

Abstract
The effects of bombesin receptor subtype-3 (BRS-3) agonists were investigated on lung cancer cells. The BRS-3 agonist (DTyr6, βAla11, Phe13, Nle14)bombesin6-14 (BA1), but not gastrin releasing peptide (GRP) or neuromedin B (NMB) increased significantly the clonal growth of NCI-H1299 cells stably transfected with BRS-3 (NCI-H1299-BRS-3). Also, BA1 addition to NCI-H727 or NCI-H1299-BRS-3 cells caused Tyr1068 phosphorylation of the epidermal growth factor receptor (EGFR). Similarly, (DTyr6, R-Apa11, Phe13, Nle14)bombesin6-14 (BA2) and (DTyr6, R-Apa11, 4-Cl,Phe13, Nle14)bombesin6-14 (BA3) but not gastrin releasing peptide (GRP) or neuromedin B (NMB) caused EGFR transactivation in NCI-H1299-BRS-3 cells. BA1-induced EGFR or ERK tyrosine phosphorylation was not inhibited by addition of BW2258U89 (BB2R antagonist) or PD168368 (BB1R antagonist) but was blocked by (DNal-Cys-Tyr-DTrp-Lys-Val-Cys-Nal)NH2 (BRS-3 ant.). The BRS-3 ant. reduced clonal growth of NCI-H1299-BRS-3 cells. BA1, BA2, BA3 and BRS-3 ant. inhibit specific 125I-BA1 binding to NCI-H1299-BRS-3 cells with an IC50 values of 1.1, 21, 15 and 750 nM respectively. The ability of BRS-3 to regulate EGFR transactivation in NCI-H1299-BRS-3 cells was reduced by AG1478 or gefitinib (EGFR tyrosine kinase inhibitors), GM6001 (matrix metalloprotease inhibitor), PP2 (Src inhibitor), N-acetylcysteine (anti-oxidant), Tiron (superoxide scavenger) and DPI (NADPH oxidase inhibitor). These results demonstrate that BRS-3 agonists may stimulate lung cancer growth as a result of EGFR transactivation and that the transactivation is regulated by BRS-3 in a Src-, reactive oxygen and matrix metalloprotease-dependent manner.
Keywords: bombesin receptor subtype-3, epidermal growth factor receptor, tyrosine phosphorylation, lung cancer, proliferation
1. Introduction
Three G-protein coupled receptors (GPCR) comprise the mammalian bombesin (BB) receptor family of peptides including the BB1 receptor [46], which binds neuromedin B (NMB) with high affinity, the BB2 receptor [1, 41], which binds gastrin releasing peptide (GRP) with high affinity and the orphan receptor bombesin receptor subtype-3 (BRS-3). BRS-3 contains 399 amino acids and has 51% and 47% sequence homology with BB2R and BB1R respectively [8]. While no endogenous ligands have been identified for BRS-3, it binds the synthetic BB analog (D-Tyr6, β-Ala11, Phe13, NLeu14)BB6-14 (BA1) with high affinity [29,36]. Because BRS-3 does not bind BB, GRP or NMB with high affinity, it has a unique pharmacological profile. Recently a synthetic somatostatin analog, (DNal-Cys-Tyr-DTrp-Lys-Val-Cys-Nal)NH2 (BRS-3 ant.), was identified as a BRS-3 antagonist and (DTyr6, R-Apa11, Phe13, Nle14)bombesin6-14 (BA2) and (DTyr6, RApa11, 4-Cl,Phe13, Nle14)bombesin6-14 (BA3) were characterized as selective BRS-3 agonists [13]. Even though the endogenous ligand for BRS-3 is unknown, synthetic agonists and antagonists are available to characterize BRS-3.
Because knockout BRS-3 mice developed obesity and diabetes, BRS-3 may be important in the regulation of energy homeostasis [34]. Also BRS-3 knockout mice are hypertensive, have a reduced metabolic rate, 2-fold increase in plasma insulin and a 5-fold increase in serum leptin [30]. This has led to the investigation of BRS-3 agonists as anti-obesity agents [15].
An additional area of interest is the role of BB agonists as regulators of neoplastic proliferation [3, 5, 11, 19]. BRS-3 is present in many human tumors including small cell lung cancer (SCLC) and non-SCLC (NSCLC), lung carcinoids, renal cell cancers, Ewing sarcomas, pancreatic cancer, ovarian cancer and prostate cancer [37]. Addition of BA1 to NCI-N417 cells enhanced cellular adhesion [17]. While it is not known if BRS-3 activation, similar to BB1R or BB2R activation [18], results in tumor growth, BRS-3 causes proliferation of normal bronchiolar epithelial cells [42]. Like BRS-3, BB1R and BB2R are present in many lung cancer cells [6, 9, 21, 33, 45] Similar to BB1R or BB2R signal transduction, BRS-3 activation stimulates phospholipase C [38], ERK tyrosine phosphorylation, Elk-1 and c-fos expression [48].
In addition to BBR, non-small cell lung cancer (NSCLC) cells have high levels of tyrosine kinase receptors such as epidermal growth factor receptor (EGFR) [32]. The EGFR can be activated directly by agonists such as transforming growth factor α (TFG α) [7]. Alternatively the EGFR can be regulated by GPCR such as the BB1R or BB2R [18, 19, 31]. Recent studies show activation of the BB2R regulates the rapid tyrosine phosphorylation of the EGFR and ERK by stimulating matrix metalloproteases to release TGFα and amphiregulin from head and neck cancer cells, by a Src-dependent mechanism [24, 44, 52]. Transactivation of the EGFR due to BB2R activation occurs in a number of head/neck, lung and prostate cancer cells [24, 49, 51], as well as a number of other BB2R-containing cells [40]. The EGFR transactivation regulated by BB1R or BB2R may be important in the proliferation of cancer cells.
In this study we found that BRS-3 agonists stimulated the proliferation of lung cancer cells. BRS-3 activation by (DTyr6, βAla11, Phe13, Nle14)bombesin6-14 (BA1) increased Tyr1068 phosphorylation of the EGFR in lung cancer cells. Similarly, (DTyr6, R-Apa11, Phe13, Nle14)bombesin6-14 (BA2) and (DTyr6, R-Apa11, 4-Cl,Phe13, Nle14)bombesin6-14 (BA3) but not GRP or NMB caused EGFR transactivation in NCI-H1299-BRS-3 cells. BA1-induced EGFR or ERK tyrosine phosphorylation was not inhibited by addition of BW2258U89 (BB2R antagonist) or PD168368 (BB1R antagonist) but was blocked by (DNal-Cys-Tyr-DTrp-Lys-Val-Cys-Nal)NH2 (BRS-3 ant.). Also, BRS-3 ant. inhibited the proliferation of lung cancer cells. These results suggest that BRS-3 may regulate lung cancer growth through EGFR transactivation.
2. Materials and Methods
2.1 Cell culture
NSCLC NCI-H1299 or NCI-H727 cells, which are known to contain BRS-3 and wild type EGFR [9], were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium containing 10% heat-inactivated fetal bovine serum (FBS; Invitrogen, Grand Island, NY). NCI-H1299; the cancer cells had approximately 1000 BRS-3/cell. Balb-3T3 cells stably transfected with BRS-3 (NCI-H1299-BRS-3 and Balb/3T3-BRS-3) were cultured in RPMI 1640 containing 10% FBS supplemented with 300 mg/l G418 sulfate (Sigma-Aldrich, St. Louis, MO); Balb/3T3 cells have EGFR [14]; the transfected cells had approximately 50,000 BRS-3/cell. As a control, Balb/3T3 cells which lack BRS-3 were cultured in DMEM containing 10% FBS. The cells were split weekly 1/20 with trypsinethylenediaminotetraacetic acid (EDTA). The cells were mycoplasma free and were used when they were in exponential growth phase after incubation at 37°C in 5% CO2/95% air.
2.2. Receptor binding
BA1, which binds with high affinity to all human BB receptors, was radiolabeled using iodogen and HPLC purified as reported previously [28]. The ability of BRS-3 ligands to inhibit specific 125I-BA1 binding to NCI-H1299-BRS-3 cells (80,000 BRS-3/cell) was investigated. GRP, NMB and BB were purchased from Phoenix Pharmaceuticals, Belmont, CA. BW2258U89 was a gift from Dr. J. McDermed and PD168368 was a gift from Dr. J. Hughes. The NCI-H1299-BRS-3 cells were washed 3 times in SIT medium (RPMI-1640 containing 3 × 10-8 M sodium selenite, 5 μg/ml bovine insulin and 10 μg/ml transferrin (Sigma-Aldrich, St. Louis, MO)). The cells were incubated in SIT buffer containing 0.25% bovine serum albumin and 250 μg/ml bacitracin (Sigma-Aldrich, St. Louis, MO) and 125I-BA1 (100,000 cpm) added as well as various concentrations of unlabelled competitor. After incubation at 37°C for 30 min, free 125I-BA1 was removed by washing 3 times in buffer and the cells which contained bound 125I-BA1 dissolved in 0.2 N NaOH and counted in a gamma counter. The half-maximal inhibitory concentration (IC50) was calculated for each unlabeled competitor.
2.3. Western Blot
The ability of BA1 to stimulate tyrosine phosphorylation of EGFR or ERK (p42/p44 MAP kinase) was investigated by Western blotting. NCI-H1299-BRS-3, Balb/3T3/BRS-3 or NCI-H727 cells were cultured in 15 cm dishes. When a monolayer of cells formed they were placed in SIT media for 3 hours. Routinely, cells were pre-treated with gefitinib, GM6001, PP2, N-acetylcysteine, Tiron or diphenylene iodonium (DPI, Sigma-Adrich, St. Louis, MO) for 30 min. Then cells were treated with 100 nM BA1 for 2 min, washed twice with PBS and lysed in buffer containing 50 mM Tris.HCl (pH 7.5), 150 mM sodium chloride, 1% Triton X-100, 1% deoxycholate, 1% sodium azide, 1 mM ethyleneglycoltetraacetic acid, 0.4 M EDTA, 1.5 μg/ml aprotinin, 1.5 μg/ml leupeptin, 1 mM phenylmethylsulfonylfluoride and 0.2 mM sodium vanadate (Sigma-Aldrich, St. Louis, MO). The lysate was sonicated for 5 s at 4°C and centrifuged at 10000 × g for 15 min. Protein concentration was measured using the BCA reagent (Pierce Chemical Co., Rockford, IL), and 400 μg of protein was incubated with 4 μg of anti-phosphotyrosine (PY) monoclonal antibody, 4 μg of goat anti-mouse immunoglobulin IgG and 30 μl of immobilized protein G (Pierce Chemical Co., Rockford, IL) overnight at 4°C. The immunoprecipitates were washed 3 times with phosphate buffered saline and analyzed by sodium dodecyl sulfate/polyacrylamide gel electrophoresis and Western blotting. Immunoprecipitates were fractionated using 4-20% polyacrylamide gels (Novex, San Diego, CA). Proteins were transferred to nitrocellulose membranes and the membranes were blocked overnight at 4°C using blotto (5% non-fat dried milk in solution containing 50 mM Tris/HCl (pH 8.0), 2 mM CaCl2, 80 mM sodium chloride, 0.05% Tween 20 and 0.02% sodium azide (Sigma-Aldrich, St. Louis, MO) and incubated for 16 h at 4°C with 1 μg/ml anti-EGFR antibody (Cell Signaling Technologies, Danvers, MA) followed by anti-rabbit immunoglobulin G-horseradish peroxidase conjugate (Upstate Biotechnologies, Lake Placid, NY). The membrane was washed for 10 min with blotto and twice for 10 min with washing solution (50 mM Tris/HCl (pH 8.0), 2 mM CaCl2, 80 mM sodium chloride, 0.05% Tween 20 and 0.02% sodium azide (Sigma-Aldrich, St. Louis, MO). The blot was incubated with enhanced chemiluminescence detection reagent for 5 min and exposed to Kodak XAR film. The intensity of the bands was determined using a densitometer.
Alternatively, 20 μg of cellular extract was loaded onto a 15 well 4-20% polyacrylamide gels. After transfer to nitrocellulose, the blot was probed with anti PY1068-EGFR, anti-EGFR, anti-PY-ERK, anti-ERK or anti-tubulin (Cell Signaling Technologies, Danvers, MA).
2.4 Proliferation assays
In the 3H-Thymidine assay, Balb/3T3-BRS-3 cells, which can rest in Go growth phase, were trypsinized and 50,000 cells placed in 24 well plates containing DMEM with 10% fetal bovine serum and 0.3 mg/ml G418 sulfate. After 3 days, the confluent cells were placed in DMEM containing 30 mM sodium selenite, 5 μg/ml insulin and 10 μg/ml transferin and varying concentrations of BA1 in the presence or absence of BRS-3 ant. After 24 hr the cells were incubated with 3H-Thymidine (106 cpm/ml) for 3 hr. The 24 well plates were washed 3 times with 1 ml of PBS. The cells were treated with 0.2 N HCl (0.25 ml) and the solution placed in a scintillation vial. The cells were treated with 0.2 N NaOH (0.25 ml) and added to the scintillation vial. Then 10 ml of scintillation fluid were added to the vial and after shaking, the vial was counted in a β-counter.
In the clonogenic assay, NCI-H1299-BRS-3 cells, which cannot rest in Go growth phase, were treated with BA1 in the presence or absence of BRS-3 ant. or gefitinib (Tocris Bioscience, Ellisville, MO). The bottom layer contained 0.5% agarose in SIT medium containing 5% FBS in 6 well plates in 2 ml. The top layer consisted of 2 ml of SIT medium in 0.3% agarose, gefitinib, BRS-3 ant. or BA1 as well as 5 × 104 NCI-H1299-BRS3 cells. Triplicate wells were plated and after 2 weeks, 1 ml of 0.1% p-iodonitrotetrazolium violet (Sigma-Aldrich, St. Louis, MO) was added and after 16 hours at 37°C, the plates were screened for colony formation; the number of colonies larger than 50 μm in diameter were counted using an Omnicon image analysis system.
In the 3-(4,5-dimethylthiazol-2-yl)-2.5-diphenyl-2H-tetrazolium bromide (MTT) assay, Balb/3T3 or Balb/3T3-BRS-3 cells (104/well) were placed in SIT medium and 10 nM BA1, 10 uM PD98059, or 30 ug/ml gefitinib added. After 16 hours, 15 μl of 0.1 % MTT solution added. After 3 hours, 150 μl of dimethylsulfoxide was added. The optical density at 570 nm was determined.
3. Results
3.1 BRS-3 agonists stimulate and antagonists inhibit proliferation
The effects of BRS-3 agonists and antagonists on cellular proliferation were investigated. Table I shows that addition of 10 nM BA1, but not GRP or NMB, increased significantly NCI-H1299-BRS-3 colony number. The increase in NCI-H1299-BRS-3 colony number caused by BA1 was reversed by 3 or 10 μg/ml but not 0.3 or 1 μg/ml gefitinib. BRS-3 ant. decreased significantly NCI-H1299-BRS-3 colony number. In contrast, NCI-H727 cells, Balb/3T3-BRS-3 cells or Balb/3T3 cells did not form colonies (data not shown).
Table I.
Clonal growth of NCI-H1299-BRS-3 cells
| Addition | Colony number |
|---|---|
| None | 40 ± 6aa |
| BA1, 10 nM | 75 ± 3** |
| BA1 + Gef 0.3 μg/ml | 72 ± 6** |
| BA1 + Gef 1 μg/ml | 65 ± 8* |
| BA1 + Gef 3 μg/ml | 47 ± 5a |
| BA1 + Gef 10 μg/ml | 20 ± 3**aa |
| BRS3 ant., 10 μM | 30 ± 3*aa |
| GRP, 10 nM | 41 ± 5aa |
| NMB, 10 nM | 46 ± 5a |
The mean colony number ± S.D. of 3 determinations each repeated in triplicate is indicated; relative to no additions; relative to 10 nM BA1 using the Student's t-test.
p < 0.05
p < 0.01
p < 0.05
p < 0.01
Fig. 1A shows that BA1 caused increase 3H-thymidine uptake in Balb/3T3-BRS-3 cells in a dose-dependent manner. BA1, 0.01 nM, had little effect whereas 100 nM BA1 maximally increased 3H-thymidine uptake; the IC50 value was 0.3 nM. BRS-3 ant., 10 μM, shifted the BA1 dose-response curve to the right with an IC50 value of 5 nM. In contrast BA1 did not stimulate 3H-thymidine uptake in Balb/3T3 cells (data not shown).
Fig. 1.

BRS-3 and proliferation. (A) The ability of BA1 to stimulate 3H-Thymidine uptake into Balb/3T3-BRS-3 cells was determined in the absence (●) and presence (○) of 10 μM BRS-3 ant. The mean value ± S.D. of 4 determinations is indicated. (B). The ability of gefitinib to alter colony formation in NCI-H1299-BRS-3 cells was determined in the absence (■) and presence (□) of 10 μM BRS-3-ant; exogenous BA1 was not added. The mean value ± S.D. of 3 determinations is indicated. These experiments are representative of 3 others.
Figure 1B shows that gefitinib addition to NCI-H1299-BRS-3 cells reduced colony number in a dose-dependent manner. BRS-3 ant. shifted the gefitinib dose-response curve to the left, resulting in an increase of gefitinib cytotoxicity. The results show that BA1 increases the proliferation of BRS-3 containing cells, whereas gefitinib and BRS-3 ant. reduces proliferation.
Table II shows that using the MTT assay, 10 nM BA1 stimulated significantly the growth of Balb/3T3-BRS-3 but not control Balb/3T3 cells. Because each of the cell lines have EGFR, 10 μg/ml gefitinib significantly inhibited proliferation in the absence or presence of BA1. BRS-3 ant. inhibited the growth of Balb/3T3-BRS-3 but not Balb/3T3 cells (data not shown).
Table II.
Proliferation of 3T3 cells.
| Addition | % Growth | |
|---|---|---|
| Balb/3T3 | Balb/3T3-BRS-3 | |
| None | 99 ± 4 | 101 ± 3 |
| BA1, 10 nM | 90 ± 5 | 138 ± 11* |
| Gefitinib, 30 μg/ml | 31 ± 4** | 41 ± 6** |
| BA1 + Gefitinib | 37 ± 5** | 73 ± 7* |
Balb/3T3 or Balb/3T3-BRS-3 cells (10,000/well) were placed in 96 well plates in 100 μl of SIT medium. BA1, Gefitinib or PD98059 were added for 16 hours. After addition of MTT, the cells were incubated for 3 hours, then 150 ul of DMSO added and the absorbance determined at 570 nm. The mean value ± S.E. of 3 determinations, each based on 8 data points, was calculated; using the Student's t-test.
p < 0.05
p < 0.01
3.2 BRS-3 agonists but not antagonists increase EGFR tyrosine phosphorylation
The ability of BB analogues to cause transactivation of the EGFR was investigated. Figure 2 shows that 100 nM BA1, but not GRP or NMB, caused tyrosine phosphorylation of Tyr1068 of the EGFR using NCI-H727 cells (Fig. 2A). Equal amounts of total EGFR were seen in all lanes. Similar results were obtained with NCI-H1299-BRS-3 cells (Fig. 2B). These results demonstrate that BRS-3 regulates EGFR tyrosine phosphorylation in lung cancer cells.
Fig. 2.
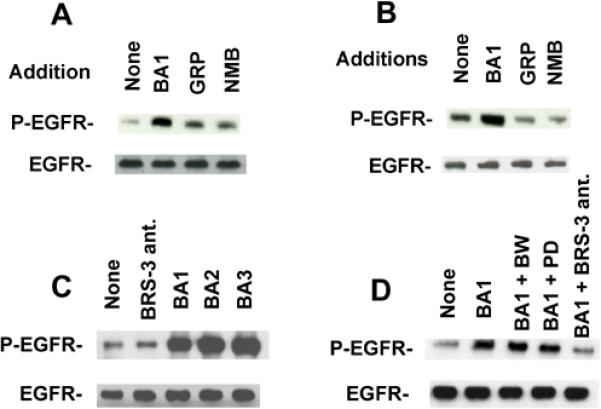
Specificity of EGFR tyrosine phosphorylation. (A) The ability of 100 nM BA1, 100 nM GRP or 100 nM NMB to alter EGFR tyrosine phosphorylation in NCIH727 cells was investigated. (B). The ability of BA1, GRP and NMB to alter EGFR tyrosine phosphorylation in NCI-H1299-BRS-3 cells was investigated. (C) The ability of 10 μM BRS-3 ant., 100 nM BA1, 100 nM BA2 or 100 nM BA3 to alter EGFR tyrosine phsophorylation in NCI-H1299-BRS-3 cells was investigated. (D) The ability of 10 μM BW2258U889 (BW), 10 μM PD168368 (PD) or 10 μM BRS-3 ant. to inhibit the increase in EGFR tyrosine phosphorylation caused by BA1 addition to NCI-H1299-BRS-3 cells was determined. As a control, equal amounts of total EGFR were loaded onto the gel. These experiments are representative of 3 others.
The ability of BRS-3 selective agonists and antagonists to alter EGFR transactivation was investigated. Figure 2C shows that BRS-3 ant. had little effect of basal EGFR tyrosine phosphorylation. In contrast, BA1, BA2 or BA3 when added to NCI-H1299-BRS-3 cells increased EGFR tyrosine phosphoryation. Figure 2D shows that the increase in EGFR tyrosine phosphorylation caused by addition of BA1 to NCI-H1299-BRS-3 cells was inhibited by BRS-3 ant. but not BW2258U89, a BB2R ant., or PD168368, a BB1R ant. These results indicate the BA1, BA2 and BA3 are agonists for BRS-3 whereas BRS-3 ant. blocks BRS-3. Table III shows that BRS-3 ant. inhibited specific 125I-BA1 binding to NCI-H1299-BRS-3 cells with an IC50 value of 750 nM. BRS-3 agonists such as BA1, BA2 and BA3 had IC50 values of 1.1, 21 and 15 nM, respectively. In contrast, BB, BW2258U89, gefitinib, GRP, NMB and PD168368 had IC50 values >2000 nM. These results show that BRS-3 ant. and BA1, BA2 as well as BA3, but not PD168368 or BW2258U89, interact with lung cancer BRS-3.
Table III.
Binding of BB ligands
| Addition | IC50, nM |
|---|---|
| BA1 | 1.1 ± 0.2 |
| BA2 | 21± 3 |
| BA3 | 15 ± 2 |
| BB | >2000 |
| BRS3 ant. | 750 ± 82 |
| BW2258U89 | >2000 |
| Gefitinib | >2000 |
| GRP | >2000 |
| NMB | >2000 |
| PD168368 | >2000 |
The ability of ligands to half maximally (IC50) inhibit specific 125I-BA1 binding to NCI-H1299-BRS-3 cells was determined at 37°C. The mean value ± S.E. of 4 determinations is shown.
The peptide structures are:
BB- Pyr-Gln-Arg-Leu-Gly-Asn-Leu-Trp-Ala-Val-Gly-His-Leu-Met-NH2
BA1- (DTyr6, βAla11, Phe13, Nle14)BB6-14
BA2- (DTyr6, R-Apa11, Phe13, Nle14)BB6-14
BA3- (DTyr6, R-Apa11, 4-Cl,Phe13, Nle14)BB6-14
BRS-3 ant. (DNal-Cys-Tyr-DTrp-Lys-Val-Cys-Nal)NH2.
3.3. BRS-3 agonists cause tyrosine phosphorylation of ERK
The ability of BA1 to cause ERK tyrosine phosphorylation was investigated. BA1 addition to NCI-H1299-BRS-3 cells caused tyrosine phosphorylation of the 170 KDa EGFR moderately after 0.5 min but strongly after 1 or 2 min (Fig. 3A). Figure 3B shows that BA1 significantly increased EGFR tyrosine phosphorylation 4.5-fold after 2 min. BA1 addition to NCI-H1299-BRS-3 cells increased phosphorylation of the 42 and 44 KDa ERK after 2 but not 0.5 or 1 min (Fig 3A). ERK tyrosine phosphorylation was significantly increased 2.8-fold two min after BA1 addition to lung cancer cells (Fig. 3B). The results indicate that BRS-3 agonists activate the EGFR and ERK.
Fig. 3.

BRS-3 regulates EGFR and ERK tyrosine phosphorylation. (A) The ability of 100 nM BA1 to cause tyrosine phosphorylation of the EGFR and ERK was investigated as a function of time after addition to NCI-H1299-BRS-3 cells. As controls equal amounts of EGFR and ERK were loaded onto the gel. (B) Densitometry analysis. The mean value ± S.D. of 4 experiments is indicated; p < 0.05 relative to control, *; p < 0.01, ** using the Student's t-test (C) The ability of various doses of gefitinib (Gef) to reverse the EGFR tyrosine phosphorylation caused by BA1 was investigated using NCI-H1299-BRS-3 cells. (D) Densitometry analysis. The mean value ± S.D. of 4 experiments is indicated; p < 0.01 relative to control, **; p < 0.05 relative to BA1, a; p < 0.01 relative to BA1, aa; using the Student's t-test.
3.4 Tyrosine kinase inhibitors block EGFR transactivation
The ability of BRS-3 agonists to cause EGFR transactivation was investigated using the tyrosine kinase inhibitor gefitinib. Gefitinib at 1 or 10 μg/ml but not 0.1 μg/ml inhibited the ability of BA1 to increase EGFR tyrosine phosphorylation in NCI-H1299-BRS-3 cells (Fig. 3C). BA1 significantly increased EGFR tyrosine phosphorylation 4.7-fold and the increase caused by BA1 was inhibited significantly by 1 or 10 μg/ml, but not 0.1 μg/ml gefitinib (Fig. 3D). Similarly the tyrosine kinase inhibitor AG1478 blocked EGFR or ERK tyrosine phosphorylation caused by BA1 (data not shown).
3.5. Other inhibitors of EGFR transactivation
Growth factors such as amphiregulin, EGF, heparin binding-EGF or transforming growth factor α bind to and activate the EGFR [7]. Src and matrix metalloproteases (MMP) are essential for release of endogenous TGFα from lung cancer cells and subsequent EGFR tyrosine phosphorylation [31]. Fig. 4A shows that BA1 addition to NCI-H1299-BRS-3 cells caused EGFR transactivation, which was inhibited if the cells were pretreated with GM6001 (MMP inhibitor), PP2 (Src inhibitor) or DPI (NADPH oxidase inhibitor). Figure 4B shows that BA1 addition to NCI-H1299-BRS-3 cells significantly increased EGFR tyrosine phosphorylation 4-fold whereas the increase caused by BA1 was significantly reversed if lung cancer cells were pretreated with GM60001, PP2 or DPI. Figure 4C shows that 5 mM N-acetylcysteine (NAC) or 5 mM Tiron (Tir) reversed the EGFR transactivation of the EGFR caused by BA1 to NCIH1299-BRS-3 cells. BA1 significantly increased EGFR tyrosine phosphorylation 3.9-fold and the increase caused by BA1 was significantly inhibited by NAC or tiron (Fig. 4D). The results suggest that BA1 causes transactivation of the EGFR in lung cancer cells in a Src-, MMP- and oxygen-dependent manner.
Fig. 4.

Inhibition of EGFR transactivation. (A) The ability of 10 μM PP2, 15 μM DPI or 10 μM GM6001 to inhibit EGFR tyrosine phosphorylation caused by 100 nM BA1 addition to NCI-H1299-BRS-3 cells was investigated. (B) Densitometry analysis. The mean value ± S.D. of 4 experiments is indicated; p < 0.01, ** relative to control; p < 0.05 relative to BA1, a; p < 0.01 relative to BA1, aa; using the Student's t-test. (C) The ability of 5 mM N-acetylcysteine (NAC) or 5 mM Tiron (Tir) to inhibit EGFR transactivation was investigated. (D) Densitometry analysis. The mean value ± S.D. of 4 experiments is indicated; p < 0.01, ** relative to control; p < 0.05 relative to BA1, a; p < 0.01 relative to BA1, aa; using the Student's t-test.
4. Discussion
The EGFR plays a proliferative role in NSCLC. The proliferation of NSCLC cells is stimulated by EGF or TGFα but is inhibited by the addition of EGFR monoclonal Abs [20, 22]. Recently, EGFR tyrosine kinase inhibitors (erlotininb and gefitinib) were approved for treating NSCLC patients who fail chemotherapy [16, 25, 35]. EGFR mutations of the ATP binding site are associated with increased response to EGFR tyrosine kinase inhibitors and a reduction in tumor size [16, 25, 35]. Approximately 90% of the NSCLC patients, however, have wild type EGFR with reduced sensitivity to gefitinib suggesting the need for new treatment modalities.
One area receiving considerable attention is the role of BB-like peptides in stimulating lung cancer growth in an autocrine manner. High levels of GRP and NMB are present in lung cancer cell lines [11]. NMB or GRP can be secreted from lung cancer cells and activate BB1R or BB2R respectively resulting in increased proliferation [3, 21, 33]. PD168368 and BW2258U89 are BB1R and BB2R antagonists which inhibit the growth of lung cancer cells. The BB2R antagonist PD176252 was synergistic with the erlotinib at inhibiting the growth of head and neck squamous carcinoma cells [51]. Recently, the BB1R antagonist PD168368 was found to be synergistic with gefitinib at inhibiting the growth of NSCLC cells [31]. The results suggest that the activation of the EGFR and BB family of receptors have important synergistic effects on the growth of some lung cancer cells.
Previously BRS-3 agonists stimulated the proliferation of bronchial epithelial cells [42] and we demonstrate that BRS-3 agonists increased NSCLC proliferation. BA1 increased the growth of Balb/3T3-BRS-3 cells but not control Balb/3T3 cells. A general problem with lung cancer cells, however, is that they have low densities of BRS-3 receptors in addition to BB1R and/or BB2R (1000/cell). Signals are amplified, however, when the cells are transfected with BRS-3; NCI-H1299-BRS-3 has 80,000 receptors/cell. BA1 but not GRP or NMB increased significantly NCI-H1299-BRS-3 colony number. BRS-3 ant. reduced NCI-H1299-BRS-3 colony number suggesting that NSCLC cells release an endogenous ligand which activates BRS-3. NCI-H1299-BRS-3 cellular proliferation had little effect upon treatment with PD168368 or BW2258U89 (T. Moody, unpublished). Gefitinib reduced the number of NCI-H1299-BRS-3 colonies in the presence or absence of BA1. BRS-3 ant. shifted the gefitinib dose-response curve to the left. The results indicate that BA1 stimulates growth of cells containing BRS-3. BRS-3 ant. addition to NSCLC cells increases gefitinib cytotoxicity.
Even though the natural ligand for BRS-3 remains unknown, it is possible to study the effects of BRS-3 using synthetic agonists and antagonists. The agonist BA1 binds with high affinity to BRS-3 (IC50 = 0.54 nM), however, it also binds with high affinity to human BB1R and BB2R (IC50 = 7.4 and 0.53 nM) respectively [29]. A related agonist Ac-Phe-Trp-Ala-His(γBzl)-Nip-Gly-Arg-NH2 was more selective for BRS-3 ( IC50 = 1300 nM) than BB2R (IC50 = >10000 nM [2, 28]. The BA1 related analog, Ac-Phe-Trp-Ala-His(γBzl)-Nip-Gly-Arg-NH2, preferred BRS-3 (IC50 = 80 nM) relative to BB2R (IC50 = 2800 nM) [26, 27]. Structure-function studies showed that extracellular domains 2 and 3 were important for BA3 binding with high affinity to BRS-3. In particular amino acids Val101, Thr106 and His107 of BRS-3 were important to bind BA3 with high affinity [10]. Leu123, Tyr291 and Arg316 were important for high affinity binding of BA2 to BRS-3 [12]. The peptoid 16a, N1-(2-Phenylethyl)-2R)-2-{[(1S)-1-(benzylcarboxamido)ethyl]carboxamido}-3-(1H-3-indolyl)propanamide, was reported to prefer BRS-3 relative to BB1 or BB2R [47, 50]. Unfortunately, 16a binds to human BRS-3 with low affinity (IC50 = 3160 nM) [39]. Subsequently BA2 and BA3 were described which bound with high affinity to hBRS-3 (IC50 = 2.8-82 nM) and had over 60-fold selectivity for hBRS-3 over hNMBR or hGRPR [28, 29]. In this communication BA1, BA2 or BA3 but not NMB or GRP addition to NCI-H1299-BRS-3 cells significantly increased tyrosine phosphorylation of the EGFR. The transactivation of the EGFR caused by BA1 addition to lung cancer cells was time-dependent (being maximal at 1 min) and dose-dependent (being maximal at 100 nM BA1). The results support the hypothesis that BRS-3 regulates EGFR transactivation in lung cancer cells.
Specific antagonists for BB1R and BB2R exist. PD168368 binds to BB1R, BB2R and BRS-3 transfected Balb 3T3 cells with IC50 values of 0.5, 213 and >10000 nM respectively [13]. BW2258U89 binds to BB1R, BB2R and BRS-3 with IC50 values of 4899, 0.2 and 6820 nM respectively [13]. Highly selective antagonists have not been developed for BRS-3. The substance P (SP) analog (D-Arg1, D-Pro2, D-Trp7,9, Leu11) SP and the somatostatin analog (BRS-3 ant.) antagonize BRS-3 [12, 48]. For Balb/3T3-BRS-3 cells, BB1R or BB2R the IC50 values for (DNal-Cys-Tyr-DTrp-Lys-Val-Cys-Nal)NH2 are 338, 605 and 4570 nM respectively. These results indicate that (DNal-Cys-Tyr-DTrp-Lys-Val-Cys-Nal)NH2 binds with higher affinity to BRS-3 and BB1R relative to BB2R. In contrast, (D-Arg1, D-Trp7,9, Leu11)SP bound with higher affinity to BB2R than BB1R or BRS-3. The addition of BRS-3 ant. to NCI-H1299-BRS-3 cells had little effect on basal EGFR tyrosine phosphorylation but decreased the EGFR tyrosine phosphorylation caused by BA1. In contrast, BW2258U89 or PD168368 did not antagonize the EGFR transactivation caused by BA1. BRS-3 ant. bound with moderate affinity (IC50 = 750 nM) to lung cancer cells. The results suggest that (DNal-Cys-Tyr-DTrp-Lys-Val-Cys-Nal)NH2 functions as a lung cancer BRS-3 antagonist. (DNal-Cys-Tyr-DTrp-Lys-Val-Cys-Nal) may function as a somatostatin receptor agonist, however, NSCLC cells do not express abundant somatostatin receptors [43], especially NCI-H1299 cells (T. Moody, unpublished).
BRS-3 regulates PI turnover in lung cancer cells [48] leading to ERK tyrosine phosphorylation. Addition of BA1 to NCI-H1299-BRS-3 cells increased ERK tyrosine phosphorylation 2.9-fold after 2 min. Also, BA1 addition to lung cancer cells increases c-fos mRNA after 1 hour. The increase in ERK phosphorylation caused by BA1 addition to NCI-H1299-BRS-3 cells was reversed by PD98059, a MEK-1 inhibitor, whereas PD98059 had no effect on EGFR transactivation (T. Moody, unpublished). These results suggest that EGFR tyrosine phosphorylation is upstream from ERK tyrosine phosphorylation. Preliminary data (T. Moody, unpublished) indicate that 10 μM PD98059 had little effect of the growth of all cells tested.
The increase in EGFR or ERK tyrosine phosphorylation caused by BA1 addition to NCI-H1299-BRS-3 cells was blocked by gefitinib or AG1478. Previously, we found that the NMB receptor activation increased the release of TGFα from NCI-H1299 cells [31]. Similarly, the increase in EGFR tyrosine phosphorylation caused by addition of NMB to NCI-H1299 cells was inhibited by addition of anti-TGFα antibodies. Preliminary data (T. Moody, unpublished) indicate that addition of TGFα but not amphiregulin, EGF or heparin binding EGF antibodies to NCI-H1299-BRS-3 cells inhibited the ability of BA1 to increased EGFR tyrosine phosphorylation. These results suggest that the generation of endogenous TGFα by BRS-3 activation is mediating EGFR tyrosine phosphorylation in lung cancer cells.
The ability of other agents to inhibit BRS-3 regulation of EGFR transactivation was examined. Galardin (GM 6001) is a broad-spectrum MMP inhibitor which blocks bombesin- and LPA-induced EGF receptor transactivation and DNA synthesis [40]. We found that GM6001 and PP2 inhibited the ability of BRS-3 to regulate EGFR transactivation in lung cancer cells. These results indicate that EGFR transactivation regulated by BRS-3 is dependent upon both Src and MMP activation. It remains to be determined if BRS-3 increases MMP activity causing metabolism of pro-TGFα to TGFα in lung cancer cells.
In some cells transactivation of the EGFR requires reactive oxygen species [31]. BRS-3 containing cells, similar to the BB1R [31], increased reactive oxygen species in NCI-H1299 cells that was reversed by tiron, a superoxide scavenger (T. Moody, unpublished). N-acetyl cysteine or tiron reversed the EGFR transactivation caused by addition of BA1 to NCI-H1299-BRS-3 cells. Because EGFR transactivation was reversed by DPI, the ROS may be due to NADPH oxidase activation. Oxidation of cysteine in the catalytic Src homology 2-containing tyrosine phosphastase may lead to reduced phosphatase enzymatic activity leading to an increase in the phosphorylation of the EGFR [4]. It remains to be determined if BRS-3 activation reduces phosphatase enzymatic activity.
In summary, BRS-3 similar to BB1R or BB2R, can regulate the growth of NSCLC cells. The BB family of receptors have similar signal transduction mechanisms, including phosphatidylinositol turnover and ERK tyrosine phosphorylation [18]. Peptide receptor antagonists such as BRS-3 ant., PD168368 or BW2258U89 have been shown to inhibit the growth of NSCLC cells in a cytostatic manner. Many of the growth effects of BBR antagonists in vitro, however, may result because EGFR tyrosine kinase activity is reduced. BRS-3 ant., PD168368 or PD176252 have been shown to increase the cytotoxicity of gefitinib using NSCLC cells. The results suggest that if more potent and selective BRS-3 antagonists were developed they may have important clinical applications.
Acknowledgments
This research is partially supported by the intramural research program of the NIDDK and NCI, of NIH.
References
- 1.Battey JF, Way JM, Corjay MH, Shapira H, Kusano K, Harkins R, et al. Molecular cloning of the bombesin/gastrin-releasing peptide receptor from Swiss 3T3 cells. Proc Natl Acad Sci USA. 1991;88:395–9. doi: 10.1073/pnas.88.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boyle RG, Humphries J, Mitchell T, Showell GA, Apaya R, Iijima H, et al. The design of a new potent and selective ligand for the orphan bombesin receptor subtype 3 (BRS-3). J Pept Sci. 2005;11:136–41. doi: 10.1002/psc.599. [DOI] [PubMed] [Google Scholar]
- 3.Carney DN, Cuttitta F, Moody TW, Minna JD. Selective stimulation of small cell lung cancer clonal growth by bombesin and gastrin-releasing peptide. Cancer Res. 1987;47:821–5. [PubMed] [Google Scholar]
- 4.Cheng-Hsien C, Yung-Ho H, Yuh-Mou S, Chun-Cheng H, Horng-Mo L, Huei-Mei H, Tso-Hsiao C. Src homology 2-containing phosphotyrosine phosphatase regulates endothelin-1-induced epidermal growth factor receptor transactivation in rat renal tubular cell NRK-52E. Pflugers Arch. 2006;452:16–24. doi: 10.1007/s00424-005-0006-9. [DOI] [PubMed] [Google Scholar]
- 5.Cuttitta F, Carney DN, Mulshine J, Moody TW, Fedorko J, Fischler A, Minna JD. Bombesin-like peptides can function as autocrine growth factors in human small-cell lung cancer cells. Nature. 1985;316(6031):823–6. doi: 10.1038/316823a0. [DOI] [PubMed] [Google Scholar]
- 6.DeMichele MA, Davis AL, Hunt JD, Landreneau RJ, Siegfried JM. Expression of mRNA for three bombesin receptor subtypes in human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;11(1):66–74. doi: 10.1165/ajrcmb.11.1.8018339. [DOI] [PubMed] [Google Scholar]
- 7.Draoui M, Siegall CB, FitzGerald D, Pastan I, Moody TW. TGFα-PE40 inhibit non-small cell lung cancer growth. Life Sci. 1994;54:445–53. doi: 10.1016/0024-3205(94)00403-x. [DOI] [PubMed] [Google Scholar]
- 8.Fathi Z, Corjay MH, Shapira H, Wada E, Benya R, Jensen R, et al. BRS-3: A novel bombesin receptor subtype selectively expressed in testis and lung carcinoma cells. J Biol Chem. 1993;268(8):5979–84. [PubMed] [Google Scholar]
- 9.Fathi Z, Way JW, Corjay MH, Viallet J, Sausville EA, Battey JF. Bombesin receptor structure and expression in human lung carcinoma cell lines. J Cell Biochem Supp. 1996;24:237–46. doi: 10.1002/jcb.240630519. [DOI] [PubMed] [Google Scholar]
- 10.Gbahou F, Holst B, Schwartz TW. Molecular basis for agonism in the BB3 receptor: an epitope located on the interface of transmembrane-III, -VI and –VII. J Pharmacol Exp Ther. 2010;333(1):51–9. doi: 10.1124/jpet.109.162131. [DOI] [PubMed] [Google Scholar]
- 11.Giaccone G, Battey J, Gazdar AF, Oie H, Draoui M, Moody TW. Neuromedin B is present in lung cancer cell lines. Cancer Res. 1992;52:2732–6s. [PubMed] [Google Scholar]
- 12.Gonzalez N, Hocart S, Portal-Nufiez S, Mantey SA, Nakagawa T, Zudaire E, et al. Molecular basis for agonist selectivity and activation of the orphan BRS-3-receptor. J Pharmacol Exp Ther. 2008;324(2):463–74. doi: 10.1124/jpet.107.132332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez N, Mantey SA, Pradhan TK, Sancho V, Moody TW, Coy DH, Jensen RT. Characterization of putative GRP and NMB-receptor antagonist's interaction with human receptors. Peptides. 2009;30:1473–86. doi: 10.1016/j.peptides.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gopas J, Ono M, Princler G, Smith MR, Tainsky MA, Siddiqui MA, et al. EGF receptor activity and mitogenic response of Balb/3T3 cells expressing Ras and Myc oncogenes. EGF receptor activity in oncogene transformed cells. Cell Mol Biol. 1992;38:605–14. [PubMed] [Google Scholar]
- 15.Guan XM, Chen H, Dobbelaar PH, Dong Y, Fong TM, Gagen K, et al. Regulation of energy homeostasis by bombesin receptor subtype-3: Selective receptor agonists for the treatment of obesity. Cell Metabolism. 2010;11:101–12. doi: 10.1016/j.cmet.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Helfrich BA, Raben D, Varella-Garcia M, Gustafson D, Chan DC, Bemis L, et al. Antitumor activity of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor gefitinib (ZD1839, Iressa) in non-small cell lung cancer cell lines correlates with gene copy number and EGFR mutations but not EGFR protein levels. Clin Cancer Res. 2006;12(23):7117–25. doi: 10.1158/1078-0432.CCR-06-0760. [DOI] [PubMed] [Google Scholar]
- 17.Hou X, Wei L, Harada A, Tatamoto K. Activation of bombesin receptor subtype-3 stimulates adhesion of lung cancer cells. Lung Cancer. 2006;54:143–8. doi: 10.1016/j.lungcan.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Jensen RT, Battey JF, Spindel ER, Benya RV. International Union of Pharmacology. LVIII. Mammalian Bombesin Receptors: Nomenclature, distribution, pharmacology, signaling and functions in normal and disease states. Pharmacol Rev. 2008;60:1–42. doi: 10.1124/pr.107.07108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen RT, Moody TW. Bombesin-related peptides and neurotensin: Effects on cancer growth/proliferation and cellular signaling in cancer. In: Kastin AJ, editor. Handbook of Biologically Active Peptides. Elsevier; Amsterdam: 2006. pp. 429–434. [Google Scholar]
- 20.Kollmannsberger C, Schittenheilm M, Honecker F, Tillner J, Weber D, et al. A. phase I study of the humanized monoclonal anti-epidermal growth factor receptor (EGFR) antibody EMD 72000 (matuzumab) in combination with paclitaxel in patients with EGFR-positive advancer non-small cell lung cancer (NSCLC). Ann Oncol. 2006;17(6):1007–13. doi: 10.1093/annonc/mdl042. [DOI] [PubMed] [Google Scholar]
- 21.Korman L, Carney DN, Citron ML, Moody TW. Secretin/VIP stimulated secretion of bombesin-like peptides from human small cell lung cancer. Cancer Res. 1986;46:1214–18. [PubMed] [Google Scholar]
- 22.Lee M, Draoui M, Zia F, Gazdar A, Oie H, Bepler G, et al. Epidermal growth factor receptor monoclonal antibodies inhibit the growth of lung cancer cell lines. J. Natl Cancer Inst. Monogr. 1992;(13):117–23. [PubMed] [Google Scholar]
- 23.Liu X, Carlisle DL, Swick MC, Gaither-Davis A, Grandis JR, Siegfried JM. Gastrin-releasing peptide activates Akt through the epidermal growth factor receptor pathway and abrogates the effect of gefitinib. Exp Cell Res. 2007;313(7):1361–72. doi: 10.1016/j.yexcr.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Lui VW, Thomas SM, Zhang Q, Wentzel AM, Siegfried JM, Li JY, Grandis JR. Mitogenic effects of gastrin-releasing peptide in head and neck squamous cancer cells are mediated by activation of the epidermal growth factor receptor. Oncogene. 2003;22(40):6183–93. doi: 10.1038/sj.onc.1206720. [DOI] [PubMed] [Google Scholar]
- 25.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 26.Mantey SA, Coy DH, Entsuah LK, Jensen RT. Development of bombesin analogs with conformationally restructed amino acid substitutions with enhance selectivity for orphan receptor human bombesin receptor subtype 3. J Pharm Exp Ther. 2004;310:1161–70. doi: 10.1124/jpet.104.066761. [DOI] [PubMed] [Google Scholar]
- 27.Mantey SA, Coy DH, Pradhan TK, Igarashi H, Rizo IM, Shen L, et al. Rational design of a peptide angonist that interacts selectively with the orphan receptor, bombesin receptor subtype 3. J Biol Chem. 2001;276:9219–29. doi: 10.1074/jbc.M008737200. [DOI] [PubMed] [Google Scholar]
- 28.Mantey SA, Gonzalez N, Schumann M, Pradhan TK, Shen L, Coy DH, Jensen RT. Identification of bombesin receptor subtype-specific ligands: Effect of N-methyl scanning, truncation, substitution and evaluation of putative reported selective ligands. J. Pharm Exp Ther. 2006;319:980–9. doi: 10.1124/jpet.106.107011. [DOI] [PubMed] [Google Scholar]
- 29.Mantey SA, Weber HC, Sainz E, Akeson M, Ryan RR, Pradhan TK, Searles RP, et al. Discovery of a high affinity radioligand for the human orphan receptor, bombesin receptor subtype 3, which demonstrates it has a unique pharmacology compared to other mammalian bombesin receptors. J Biol Chem. 1997;272(41):26062–71. doi: 10.1074/jbc.272.41.26062. [DOI] [PubMed] [Google Scholar]
- 30.Matsumoto K, Yamada K, Wada E, Hasegawa T, Usui Y, Wada K. Bombesin receptor subtype-3 modulates plasma insulin concentration. Peptides. 2003;24:83–90. doi: 10.1016/s0196-9781(02)00279-6. [DOI] [PubMed] [Google Scholar]
- 31.Moody TW, Berna MJ, Mantey S, Sancho V, Ridnour L, Wink DA, et al. Neuromedin B receptors regulate EGF receptor tyrosine phosphorylation in lung cancer cells. Eur J Pharm. 2010;637:38–45. doi: 10.1016/j.ejphar.2010.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moody TW, Lee M, Kris RM, Bellot F, Bepler G, Oie H, Gazdar AF. Lung carcinoid cell lines have bombesin-like peptides and EGF receptors. J. Cellular Biochem. 1990;43:139–47. doi: 10.1002/jcb.240430205. [DOI] [PubMed] [Google Scholar]
- 33.Moody TW, Staley J, Zia F, Coy DH, Jensen RT. Neuromedin B binds with high affinity, elevates cytosolic calcium and stimulates the growth of small-cell lung cancer cell lines. J Pharm Exp Ther. 1992;263:311–7. [PubMed] [Google Scholar]
- 34.Ohki Hamazaki H, Watase K, Yamamoto K, Ogura H, Yamano M, Yanada K, et al. Mice lacking bombesin receptor subtrype-3 develop metabolic defects and obesity. Nature. 1997;656:165–9. doi: 10.1038/36568. [DOI] [PubMed] [Google Scholar]
- 35.Paez JG, Janne PA, Lee JC, Tracey S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 36.Pradhan TK, Katsuno T, Taylor JE, Kim SH, Ryan RR, Mantey SA, et al. Identificatin of a unique ligand which has high affinity for all four bombesin receptor subtypes. Eur J Pharmacol. 1998;343(2-3):275–87. doi: 10.1016/s0014-2999(97)01527-6. [DOI] [PubMed] [Google Scholar]
- 37.Reubi JC, Wenger S, Schumuckli-Maurer J, Schaer JC, Gugger M. Bombesin receptor subtypes in human cancers: Detection with the universal radoligand (125)I-[D-TYR(6), beta-ALA(11),PHE(13), NLE(14)] bombesin(6-14). Clin Cancer Res. 2002;8(4):1139–46. [PubMed] [Google Scholar]
- 38.Ryan RR, Weber HC, Mantey SA, Hou W, Hilburger ME, Pradhan TK, et al. Pharmacology and intracellular signaling mechanisms of the native human orphan receptor BRS-3 in lung cancer cells. J Pharm Exp Ther. 1998;273:13613–24. [PubMed] [Google Scholar]
- 39.Sancho V, Moody TW, Mantey SA, Di Florio A, Uehara H, Coy DH, Jensen RT. Pharmacology of putative selective hBRS-3 receptor agonists for human bombesin receptors (BnR): Affinities, potencies and selectivity in multiple native and BnR transfected cells. Peptides. 2010;31:1569–78. doi: 10.1016/j.peptides.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santiskulvong C, Rozengurt E. Galardin (GM 6001), a broad-spectrum matrix metalloproteinase inhibitor, blocks bombesin- and LPA-induced EGF receptor transactivation and DNA synthesis in rat-1 cells. Exp Cell Res. 2003;290(2):437–46. doi: 10.1016/s0014-4827(03)00355-0. [DOI] [PubMed] [Google Scholar]
- 41.Spindel ET, Giladi E, Brehm TP, Goodman RH, Segerson TP. Cloning and functional characterization of a cDNA encoding the murine fibroblast bombesin/GRP receptor. Mol. Endocrin. 1990;4:1956–63. doi: 10.1210/mend-4-12-1956. [DOI] [PubMed] [Google Scholar]
- 42.Tan YR, Qi MM, Qin XQ, Xiang Y, Xiang L, Wang Y, et al. Wound repair and proliferation of bronchial epithelial cells enhance by bombesin receptor subtype 3 activation. Peptides. 2006;27:1952–8. doi: 10.1016/j.peptides.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 43.Taylor JE, Theveniau MA, Bashirzadeh R, Resine T, Eden PA. Detection of somatostatin receptor sybtype 2 (SSTR2) in established tumors and tumor cell lines : Evidence for SSTR2 heterogeneity. Peptides. 1994;15(7):1229–36. doi: 10.1016/0196-9781(94)90146-5. [DOI] [PubMed] [Google Scholar]
- 44.Thomas SM, Grandis JR, Wentzel AL, Gooding WE, Lui VW, Siegfried JM. Gastrin-releasing peptide receptor mediates activation of the epidermal growth factor receptor in lung cancer cells. Neoplasia. 2005;7:426–31. doi: 10.1593/neo.04454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toi-Scott M, Jones CLA, Kane MA. Clinical correlates of bombesin-like peptide receptor subtype expression in human lung cancer cells. Lung Cancer. 1996;15:341–54. doi: 10.1016/0169-5002(95)00597-8. [DOI] [PubMed] [Google Scholar]
- 46.Wada E, Way J, Shapira H, Kusamo K, Lebacq-Verheyden AM, Coy D, Jensen RT, Battey J. cDNA cloning, characterization and brain region-specific expression of a neuromedin-B preferring bombesin receptor. Neuron. 1991;6:421–30. doi: 10.1016/0896-6273(91)90250-4. [DOI] [PubMed] [Google Scholar]
- 47.Weber D, Berger C, Eickelmann P, Antel J, Kessler H. Design of selective peptidomimetic agonists for the human orphan receptor BRS-3. J Med Chem. 2003;146:1918–30. doi: 10.1021/jm0210921. [DOI] [PubMed] [Google Scholar]
- 48.Weber HC, Walters J, Leyton J, Casibang M, Purdom S, Jensen RT, et al. A bombesin receptor subtype-3 peptide increases nuclear oncogene expression in a MEK-1 dependent manner in human lung cancer cells. Eur J Pharm. 2001;412:13–20. doi: 10.1016/s0014-2999(00)00941-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiao D, Qu X, Weber HC. Activation of extracellular signal-regulated kinase mediates bombesin-induced mitogenic responses in prostate cancer cells. Cell Signal. 2003;15(10):945–53. doi: 10.1016/s0898-6568(03)00059-7. [DOI] [PubMed] [Google Scholar]
- 50.Zhang L, Nothacker HP, Wang Z, Bohn LM, Civelli O. Pharmacological characterization of a selective agonist for bombesin receptor subtype 3. Biochem Biophys Res Commun. 2009;387(2):183–8. doi: 10.1016/j.bbrc.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Q, Bhola NE, Lui VW, Siwak DR, Thomas SM, Gubish CT, et al. Antitumor mechanisms of combined gastrin-releasing peptide receptor and epidermal growth factor receptor targeting in head and neck cancer. Mol Cancer Ther. 2007;6(4):1414–24. doi: 10.1158/1535-7163.MCT-06-0678. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Q, Thomas SM, Smithgall TE, Siegfried JM, Kamens J, Gooding WE, Grandis JR. SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res. 2004;64(17):6166–73. doi: 10.1158/0008-5472.CAN-04-0504. [DOI] [PubMed] [Google Scholar]