

Abstract
West Nile virus (WNV) has become firmly established in northeastern U.S., reemerging every summer since its introduction into North America in 1999. To determine whether WNV overwinters locally or is reseeded annually, we examined the patterns of viral lineage persistence and replacement in Connecticut over 10 consecutive transmission seasons by phylogenetic analysis. In addition, we compared the full protein coding sequence among WNV isolates to search for evidence of convergent and adaptive evolution. Viruses sampled from Connecticut segregated into a number of well-supported subclades by year of isolation with few clades persisting ≥2 years. Similar viral strains were dispersed in different locations across the state and divergent strains appeared within a single location during a single transmission season, implying widespread movement and rapid colonization of virus. Numerous amino acid substitutions arose in the population but only one change, V→A at position 159 of the envelope protein, became permanently fixed. Several instances of parallel evolution were identified in independent lineages, including one amino acid change in the NS4A protein that appears to bepositively selected. Our results suggest that annual reemergence of WNV is driven by both reintroduction and local-overwintering of virus. Despite ongoing evolution of WNV, most amino acid variants occurred at low frequencies and were transient in the virus population.
Keywords: Phylogeny, Flaviviridae, Flavivirus, West Nile virus, Molecular epidemiology, Viral evolution
INTRODUCTION
Invasive pathogens threaten the health of immunologically-naïve human and wildlife populations as illustrated by the introduction of West Nile virus (WNV; Flaviviridae, Flavivirus) into North America in 1999. Since that time, this virus has spread throughout the Western Hemisphere where it has caused more than 30,000 confirmed human cases and 1,200 deaths in the US, and imposed substantial mortality on native bird populations. WNV has become firmly established across the continental U.S. by perpetuating in an enzootic cycle involving ornithophilic mosquitoes (mainly Culex species) and passerine bird hosts (Komar, 2003; Kramer, Styer, and Ebel, 2008). Humans and other mammals are dead-end hosts in the transmission cycle, becoming infected when mosquito vectors feed opportunistically on both viremic birds and mammalian hosts (Molaei et al., 2006; Weaver and Barrett, 2004).
The introduction of WNV as a point source into the New York City area, and its continued perpetuation for over a decade in this region, provide an opportunity to evaluate the evolutionary processes acting on an invading virus when it enters a new environment. WNV is a single-stranded, positive-sense RNA virus that exhibits higher mutation rates than DNA-based organisms (May et al., 2011). The viral genome is relatively small, approximately 11 kb in length, making genomic sequencing and analyses from a large number of samples feasible. The acquisition and sequencing of virus isolates during the onset of the outbreak gives us access to the ancestral genotype (Anderson et al., 1999; Lanciotti et al., 1999). The first isolates of WNV (designated as NY99) were shown to be genetically similar to a strain isolated from Israel in 1998 (Lanciotti et al., 2002; Lanciotti et al., 1999). Initial analysis of WNV from Connecticut revealed a homoplasy free phylogeny with low genetic variability during the first two years of the outbreak (Anderson et al., 2001). In 2002, another variant (designated as WN02) arose, rapidly displaced the NY99 strain, and spread throughout North America (Davis et al., 2003; Davis et al., 2005; Ebel et al., 2004; Grinev et al., 2008; Herring et al., 2007). The mechanistic basis for this genotype replacement is related to viral fitness differences. WN02 variants were shown to replicate and disseminate more rapidly in colonies of Culex pipiens collected from New York and Pennsylvania, and Culex tarsalis from California (Ebel et al., 2004; Kilpatrick et al., 2008; Moudy et al., 2007), perhaps due to the fixation of a single amino acid substitution in the envelope protein (Davis et al., 2003; Davis et al., 2005; Ebel et al., 2004).
Phylogenetic comparisons of WNV also indicate an overall lack of geographic structure in North America (Bertolotti, Kitron, and Goldberg, 2007; Davis et al., 2005; Grinev et al., 2008), implying extensive movement of viral strains throughout this region. Birds could serve as an effective vehicle for dispersing viruses over long distances, thereby mixing strains from different geographic regions. Nevertheless, regional variants of WNV have arisen in South Texas (Davis et al., 2003; Davis et al., 2005), southwestern US(Herring et al., 2007; McMullen et al., 2011), and on the Pacific coast (Herring et al., 2007). These findings suggest that virus may perpetuate and evolve in relative isolation under certain circumstances. Viruses sampled from Chicago Illinois, in contrast, were shown to contain a mixture of both locally-derived and exogenous virus strains over a three-year period (Amore et al., 2010).
In the northeastern US, WNV transmission is highly seasonal, re-emerging every summer and continuing into fall until mosquito feeding ceases. The primary mechanism(s) for reinitiating and sustaining transmission in this and other temperate regions is not well understood. WNV may persist and evolve locally, perhaps surviving through winter in vertically-infected, hibernating mosquitoes (Anderson and Main, 2006; Andreadis, Armstrong, and Bajwa, 2010; Bugbee and Forte, 2004; Farajollahi et al., 2005; Nasci et al., 2001). Alternatively, virus transmission may be reseeded annually by the reintroduction of new viral strains from other geographic regions. By intensively sampling virus on both local and statewide scales, we examined the molecular evolution of WNV in Connecticut over 10 consecutive years by phylogenetic analysis. The full coding region of the WNV genome was sequenced and analyzed to differentiate WNV strains, track their distribution and persistence, and monitor evolutionary divergence. In addition, we analyzed the patterns of amino acid substitution to search for evidence of convergent and adaptive evolution within this geographic region.
RESULTS
Nucleotide sequence analysis
Our analysis included the entire coding sequence and flanking portions of the 5' and 3' un-translated regions from 100 WNV isolates from Connecticut, 33 from other US states, one isolate from Mexico and one from Israel (Supplementary Table). Within Connecticut, 53 WNV sequences originated from the town of Stratford during 1999 and 2001–2008, 42 viral sequences came from 21 other towns during 2003, and the remaining sequences were from Greenwich 1999 (N=1), Milford 2000 (N=3), and Shelton 2000 (N=1) (Figure 1). Thus, our sample represented a large number of sequences from the same location in Connecticut (Stratford) over many consecutive years, as well as sequences from a large number of towns during a single year (2003). WNV was not detected in Stratford in 2000 so we included strains from the nearby towns of Milford and Shelton during that year. The resulting alignment comprised a total of 10,393 nucleotide positions representing 94.2% of the genome, 977 variable sites, and 407 parsimony informative sites. Mean nucleotide distance over all sequence pairs was 0.3%. The majority of virus sequences were genetically unique, except for three sequence pairs and one group of four viruses that were identical to each other. The mean nucleotide substitution rate was 5.83 ×10−4 substitutions/site/year which is consistent with previous estimates for WNV (Amore et al., 2010; Bertolotti, Kitron, and Goldberg, 2007; May et al., 2011).
Figure 1.
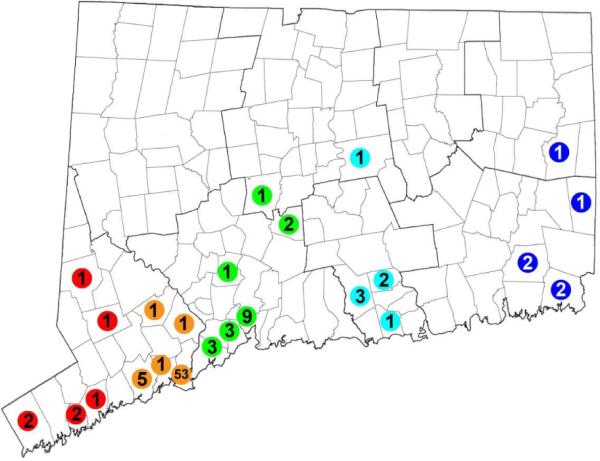
Map of Connecticut showing the geographic location and number of WNV isolates analyzed in this study. Collection sites are color coded from west (warm colors) to east (cool colors) along a longitudinal gradient.
Phylogenetic analysis
Figure 2 depicts the phylogenetic relationships among WNV isolates based on maximum likelihood analysis of nucleotide sequences. Viruses segregated into three major groups as previously defined: NY99, intermediate (INT), and WN02 genotypes (Davis et al., 2005; Ebel et al., 2004). Earlier WNV isolates sampled from northeastern US (1999–2003) and one isolate from Texas during 2002 formed the ancestral NY99 genotype. The INT genotype contained six isolates from Connecticut, Florida, Ohio, New York, and Mexico (2000– 2003). The remaining viruses clustered together to form the WN02 genotype and had originated from sites throughout the US (2002–2008). The WN02 genotype appears to have completely supplanted the NY99 genotype in support of previous findings (Davis et al., 2005; Ebel et al., 2004; Grinev et al., 2008; Herring et al., 2007). WNV isolates from Stratford, Connecticut are highlighted with a black dot in Figure 2. These viruses were genetically diverse with many strains grouping into well-supported subclades. Most of these subclades were defined by year of isolation with the exception of two clades that were detected from 2002–03 and a larger group sampled from 2003–06. Viruses sampled from Stratford, other Connecticut towns and US states were distributed throughout the phylogeny suggesting virus dispersal among these geographic regions.
Figure 2.

Phylogenetic tree depicting relationships of 135 WNV isolates based on maximum likelihood analysis of 10,393 nucleotides sites. Viruses are labeled by the state or country of origin followed by the year of isolation. Numbers at nodes indicate bootstrap values >50%. Branch lengths are proportional to the number of nucleotide substitutions. Stratford taxa are highlighted with a black dot. Asterisk indicates bootstrap values with and without recombinant taxon TX04.
To evaluate the spatial distribution of WNV in Connecticut, we restricted our phylogenetic analysis to 46 isolates obtained statewide during 2003 (Figure 3). We focused on this year because virus activity was more widely distributed throughout the state in comparison to other years(Andreadis et al., 2004). Taxa were color coded according to their geographic location in the state and did not appear to be structured by region. However, viruses often segregated into subclades on a finer geographic scale that corresponded to a particular trapping location. These clades were generally detected transiently within a single location followed by the appearance of new variants in the same location, as seen in Darien, Fairfield, New Haven, Stratford, and West Haven, Connecticut. In addition, similar WNV strains were sometimes dispersed in different regions of Connecticut, indicating widespread migration across the state.
Figure 3.

Phylogenetic tree generated by maximum likelihood analysis of 10,393 nucleotide sites from WNV isolates collected in Connecticut during 2003. Taxon names specify the Genbank number, mosquito species, town, and date of collection and are color-coded according to their location shown in figure 1. Numbers at nodes indicate bootstrap values >50%. Branch lengths are proportional to the number of nucleotide substitutions.
Detection of recombination
To evaluate the potential contribution of recombination to WNV evolution, we analyzed our dataset by the SBP and GARD recombination detection methods. These analyses search for evidence of phylogenetic incongruence among fragments in the alignment to identify potential recombination breakpoints and then compare goodness of fit scores for recombination versus non-recombination models. The GARD method detected evidence for recombination at a single breakpoint predicted at nucleotide position 5979 (cAIC=15.5, p=0.0002). These results were supported by the SBP method with evidence of recombination at breakpoint position 5985 and a cAIC= 15.4. Phylogenetic trees were generated from each side of the proposed recombination breakpoint and compared to identify inconsistent relationships among taxa (data not shown). One virus isolated from Harris County Texas in 2004 (TX04; Genbank no. DQ164206) displayed the genetic signature of hybridization by recombination among parental strains in the NY99 and WN02 genotypes (Table 1). A total of 9 nucleotide differences defined the NY99 and WN02 genotypes. The TX04 strain shared the WN02 sequence at five of these positions: 1442, 2466, 4146, 4803, and 9352, whereas it contained the NY99 sequence at positions 6138, 6426, 6996, and 7938. No other recombinant sequences were identified using the GARD and SBP methods when the TX04 isolate was removed from our dataset. Removal of this sequence also affected the bootstrap values on the full-length maximum likelihood tree. Bootstrap support increased at nodes defining the INT and WN02 genotypes from 74 to 90% and 54 to 86% after removing the TX04 isolate (Figure 2).
Table 1.
Nucleotide differences among the NY99 and WN02 genotypes, and recombinant sequence- TX04.
| Position | NY99 | WN02 | TX04 |
|---|---|---|---|
|
| |||
| 1442 | U | C | C |
| 2466 | C | U | U |
| 4146 | A | G | G |
| 4803 | C | U | U |
| 6138 | C | U | C |
| 6426 | C | U | C |
| 6996 | C | U | C |
| 7938 | U | C | U |
| 9352 | C | U | U |
Amino acid analysis
We then analyzed the patterns of amino acid diversity among WNV isolates in our sample. The open reading frame translated into a full-length polyprotein alignment of 3433 amino acids which varied at 182 positions. Most amino acid variants occurred at low frequencies in this dataset. A total of 118 amino acid variants occurred only once in the sample and of the remaining 67 amino acid polymorphisms, most were shared by only two (n=45) or three taxa (n=12). Only one substitution, V→A at position 159 of the envelope protein, became fixed after it first appeared in 2002. This change was mapped on to our phylogenetic tree (Figure 2) and appears to have occurred in two separate lineages: once to give rise to the WN02 genotype and again within the INT genotype.
To search for evidence of convergent evolution, we mapped all the amino acid substitutions onto our phylogenetic tree. For this and all subsequent analyses, the aforementioned recombinant sequence (TX04) was excluded from the dataset. A total of 19 parallel amino substitutions were identified in unrelated lineages (Table 2). Five of these changes arose in both NY99 and WN02 genotypes, two in the intermediate and WN02 genotypes, and one change in all three genotypes. The remaining parallel substitutions were mapped along different branches within the WN02 genotype. In addition, one change was identified as a possible reversion back to the sequence of the 1998 Israel strain, S→P→S at position 54 of the NS5 protein. Only one of these changes Y→H at position 355 of the NS3 gene resulted in a charge difference.
Table 2.
Predicted parallel amino acid changes.
| Protein | Site | Root | Inferred substitution | No. viruses w/substitution | Location-Year of amino acid variants | Genotype of amino acid variants |
|---|---|---|---|---|---|---|
| Capsid | 44 | I | I-->2T | 2 | CT03, CT04 | WN02 |
| Envelope | 159 | V | V-->2A | 104 | Widespread after 2002 | INT, WN02 |
| NS2A | 34 | M | M-->2L | 4 | MD00, NY02–03, CT03 | NY99, WN02 |
| 43 | V | V-->2A | 2 | CT03, CT07 | WN02 | |
| 90 | M | M-->2V | 3 | CT01, CT06 | NY99, WN02 | |
| 119 | H | H-->2Y | 2 | FL03, CT03 | INT, WN02 | |
| NS2B | 119 | V | V-->2I | 3 | CT01, CT03 | NY99, WN02 |
| NS3 | 106 | V | V-->2A | 2 | CT03, NY03 | NY99, WN02 |
| 355 | Y | Y-->3H | 3 | CT03, CT06, CT08 | WN02 | |
| 466 | P | P-->2S | 2 | CT03, CT07 | WN02 | |
| NS4A | 135 | V | V-->6M | 10 | CT02–03, CT05–06, TX03 | WN02 |
| NS4B | 202 | I | I-->2T | 2 | CO03, CT06 | WN02 |
| 240 | I | I-->3M | 11 | CT00, CT04, CT08, NY03, TX02 | NY99, WN02 | |
| 241 | T | T-->2A | 4 | TX03, CT05–06 | WN02 | |
| 245 | I | I-->3V | 4 | CO03, CT00, FL03, MEX03 | INT, NY99, WN02 | |
| NS5 | 54 | S | S-->P-->S | 1-->132-->1 | ISL98-->N. America-->CT03 | Reversion in WN02 |
| 258 | V | V-->2A | 2 | CT03, CT06 | WN02 | |
| 312 | D | D-->2E | 2 | CT07, TX03 | WN02 | |
| 314 | K | K-->2R | 2 | AZ04, CT03 | WN02 |
Selection analysis
To determine whether amino acid positions are subject to negative or positive selection pressures, we used maximum likelihood methods to estimate and compare rates of synonymous (dS) versus non-synonymous (dN) substitution. Significant departures in these rates provide strong evidence for either negative selection against amino acid change (when dS>dN) or positive selection to change the protein sequence (when dN>dS). The mean dN/dS ratio for the entire coding sequence was 0.07, indicating that the vast majority of nucleotide substitutions are silent changes and that, overall, the WNV genome is subject to strong purifying (negative) selection. There was strong support for negative selection in 407 codons by FEL analysis and 204 codons by SLAC analysis. Positive selection was identified by FEL (p=0.02) and SLAC (p=0.09) analysis at position 135 of the NS4A gene, whereby a V→ M substitution was inferred in 6 separate instances (Table 2).
DISCUSSION
In this study, we examined the molecular evolutionof WNV in Connecticut by intensively sampling virus over 10 successive years. Our sampling was stratified by location during 2003 and by year in the town of Stratford to discern patterns of lineage turnover in a stable WNV focus. Viruses from Stratford were genetically diverse as evidenced by their positions throughout the phylogram. These isolates tended to group by year of isolation with 2 or more distinct viral strains or subclades appearing in a given year. Viruses sampled from a range of locations across Connecticut grouped into subclades that were detected transiently within a single location and sometimes contained viruses from different regions of the state. This indicates high rates of WNV dispersal in the environment and supports epidemiologic observations of rapid viral spread across North America. The population from Connecticut appears to be constantly turning over with an influx of new strains between years and within a given transmission season.
Our analysis also provides support for the occurrence of local overwintering of WNV in this region. One metric for the contribution of virus overwintering versus reintroduction is indicated by the degree of viral clade localization. We observed evidence of year-to-year continuity with two viral subclades persisting in Connecticut from 2002–2003 and one from 2003–2006. The virus could survive through winter in resident birds(Garmendia et al., 2000) or mosquitoes, as previously shown for Cx.pipiens(Andreadis, Armstrong, and Bajwa, 2010; Bugbee and Forte, 2004; Farajollahi et al., 2005; Nasci et al., 2001). WNV has been detected in hibernating Cx. pipiens from nearby New York City (Andreadis, Armstrong, and Bajwa, 2010; Nasci et al., 2001) and was shown to persist in unfed vertically-infected mosquitoes from Connecticut (Anderson et al., 2006; Anderson and Main, 2006). This could provide a plausible mechanism for local overwintering of virus as indicated in our analysis. Alternatively, the occasional instances of viral lineage persistence could represent the annual return of the same WNV strains to Connecticut from another geographic region. Given this scenario, the virus would still require a mechanism to overwinter during periods of mosquito inactivity, unless it was derived from a distant southern source where transmission could be continuous. This seems unlikely given the lack of evidence for stable year-around WNV transmission in the southeastern US, Caribbean Basin, or Central America. Moreover, migratory birds infected by WNV in any of these locations would likely clear the infection before arriving in the Northeast.
Our results indicate that there are multiple opportunities for WNV to reestablish transmission within a given locale by either re-introduction or local overwintering of virus. The population from Stratford appears to derive from a mixture of both local and exogenous virus strains in support of previous findings in Chicago, Illinois (Amore et al., 2010). However, none of the local clades appeared to be permanently established. Given the constant influx of new virus strainsby an avian host, WNV transmission will become quickly reinstated once favorable environmental condition return. Attempts to extinguish WNV transmission by vector control efforts will be temporary if applied focally or during a limited time period even if the measures are completely effective. Together, our findings suggest that WNV transmission is resilient to control attempts or unfavorable weather conditions because sites will quickly become recolonized by virus.
Phylogenetic patterns described in this study are strikingly similar that of other avian, mosquito-borne viruses found in this region of the U.S. Eastern equine encephalitis virus (EEEV; Togaviridae; Alphavirus) and Highlands J virus (Togaviridae: Alphavirus) exhibit limited spatial structure in eastern North America, tending to group by year of isolation (Cilnis, Kang, and Weaver, 1996; Weaver, Scott, and Rico-Hesse, 1991). EEEV strains from northeastern US were shown to segregate into distinct clades that were detected regionally from 1–5 years (Armstrong et al., 2008; Young et al., 2008). These clades eventually disappeared to be superseded by new virus strains. These findings contrast sharply to the phylogeography of Jamestown canyon virus (JCV; Bunyaviridae; Orthobunyavirus) that perpetuates in a deer-mosquito cycle within this region (Andreadis et al., 2008; Armstrong and Andreadis, 2007). JCV was found to be geographically structured within Connecticut rather than temporally, in contrast to EEEV and WNV (Armstrong and Andreadis, 2007). JCV variants were stably maintained in this region and included one lineage detected over a 40 year period. The genetic structure of these mosquito-borne viruses appears to be influenced by the mobility of the main vertebrate host.
WNV isolates sequenced for this study were obtained mainly from mosquitoes collected during the statewide surveillance program and from an ecological study in the town of Stratford, but also included four viruses from American Crows in 1999. Given the limited flight range of mosquitoes relative to birds, infected mosquitoes are more likely to contain viruses circulating in proximity to their respective collection sites. Thus, our mosquito-based sample should provide accurate information about the spatial distribution of WNV variants within the state. Our conclusions about viral population change were based largely on our sample from Stratford. We chose this site because it's an active focus with recurrent WNV transmission each year, allowing us to track patterns of viral clade replacement. WNV turnover is likely to be more pronounced in sites with less stable transmission but such sites are not represented over multiple years in this study.
In this paper, we tracked the frequency and distribution of amino acid substitutions that arose during 10 years of WNV evolution. Most of these changes were detected at low frequencies which would be predicted for selectively neutral or nearly-neutral mutations in a large, expanding population. Under these conditions, most amino acid changes would be lost due to genetic drift, consistent with observations in this study. One notable exception was the E159 substitution that was rapidly driven to fixation within two years of its appearance in 2002, as noted in other U.S. regions (Amore et al., 2010; Chisenhall and Mores, 2009; Davis et al., 2003; Davis et al., 2005; Ebel et al., 2004; Grinev et al., 2008; Herring et al., 2007; McMullen et al., 2011). This substitution may confer a selective advantage for the virus, allowing it to rapidly displace the NY99 genotype (Ebel et al., 2004). If this substitution is important, then we expect to observe evidence of strong negative selection acting on the codon position after the change. Negative selection is indicated by an excess of silent or synonymous changes to preserve the amino acid sequence. One such silent substitution was observed at this codon position in three viruses from Connecticut during 2003 (Genbank nos. HM756651, HM488176, and HM488227).
Evidence for convergent or parallel evolution across viral lineages was found by mapping amino acid substitutions on to the WNV phylogeny. There were a number of instances of the same amino acid substitution arising independently in different lineages. These results are consistent with an earlier analysis that included WNV isolates sampled from throughout the world and thus, with a much deeper evolutionary history (May et al., 2011). This suggests that only a limited number of amino acid changes are permitted due to functional constraints. The adaptive benefit of these convergent changes is not clear; however, positive selection was identified at one position (135 V→ M of the NS4A protein). This amino acid substitution was found in a number of lineages circulating in Connecticut and in Texas. The selection pressure forcing this change might be related to the putative functions of the NS4A protein. This includes involvement in the viral RNA replication complex (Mackenzie et al., 1998; Shiryaev et al., 2009) and immune evasion by interfering with interferon signaling pathways (Liu et al., 2004; Liu et al., 2006). Similar examples of positive selection were identified in WNV from North America, including another amino acid substitution at position 85 A→T of the NS4A protein (McMullen et al., 2011). Adaptive convergent evolution was also identified at position 249 T→P of the NS3 helicase, occurring prior to WNV introduction into North America (Brault et al., 2007). This amino acid substitution was shown to affect viral growth properties and virulence in American Crows, and was associated with outbreaks of avian disease. Future monitoring and functional analyses are needed to assess the significance of the changes identified in this study.
During the course of our analysis, we observed a possible instance of intermolecular recombination among WNV strains circulating in North America. Out of 135 viral genomes examined, a single WNV sequence from Texas displayed the genetic signature of viral recombination. Infrequent episodes of genetic recombination have been inferred for a number of flaviviruses(Twiddy and Holmes, 2003), including among WNV strains circulating in Africa (Pickett and Lefkowitz, 2009). This process could result in rapid genetic change; however, these observations should be interpreted with caution. Natural recombinants should be verified, ideally by re-sequencing plaque-purified virus to ensure against a possible mixed infection and sequencing artifacts (Rico-Hesse, 2003). The recombinant sequence identified in this study was unexpected but clearly shows the imprint of hybridization among strains in the NY99 and WN02 genotypes. This is based on the analysis of a previously submitted Genbank sequence and therefore, requires further confirmation.
In conclusion, our analysis describes the patterns of viral lineage turnover and protein evolution within a region supporting stable WNV transmission. We observed evidence of local overwintering of virus but without permanent establishment of local populations. Moreover, we documented the monthly and yearly appearance of distinct variants, implying rapid re-colonization of virus in a given locale. Numerous nucleotide changes have arisen since its introduction into North America, but negative selection appeared to constrain changes at the protein level. Finally, we identified several instances of convergent evolution, including one amino acid change that appears to be positively selected.
MATERIALS AND METHODS
Virus Strains
WNV isolates sequenced in this study were recovered from mosquitoes (n=95) or crows (n=4) collected during the statewide surveillance program in Connecticut (Andreadis et al., 2004) or during targeted mosquito trapping efforts in Stratford, Connecticut (Anderson et al., 2006) (Supplementary Table). Of these, 57 viruses were obtained each year from an active WNV focus in Stratford during 1999 and 2001–2008 or in the adjacent towns of Milford and Shelton during 2000. An additional 42 WNV isolates were derived from mosquitoes collected in 2003 from 21 towns throughout Connecticut. Bird tissues and whole mosquitoes were processed and screened for virus infection in Vero cell cultures as previously described (Andreadis et al., 2004).
RNA Isolation and Tiling PCR
Viral RNA was isolated from primary WNV cultures (QIAmp viral RNA mini kit, Qiagen) and the RNA genome reverse transcribed to cDNA with Superscript III reverse transcriptase (Invitrogen), random hexamers (Roche) and a specific oligonucleotide targeting the 3′ end of the target genome sequences. Four overlapping PCR products, each of size ~3kb, were designed to capture the WNV coding region. Four primer pairs (1F: AGTAGTTCGCCTGTGTGAGCTGAC; 1R: ATGGGCCCTGGTTTTGTGTCTTGT; 2F: CGGCAAGAGCTGAGATGTGGAAGT; 2R: CCTCAGTCCAATGGGCGAAGTT; 3F:CGCCGGTAAAACAAGGAGGATTCT; 3R: GCAGCCAGTCCTCAACCATTTCAA; 4F: KACGGTRACAGCGGCAACAC; 4R: CGGTTCTGAGGGCTTACATG) were synthesized with a 5' amino modifier C6 (Integrated DNA Technologies) to prevent ligation in the 454 library construction and allow for even coverage across the genome (ref: Harismendy O, Frazer K. Biotechniques. 2009 Mar;46(3):229–31. PMID: 19317667). PCR was performed using the high fidelity polymerase PfuUltra II Fusion HS DNA polymerase (Stratagene) with 40 cycles of amplification. For post-PCR quality control purposes the products were run on pre-cast 1% agarose E-Gels (Invitrogen). Each reaction was quantified using the Quant-iTPicoGreendsDNA assay (Invitrogen). Based on these concentrations 50 ngs of each reaction were pooled for a total of 200 ngs and the volume was brought up to 100uL with TE for shearing and library construction.
Library Construction, Sequencing, and Assembly
Whole WNV genomes were sequenced using the Broad Institute's viral genome sequencing and assembly pipeline (http://www.broadinstitute.org/annotation/viral/WNV/). Pooled PCR products were prepared for sequencing on the 454 Genome Sequencer FLX Titanium (Roche) using standard protocols with the following modifications. Each sample received a 454 library adapter that had been synthesized with an in-house designed 5–8 base molecular tag or barcode (Lennon et al. (2010) Genome Biology 11(2):R15). Post adapter ligation, sample batches of up to 48 were pooled by volume to create sequence-ready libraries. Emulsion PCR and sequencing were performed according to manufacturers' protocols. The library was loaded in to a picotiter plate (PTP) yielding ~50× coverage for each sample. Sequence reads were binned by molecular barcode and sent to their respective project directories for assembly and analysis. Resulting sequence reads were trimmed of primer sequences, filtered for high quality, assembled de novo and annotated using the Broad Institute's in-house viral assembly and annotation algorithms. All genome sequences newly determined here have been deposited in GenBank and assigned accession numbers (Supplementary Table).
Genetic analysis
WNV sequences were combined with 36 previously published sequences available on Genbank (Supplementary Table) for a total of 135 sequences and aligned by the ClustalW algorithm. Phylogenetic relationships were evaluated by maximum-likelihood (ML) analysis in Mega 5.2 (Tamura et al., 2011). The analysis employed the GTR+G+I substitution model and nearest neighbor interchange heuristic search method. The optimal nucleotide substitution model was identified and implemented after performing ML fits of 24 different models in Mega. Support for individual nodes was obtained by performing 500 bootstrap replicates. The nucleotide substitution rate was estimated by dividing the number of base substitutions from the NY99 strain (Genbank no. AF196835) by the number of years of divergence for all sequences from 2000–2008. Estimates of evolutionary divergence were conducted using the maximum composite likelihood model in Mega.
A second dataset of 94 WNV sequences was created by eliminating identical or nearly identical sequences, and contained the first 9,999 bps of the open reading frame, in order to comply with alignment size restrictions for recombination and selection detection programs. Evidence for recombination in the alignment was tested by performing single break point (SBP) and genetic algorithm recombination detection (GARD) methods using the Datamonkey web server(http://www.datamonkey.org)(Kosakovsky Pond et al., 2006). Tests for positive and negative selection were performed by the fixed effects likelihood (FEL) and single-likelihood ancestor counting (SLAC) methods on the Datamonkey web server(Kosakovsky Pond and Frost, 2005; Pond and Frost, 2005).
Supplementary Material
ACKNOWLEDGEMENTS
We thank members our support staff for their technical assistance: Angela Bransfield, Shannon Finan, Bonnie Hamid, John Shepard, and Michael Thomas. This work was supported in whole or in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract HHSN272200900018C, Centers for Disease Control and Prevention (U50/CCU116806-01-1), and the US Department of Agriculture (58-6615-1-218, CONH00768, and CONH00773). KP was supported by National Institutes of Health, National Research Service Award Institutional Training Grant 5T32-AI07538-13
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Amore G, Bertolotti L, Hamer GL, Kitron UD, Walker ED, Ruiz MO, Brawn JD, Goldberg TL. Multi-year evolutionary dynamics of West Nile virus in suburban Chicago, USA, 2005–2007. Philos Trans R Soc Lond B Biol Sci. 2010;365(1548):1871–8. doi: 10.1098/rstb.2010.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JF, Andreadis TG, Main AJ, Ferrandino FJ, Vossbrinck CR. West Nile virus from female and male mosquitoes (Diptera: Culicidae) in subterranean, ground, and canopy habitats in Connecticut. J Med Entomol. 2006;43(5):1010–9. doi: 10.1603/0022-2585(2006)43[1010:wnvffa]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Anderson JF, Andreadis TG, Vossbrinck CR, Tirrell S, Wakem EM, French RA, Garmendia AE, Van Kruiningen HJ. Isolation of West Nile virus from mosquitoes, crows, and a Cooper's hawk in Connecticut. Science. 1999;286(5448):2331–3. doi: 10.1126/science.286.5448.2331. [DOI] [PubMed] [Google Scholar]
- Anderson JF, Main AJ. Importance of vertical and horizontal transmission of West Nile virus by Culex pipiens in the Northeastern United States. J Infect Dis. 2006;194(11):1577–9. doi: 10.1086/508754. [DOI] [PubMed] [Google Scholar]
- Anderson JF, Vossbrinck CR, Andreadis TG, Iton A, Beckwith WH, 3rd, Mayo DR. A phylogenetic approach to following West Nile virus in Connecticut. Proc Natl Acad Sci U S A. 2001;98(23):12885–9. doi: 10.1073/pnas.241472398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadis TG, Anderson JF, Armstrong PM, Main AJ. Isolations of Jamestown Canyon virus (Bunyaviridae: Orthobunyavirus) from field-collected mosquitoes (Diptera: Culicidae) in Connecticut, USA: a ten-year analysis, 1997–2006. Vector Borne Zoonotic Dis. 2008;8(2):175–88. doi: 10.1089/vbz.2007.0169. [DOI] [PubMed] [Google Scholar]
- Andreadis TG, Anderson JF, Vossbrinck CR, Main AJ. Epidemiology of West Nile virus in Connecticut: a five-year analysis of mosquito data 1999–2003. Vector Borne Zoonotic Dis. 2004;4(4):360–78. doi: 10.1089/vbz.2004.4.360. [DOI] [PubMed] [Google Scholar]
- Andreadis TG, Armstrong PM, Bajwa WI. Studies on hibernating populations of Culex pipiens from a West Nile virus endemic focus in New York City: parity rates and isolation of West Nile virus. J Am Mosq Control Assoc. 2010;26(3):257–64. doi: 10.2987/10-6004.1. [DOI] [PubMed] [Google Scholar]
- Armstrong PM, Andreadis TG. Genetic relationships of Jamestown Canyon virus strains infecting mosquitoes collected in Connecticut. Am J Trop Med Hyg. 2007;77(6):1157–62. [PubMed] [Google Scholar]
- Armstrong PM, Andreadis TG, Anderson JF, Stull JW, Mores CN. Tracking eastern equine encephalitis virus perpetuation in the northeastern United States by phylogenetic analysis. Am J Trop Med Hyg. 2008;79(2):291–6. [PubMed] [Google Scholar]
- Bertolotti L, Kitron U, Goldberg TL. Diversity and evolution of West Nile virus in Illinois and the United States, 2002–2005. Virology. 2007;360(1):143–9. doi: 10.1016/j.virol.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Brault AC, Huang CY, Langevin SA, Kinney RM, Bowen RA, Ramey WN, Panella NA, Holmes EC, Powers AM, Miller BR. A single positively selected West Nile viral mutation confers increased virogenesis in American crows. Nat Genet. 2007;39(9):1162–6. doi: 10.1038/ng2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugbee LM, Forte LR. The discovery of West Nile virus in overwintering Culex pipiens (Diptera: Culicidae) mosquitoes in Lehigh County, Pennsylvania. J Am Mosq Control Assoc. 2004;20(3):326–7. [PubMed] [Google Scholar]
- Chisenhall DM, Mores CN. Diversification of West Nile virus in a subtropical region. Virol J. 2009;6:106. doi: 10.1186/1743-422X-6-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilnis MJ, Kang W, Weaver SC. Genetic conservation of Highlands J viruses. Virology. 1996;218(2):343–51. doi: 10.1006/viro.1996.0203. [DOI] [PubMed] [Google Scholar]
- Davis CT, Beasley DW, Guzman H, Raj R, D'Anton M, Novak RJ, Unnasch TR, Tesh RB, Barrett AD. Genetic variation among temporally and geographically distinct West Nile virus isolates, United States, 2001, 2002. Emerg Infect Dis. 2003;9(11):1423–9. doi: 10.3201/eid0911.030301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CT, Ebel GD, Lanciotti RS, Brault AC, Guzman H, Siirin M, Lambert A, Parsons RE, Beasley DW, Novak RJ, Elizondo-Quiroga D, Green EN, Young DS, Stark LM, Drebot MA, Artsob H, Tesh RB, Kramer LD, Barrett AD. Phylogenetic analysis of North American West Nile virus isolates, 2001–2004: evidence for the emergence of a dominant genotype. Virology. 2005;342(2):252–65. doi: 10.1016/j.virol.2005.07.022. [DOI] [PubMed] [Google Scholar]
- Ebel GD, Carricaburu J, Young D, Bernard KA, Kramer LD. Genetic and phenotypic variation of West Nile virus in New York, 2000–2003. Am J Trop Med Hyg. 2004;71(4):493–500. [PubMed] [Google Scholar]
- Farajollahi A, Crans WJ, Bryant P, Wolf B, Burkhalter KL, Godsey MS, Aspen SE, Nasci RS. Detection of West Nile viral RNA from an overwintering pool of Culex pipens pipiens (Diptera: Culicidae) in New Jersey, 2003. J Med Entomol. 2005;42(3):490–4. doi: 10.1093/jmedent/42.3.490. [DOI] [PubMed] [Google Scholar]
- Garmendia AE, Van Kruiningen HJ, French RA, Anderson JF, Andreadis TG, Kumar A, West AB. Recovery and identification of West Nile virus from a hawk in winter. J Clin Microbiol. 2000;38(8):3110–1. doi: 10.1128/jcm.38.8.3110-3111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinev A, Daniel S, Stramer S, Rossmann S, Caglioti S, Rios M. Genetic variability of West Nile virus in US blood donors, 2002–2005. Emerg Infect Dis. 2008;14(3):436–44. doi: 10.3201/eid1403.070463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring BL, Bernardin F, Caglioti S, Stramer S, Tobler L, Andrews W, Cheng L, Rampersad S, Cameron C, Saldanha J, Busch MP, Delwart E. Phylogenetic analysis of WNV in North American blood donors during the 2003–2004 epidemic seasons. Virology. 2007;363(1):220–8. doi: 10.1016/j.virol.2007.01.019. [DOI] [PubMed] [Google Scholar]
- Kilpatrick AM, Meola MA, Moudy RM, Kramer LD. Temperature, viral genetics, and the transmission of West Nile virus by Culex pipiens mosquitoes. PLoS Pathog. 2008;4(6):e1000092. doi: 10.1371/journal.ppat.1000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar N. West Nile virus: epidemiology and ecology in North America. Adv Virus Res. 2003;61:185–234. doi: 10.1016/s0065-3527(03)61005-5. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SD. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005;22(5):1208–22. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol. 2006;23(10):1891–901. doi: 10.1093/molbev/msl051. [DOI] [PubMed] [Google Scholar]
- Kramer LD, Styer LM, Ebel GD. A global perspective on the epidemiology of West Nile virus. Annu Rev Entomol. 2008;53:61–81. doi: 10.1146/annurev.ento.53.103106.093258. [DOI] [PubMed] [Google Scholar]
- Lanciotti RS, Ebel GD, Deubel V, Kerst AJ, Murri S, Meyer R, Bowen M, McKinney N, Morrill WE, Crabtree MB, Kramer LD, Roehrig JT. Complete genome sequences and phylogenetic analysis of West Nile virus strains isolated from the United States, Europe, and the Middle East. Virology. 2002;298(1):96–105. doi: 10.1006/viro.2002.1449. [DOI] [PubMed] [Google Scholar]
- Lanciotti RS, Roehrig JT, Deubel V, Smith J, Parker M, Steele K, Crise B, Volpe KE, Crabtree MB, Scherret JH, Hall RA, MacKenzie JS, Cropp CB, Panigrahy B, Ostlund E, Schmitt B, Malkinson M, Banet C, Weissman J, Komar N, Savage HM, Stone W, McNamara T, Gubler DJ. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science. 1999;286(5448):2333–7. doi: 10.1126/science.286.5448.2333. [DOI] [PubMed] [Google Scholar]
- Liu WJ, Chen HB, Wang XJ, Huang H, Khromykh AA. Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A in inhibition of beta interferon promoter-driven transcription. J Virol. 2004;78(22):12225–35. doi: 10.1128/JVI.78.22.12225-12235.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WJ, Wang XJ, Clark DC, Lobigs M, Hall RA, Khromykh AA. A single amino acid substitution in the West Nile virus nonstructural protein NS2A disables its ability to inhibit alpha/beta interferon induction and attenuates virus virulence in mice. J Virol. 2006;80(5):2396–404. doi: 10.1128/JVI.80.5.2396-2404.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie JM, Khromykh AA, Jones MK, Westaway EG. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology. 1998;245(2):203–15. doi: 10.1006/viro.1998.9156. [DOI] [PubMed] [Google Scholar]
- May FJ, Davis CT, Tesh RB, Barrett AD. Phylogeography of West Nile virus: from the cradle of evolution in Africa to Eurasia, Australia, and the Americas. J Virol. 2011;85(6):2964–74. doi: 10.1128/JVI.01963-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen AR, May FJ, Guzman H, Bueno R, Dennett JA, Tesh RB, Barrett ADT. Evolution of new genotype of West Nile Virus in North America. Emerg Infect Dis. 2011;17(5):785–793. doi: 10.3201/eid1705.101707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molaei G, Andreadis TG, Armstrong PM, Anderson JF, Vossbrinck CR. Host feeding patterns of Culex mosquitoes and West Nile virus transmission, northeastern United States. Emerg Infect Dis. 2006;12(3):468–74. doi: 10.3201/eid1203.051004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudy RM, Meola MA, Morin LL, Ebel GD, Kramer LD. A newly emergent genotype of West Nile virus is transmitted earlier and more efficiently by Culex mosquitoes. Am J Trop Med Hyg. 2007;77(2):365–70. [PubMed] [Google Scholar]
- Nasci RS, Savage HM, White DJ, Miller JR, Cropp BC, Godsey MS, Kerst AJ, Bennett P, Gottfried K, Lanciotti RS. West Nile virus in overwintering Culex mosquitoes, New York City, 2000. Emerg Infect Dis. 2001;7(4):742–4. doi: 10.3201/eid0704.010426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett BE, Lefkowitz EJ. Recombination in West Nile Virus: minimal contribution to genomic diversity. Virol J. 2009;6:165. doi: 10.1186/1743-422X-6-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond SL, Frost SD. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21(10):2531–3. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- Rico-Hesse R. Microevolution and virulence of dengue viruses. Adv Virus Res. 2003;59:315–41. doi: 10.1016/s0065-3527(03)59009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiryaev SA, Chernov AV, Aleshin AE, Shiryaeva TN, Strongin AY. NS4A regulates the ATPase activity of the NS3 helicase: a novel cofactor role of the non-structural protein NS4A from West Nile virus. J Gen Virol. 2009;90(Pt 9):2081–5. doi: 10.1099/vir.0.012864-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011 doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twiddy SS, Holmes EC. The extent of homologous recombination in members of the genus Flavivirus. J Gen Virol. 2003;84(Pt 2):429–40. doi: 10.1099/vir.0.18660-0. [DOI] [PubMed] [Google Scholar]
- Weaver SC, Barrett AD. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat Rev Microbiol. 2004;2(10):789–801. doi: 10.1038/nrmicro1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver SC, Scott TW, Rico-Hesse R. Molecular evolution of eastern equine encephalomyelitis virus in North America. Virology. 1991;182(2):774–84. doi: 10.1016/0042-6822(91)90618-l. [DOI] [PubMed] [Google Scholar]
- Young DS, Kramer LD, Maffei JG, Dusek RJ, Backenson PB, Mores CN, Benard KA, Ebel GD. Molecular epidemiology of eastern equine encephalitis virus, New York. Emerg Infect Dis. 2008;14(3):454–460. doi: 10.3201/eid1403.070816. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.