolimus counteracts VEGF induction by sunitinib and improves anti-tumor activity against gastric cancer in vivo. Cancer Lett. 2010;296:249–256. doi: 10.1016/j.canlet.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Dar AA, Zaika A, Piazuelo MB, Correa P, Koyama T, Belkhiri A, Washington K, Castells A, Pera M, El-Rifai W. Frequent overexpression of Aurora Kinase A in upper gastrointestinal adenocarcinomas correlates with potent antiapoptotic functions. Cancer. 2008;112:1688–1698. doi: 10.1002/cncr.23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada K, Yamada Y, Hirao T, Fujimoto H, Takahama Y, Ueno M, Takayama T, Naito A, Hirao S, Nakajima Y. Amplification/overexpression of Aurora-A in human gastric carcinoma: potential role in differentiated type gastric carcinogenesis. Oncol Rep. 2004;12:593–599. [PubMed] [Google Scholar]
- Traynor AM, Hewitt M, Liu G, Flaherty KT, Clark J, Freedman SJ, Scott BB, Leighton AM, Watson PA, Zhao B, Phase I dose escalation study of MK-0457, a novel Aurora kinase inhibitor, in adult patients with advanced solid tumors. Cancer Chemother Pharmacol. 2010. [DOI] [PMC free article] [PubMed]
- Blanke CD. Biomarkers in GIST: partly ready for prime-time use. Clin Cancer Res. 2009;15:5603–5605. doi: 10.1158/1078-0432.CCR-09-1563. [DOI] [PubMed] [Google Scholar]
- Marrari A, Trent JC, George S. Personalized cancer therapy for gastrointestinal stromal tumor: synergizing tumor genotyping with imatinib plasma levels. Curr Opin Oncol. 2010;22:336–341. doi: 10.1097/CCO.0b013e32833a6b8e. [DOI] [PubMed] [Google Scholar]
- Papaetis GS, Syrigos KN. Targeted therapy for gastrointestinal stromal tumors: current status and future perspectives. Cancer Metastasis Rev. 2010;29:151–170. doi: 10.1007/s10555-010-9206-7. [DOI] [PubMed] [Google Scholar]
- Zang ZJ, Ong CK, Cutcutache I, Yu W, Zhang SL, Huang D, Ler LD, Dykema K, Gan A, Tao J. et al. Genetic and structural variation in the gastric cancer kinome revealed through targeted deep sequencing. Cancer Res. 2011;71:29–39. doi: 10.1158/0008-5472.CAN-10-1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ET. Functional genomics of cancer. Curr Opin Genet Dev. 2008;18:251–256. doi: 10.1016/j.gde.2008.07.014. [DOI] [PubMed] [Google Scholar]
- Wooster R, Bachman KE. Catalogue, cause, complexity and cure; the many uses of cancer genome sequence. Curr Opin Genet Dev. 2010;20:336–341. doi: 10.1016/j.gde.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffart TE, van Grieken NC, Tijssen M, Coffa J, Ylstra B, Grabsch HI, van de Velde CJ, Carvalho B, Meijer GA. High resolution analysis of DNA copy-number aberrations of chromosomes 8, 13, and 20 in gastric cancers. Virchows Arch. 2009;455:213–223. doi: 10.1007/s00428-009-0814-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junnila S, Kokkola A, Karjalainen-Lindsberg ML, Puolakkainen P, Monni O. Genome-wide gene copy number and expression analysis of primary gastric tumors and gastric cancer cell lines. BMC Cancer. 2010;10:73. doi: 10.1186/1471-2407-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medlin MD, Staus DP, Dubash AD, Taylor JM, Mack CP. Sphingosine 1-phosphate receptor 2 signals through leukemia-associated RhoGEF (LARG), to promote smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol. 2010;30:1779–1786. doi: 10.1161/ATVBAHA.110.209395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, Wooster R. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91:355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Costanzo-Garvey DL, Pfluger PT, Dougherty MK, Stock JL, Boehm M, Chaika O, Fernandez MR, Fisher K, Kortum RL, Hong EG. et al. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab. 2009;10:366–378. doi: 10.1016/j.cmet.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty MK, Ritt DA, Zhou M, Specht SI, Monson DM, Veenstra TD, Morrison DK. KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol Cell. 2009;34:652–662. doi: 10.1016/j.molcel.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29:157–171. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WK, Cho CH, Lee CW, Wu YC, Yu L, Li ZJ, Wong CC, Li HT, Zhang L, Ren SX. et al. Macroautophagy and ERK phosphorylation counteract the antiproliferative effect of proteasome inhibitor in gastric cancer cells. Autophagy. 2010;6:228–238. doi: 10.4161/auto.6.2.11042. [DOI] [PubMed] [Google Scholar]
- Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T, Russo M, Cancelliere C, Zecchin D, Mazzucchelli L. et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010;120:2858–2866. doi: 10.1172/JCI37539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li VS, Wong CW, Chan TL, Chan AS, Zhao W, Chu KM, So S, Chen X, Yuen ST, Leung SY. Mutations of PIK3CA in gastric adenocarcinoma. BMC Cancer. 2005;5:29. doi: 10.1186/1471-2407-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaussade C, Cho K, Mawson C, Rewcastle GW, Shepherd PR. Functional differences between two classes of oncogenic mutation in the PIK3CA gene. Biochem Biophys Res Commun. 2009;381:577–581. doi: 10.1016/j.bbrc.2009.02.081. [DOI] [PubMed] [Google Scholar]
- Soung YH, Lee JW, Nam SW, Lee JY, Yoo NJ, Lee SH. Mutational analysis of AKT1, AKT2 and AKT3 genes in common human carcinomas. Oncology. 2006;70:285–289. doi: 10.1159/000096289. [DOI] [PubMed] [Google Scholar]
- Katoh M. Genetic alterations of FGF receptors: an emerging field in clinical cancer diagnostics and therapeutics. Expert Rev Anticancer Ther. 2010;10:1375–1379. doi: 10.1586/era.10.128. [DOI] [PubMed] [Google Scholar]
- Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A. et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70:2085–2094. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertotti A, Bracco C, Girolami F, Torti D, Gastaldi S, Galimi F, Medico E, Elvin P, Comoglio PM, Trusolino L. Inhibition of Src impairs the growth of met-addicted gastric tumors. Clin Cancer Res. 2010;16:3933–3943. doi: 10.1158/1078-0432.CCR-10-0106. [DOI] [PubMed] [Google Scholar]
- Okamoto W, Okamoto I, Yoshida T, Okamoto K, Takezawa K, Hatashita E, Yamada Y, Kuwata K, Arao T, Yanagihara K. et al. Identification of c-Src as a potential therapeutic target for gastric cancer and of MET activation as a cause of resistance to c-Src inhibition. Mol Cancer Ther. 2010;9:1188–1197. doi: 10.1158/1535-7163.MCT-10-0002. [DOI] [PubMed] [Google Scholar]
- Chang W, Ma L, Lin L, Gu L, Liu X, Cai H, Yu Y, Tan X, Zhai Y, Xu X. et al. Identification of novel hub genes associated with liver metastasis of gastric cancer. Int J Cancer. 2009;125:2844–2853. doi: 10.1002/ijc.24699. [DOI] [PubMed] [Google Scholar]
- Kouraklis G, Katsoulis IE, Theocharis S, Tsourouflis G, Xipolitas N, Glinavou A, Sioka C, Kostakis A. Does the expression of cyclin E, pRb, and p21 correlate with prognosis in gastric adenocarcinoma? Dig Dis Sci. 2009;54:1015–1020. doi: 10.1007/s10620-008-0464-y. [DOI] [PubMed] [Google Scholar]
- Abate-Daga D, Garcia-Rodriguez L, Sumoy L, Fillat C. Cell cycle control pathways act as conditioning factors for TK/GCV sensitivity in pancreatic cancer cells. Biochim Biophys Acta. 2010;1803:1175–1185. doi: 10.1016/j.bbamcr.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Phelps RA, Broadbent TJ, Stafforini DM, Jones DA. New perspectives on APC control of cell fate and proliferation in colorectal cancer. Cell Cycle. 2009;8:2549–2556. doi: 10.4161/cc.8.16.9278. [DOI] [PubMed] [Google Scholar]
- Ooi CH, Ivanova T, Wu J, Lee M, Tan IB, Tao J, Ward L, Koo JH, Gopalakrishnan V, Zhu Y. et al. Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet. 2009;5:e1000676. doi: 10.1371/journal.pgen.1000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A. et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- Willert J, Epping M, Pollack JR, Brown PO, Nusse R. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev Biol. 2002;2:8. doi: 10.1186/1471-213X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Chen M, Barak LS. Development of small molecules targeting the Wnt pathway for the treatment of colon cancer: a high-throughput screening approach. Am J Physiol Gastrointest Liver Physiol. 2010;299:G293–300. doi: 10.1152/ajpgi.00005.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewan K, Pajak B, Stubbs M, Todd H, Barbeau O, Quevedo C, Botfield H, Young R, Ruddle R, Samuel L. et al. A useful approach to identify novel small-molecule inhibitors of Wnt-dependent transcription. Cancer Res. 2010;70:5963–5973. doi: 10.1158/0008-5472.CAN-10-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S. et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009;9:873–886. doi: 10.2174/156652409789105570. [DOI] [PubMed] [Google Scholar]
- Wicking C, Shanley S, Smyth I, Gillies S, Negus K, Graham S, Suthers G, Haites N, Edwards M, Wainwright B, Chenevix-Trench G. Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype-phenotype correlations are evident. Am J Hum Genet. 1997;60:21–26. [PMC free article] [PubMed] [Google Scholar]
- Ma X, Chen K, Huang S, Zhang X, Adegboyega PA, Evers BM, Zhang H, Xie J. Frequent activation of the hedgehog pathway in advanced gastric adenocarcinomas. Carcinogenesis. 2005;26:1698–1705. doi: 10.1093/carcin/bgi130. [DOI] [PubMed] [Google Scholar]
- Dierks C. GDC-0449--targeting the hedgehog signaling pathway. Recent Results Cancer Res. 2010;184:235–238. doi: 10.1007/978-3-642-01222-8_17. [DOI] [PubMed] [Google Scholar]
- Amin SH, Tibes R, Kim JE, Hybarger CP. Hedgehog antagonist GDC-0449 is effective in the treatment of advanced basal cell carcinoma. Laryngoscope. 2010. [DOI] [PubMed]
- Sabbah M, Emami S, Redeuilh G, Julien S, Prevost G, Zimber A, Ouelaa R, Bracke M, De Wever O, Gespach C. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist Updat. 2008;11:123–151. doi: 10.1016/j.drup.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Katoh M. Epithelial-mesenchymal transition in gastric cancer (Review) Int J Oncol. 2005;27:1677–1683. [PubMed] [Google Scholar]
- Pohl M, Radacz Y, Pawlik N, Schoeneck A, Baldus SE, Munding J, Schmiegel W, Schwarte-Waldhoff I, Reinacher-Schick A. SMAD4 mediates mesenchymal-epithelial reversion in SW480 colon carcinoma cells. Anticancer Res. 2010;30:2603–2613. [PubMed] [Google Scholar]
- Linger RJ, Kruk PA. BRCA1 16 years later: risk-associated BRCA1 mutations and their functional implications. FEBS J. 2010;277:3086–3096. doi: 10.1111/j.1742-4658.2010.07735.x. [DOI] [PubMed] [Google Scholar]
- Shim HJ, Yun JY, Hwang JE, Bae WK, Cho SH, Lee JH, Kim HN, Shin MH, Kweon SS, Kim HJ, Chung IJ. BRCA1 and XRCC1 polymorphisms associated with survival in advanced gastric cancer treated with taxane and cisplatin. Cancer Sci. 2010;101:1247–1254. doi: 10.1111/j.1349-7006.2010.01514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wei J, Qian X, Yin H, Zhao Y, Yu L, Wang T, Liu B. ERCC1 and BRCA1 mRNA expression levels in metastatic malignant effusions is associated with chemosensitivity to cisplatin and/or docetaxel. BMC Cancer. 2008;8:97. doi: 10.1186/1471-2407-8-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro M, Inoue T, Nishioka N, Matsuoka T, Boland CR, Hirakawa K. Allelic imbalance at p53 and microsatellite instability are predictive markers for resistance to chemotherapy in gastric carcinoma. Ann Surg Oncol. 2009;16:2926–2935. doi: 10.1245/s10434-009-0590-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murr R, Vaissiere T, Sawan C, Shukla V, Herceg Z. Orchestration of chromatin-based processes: mind the TRRAP. Oncogene. 2007;26:5358–5372. doi: 10.1038/sj.onc.1210605. [DOI] [PubMed] [Google Scholar]
- Calonge TM, Eshaghi M, Liu J, Ronai Z, O'Connell MJ. Transformation/transcription domain-associated protein (TRRAP)-mediated regulation of Wee1. Genetics. 2010;185:81–93. doi: 10.1534/genetics.110.114769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campas-Moya C. Romidepsin for the treatment of cutaneous T-cell lymphoma. Drugs Today (Barc) 2009;45:787–795. doi: 10.1358/dot.2009.45.11.1437052. [DOI] [PubMed] [Google Scholar]
- Copeland A, Buglio D, Younes A. Histone deacetylase inhibitors in lymphoma. Curr Opin Oncol. pp. 431–436. [DOI] [PubMed]
- Janetka JW, Ashwell S. Checkpoint kinase inhibitors: a review of the patent literature. Expert Opin Ther Pat. 2009;19:165–197. doi: 10.1517/13543770802653622. [DOI] [PubMed] [Google Scholar]
- Kavanaugh SM, White LA, Kolesar JM. Vorinostat: A novel therapy for the treatment of cutaneous T-cell lymphoma. Am J Health Syst Pharm. 2010;67:793–797. doi: 10.2146/ajhp090247. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, Arumugarajah S, Hylander-Gans L, Morosini D, Simeone DM. et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010;70:4972–4981. doi: 10.1158/0008-5472.CAN-09-3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramowicz MJ, Duprez L, Parma J, Vassart G, Heinrichs C. Familial congenital hypothyroidism due to inactivating mutation of the thyrotropin receptor causing profound hypoplasia of the thyroid gland. J Clin Invest. 1997;99:3018–3024. doi: 10.1172/JCI119497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Jimenez C, Santisteban P. TSH signalling and cancer. Arq Bras Endocrinol Metabol. 2007;51:654–671. doi: 10.1590/S0004-27302007000500003. [DOI] [PubMed] [Google Scholar]
- van Dekken H, Geelen E, Dinjens WN, Wijnhoven BP, Tilanus HW, Tanke HJ, Rosenberg C. Comparative genomic hybridization of cancer of the gastroesophageal junction: deletion of 14Q31-32. 1 discriminates between esophageal (Barrett's) and gastric cardia adenocarcinomas. Cancer Res. 1999;59:748–752. [PubMed] [Google Scholar]
- Hanauer DA, Rhodes DR, Sinha-Kumar C, Chinnaiyan AM. Bioinformatics approaches in the study of cancer. Curr Mol Med. 2007;7:133–141. doi: 10.2174/156652407779940431. [DOI] [PubMed] [Google Scholar]
- Henkhaus RS, Gerner EW, Ignatenko NA. Kallikrein 6 is a mediator of K-RAS-dependent migration of colon carcinoma cells. Biol Chem. 2008;389:757–764. doi: 10.1515/BC.2008.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara H, Mimori K, Utsunomiya T, Barnard GF, Ohira M, Hirakawa K, Mori M. Clinicopathologic and biological significance of kallikrein 6 overexpression in human gastric cancer. Clin Cancer Res. 2005;11:6800–6806. doi: 10.1158/1078-0432.CCR-05-0943. [DOI] [PubMed] [Google Scholar]
- Lochhead PA, Wickman G, Mezna M, Olson MF. Activating ROCK1 somatic mutations in human cancer. Oncogene. 2010;29:2591–2598. doi: 10.1038/onc.2010.3. [DOI] [PubMed] [Google Scholar]
- Volk J, Parsonnet J. In: The Biology of Gastric Cancers. Wang T, Fox J, Giraud A, editor. Springer; 2009. Epidemiology od Gastric Cancer and Helicobacter pylori. [Google Scholar]
- Compare D, Rocco A, Nardone G. Risk factors in gastric cancer. Eur Rev Med Pharmacol Sci. 2010;14:302–308. [PubMed] [Google Scholar]
- Marshall BJ, McGechie DB, Rogers PA, Glancy RJ. Pyloric Campylobacter infection and gastroduodenal disease. Med J Aust. 1985;142:439–444. doi: 10.5694/j.1326-5377.1985.tb113444.x. [DOI] [PubMed] [Google Scholar]
- Israel DA, Peek RM. In: The Biology of Gastric Cancers. Wang T, Fox J, Giraud A, editor. Springer; 2009. The role of Helicobacter pylori virulence factors in rodent and primate models of disease; pp. 403–423. [Google Scholar]
- Machado AM, Figueiredo C, Seruca R, Rasmussen LJ. Helicobacter pylori infection generates genetic instability in gastric cells. Biochim Biophys Acta. 2010;1806:58–65. doi: 10.1016/j.bbcan.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Von Hoff DD, Stephenson JJ, Jr, Rosen P, Loesch DM, Borad MJ, Anthony S, Jameson G, Brown S, Cantafio N, Richards DA, Pilot Study Using Molecular Profiling of Patients' Tumors to Find Potential Targets and Select Treatments for Their Refractory Cancers. J Clin Oncol. 2010. [DOI] [PubMed]
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD. et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Stephens PJ, O'Meara S, McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman C. et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J. et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA. et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J. et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Simon R. Clinical trial designs for evaluating the medical utility of prognostic and predictive biomarkers in oncology. Per Med. 2010;7:33–47. doi: 10.2217/pme.09.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Sample characteristics.
Table S2: List of genes sequenced.
Figure S1: Concordance matrices of samples based on array and sequence data.
Table S3: Top 200 genes with amplification at the DNA levels and concordant overexpression at the mRNA level.
Figure S2: Array data evidencing focal amplifications. Top panels show mRNA expression data from arrays, bottom panels show log2 value for DNA abundance in genomic context as derived from SNP arrays.
Figure S3: Comparison of genotyping calls with sequencing data. A total of 1005 common loci were mapped between the Affymetrix 6.0 SNP microarray and the targeted regions. Concordance of genotype calls between affymetrix 6.0 SNP and SAMtools with no filters applied (top left). Application of a consensus quality filters (threshold values plotted as points) improves concordance (y-axis) but reduces the total number of calls (x-axis)(top right). A similar trend is observed for the variant quality thresholds, but at different threshold values (plotted points)(middle left). Sample concordance of genotype calls is improved with consensus quality filter >= 50 and variant quality > 0 (middle right). The total number of genotype calls stratified by reference or variant genotype, and concordance (bottom left).
Table S4: All somatic variants detected.
Figure S4: Sanger sequencing traces. Sanger sequencing traces for variants denoted by blue boxes in Figure 3 (i.e. confirmed in Illumnia and Sanger) are provided.