

Abstract
Nitric oxide (NO) is a messenger molecule that is highly diffusible and short-lived. Despite these two characteristics, seemingly unsuitable for intracellular reactions, NO modulates a variety of cellular processes via the mechanism of S-nitrosylation. An important factor that determines the specificity of S-nitrosylation as a signaling mechanism is the compartmentalization of nitric oxide synthase (NOS) with its target proteins. Endothelial NOS (eNOS) is unique among the NOS family members by being localized mainly near specific intracellular membrane domains including the cytoplasmic face of the Golgi apparatus and plasma membrane caveolae. Nitric oxide produced by eNOS localized on the Golgi apparatus can react with thiol groups on nearby Golgi proteins via a redox mechanism resulting in S-nitrosylation of these proteins. This modification influences their function as regulators of cellular processes such as protein trafficking (e.g., exocytosis and endocytosis), redox state, and cell cycle. Thus, eNOS-derived NO regulates a wide range of endothelial cell functions, such as inflammation, apoptosis, permeability, migration and cell growth.
Keywords: NOS, endothelium, Golgi apparatus, trafficking, vesicles, cardiovascular
1. Introduction
Nitric oxide (NO) produced by endothelial NO synthase (eNOS) mediates various cellular processes important for endothelial cell functions and regulates vascular response[1]. In addition to conventional NO signaling via the activation of soluble guanylyl cyclase (sGC) to produce cGMP as a second messenger[2], protein modification by NO via S-nitrosylation has also received considerable attention[3]. S-nitrosylation is the coupling of an NO moiety to a reactive thiol side chain of cysteine to form an S-nitroso-thiol and is considered an important mechanism for dynamic, post-translational regulation of many classes of proteins. A growing body of evidence suggests that eNOS-derived NO can S-nitrosylate specific proteins, influence cellular processes in endothelial cells, and modulate a variety of endothelial cell functions[4], such as inflammation[5–7], apoptosis[8], permeability[9], migration[10], and cell growth[11]. This article focuses on protein S-nitrosylation by eNOS-derived NO and discusses cellular processes regulated by S-nitrosylated proteins and the subsequent effects of these cellular processes on endothelial cell functions.
2. Unique cellular localization of eNOS
Endothelial NOS is unique among the NOS family members by being localized mainly near specific intracellular membrane domains including the cytoplasmic face of the Golgi apparatus and plasma membrane caveolae[12]. The functional significance of this unique localization of eNOS in endothelial cells remains largely unknown. However studies by Fulton and colleagues[13–15] demonstrated that these two pools of eNOS are differentially regulated. Endothelial NOS localized at the plasma membrane is activated more efficiently by calcium-dependent and Akt-dependent agonists, while eNOS on the Golgi apparatus is less responsive to both agonists[14, 15]. As a consequence, the amount of NO produced by eNOS differs at these two locations. eNOS localized on the plasma membrane releases a greater amount of NO than eNOS on the Golgi apparatus.
3. Nitric oxide is confined to the region where eNOS is localized
In comparison to the actions of NO-regulating metal-centered processes (activation of sGC and inhibition of cytochrome oxidase) or its interaction with other radicals, the primary biological reactions involving S-nitrosylation of a thiol by NO are thought to require higher concentrations of NO to sustain them in spite of its diffusible nature [16, 17] (Figure 1). The presence of a localized NO pool has been reported in human endothelial cells[16]. It was shown that the presence of vascular endothelial growth factor (VEGF) which leads to the phosphorylation and activation of eNOS was concurrent with a localized, concentrated NO pool in the perinuclear region of the cell. In this study[16], the localized NO pool was visualized by detecting diaminofluorescein-2 (DAF-2) nitrosation products in endothelial cells.
Figure 1. The target protein is supposed to be surrounded by eNOS, or have an environment enriched with NO to promote S-nitrosylation.
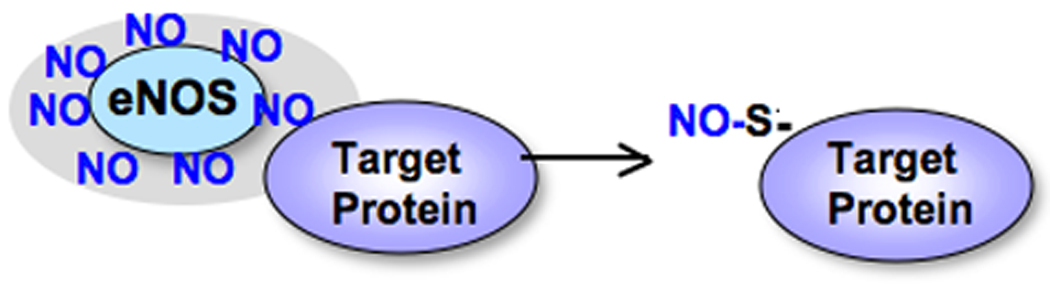
Nitric oxide pools are formed in the region where eNOS is localized and create favorable environments for target proteins to be S-nitrosylated.
To demonstrate the association of localized sites of active NOS with the presence of a concentrated NO pool, we generated red fluorescent protein (RFP)-tagged eNOS constructs and monitored the simultaneous presence of both red (for eNOS localization) and green (DAF-2, for areas of NO generated) fluorescence in transfected COS-7 cells using confocal microscopy[4]. Upon stimulation of the cells with ATP, the highest amount of DAF-2 fluorescence was detected in a perinuclear region overlapping with or adjacent to RFP-tagged eNOS WT, similar to that observed in human endothelial cells[16]. To further confirm that eNOS-derived NO acts locally, we performed identical experiments in cells transfected with an RFP-tagged eNOS-NLS mutant which localizes in the nucleus. As expected, the presence of concentrated NO (imaged by DAF-2) also co-localized in the nucleus. Thus, these data using RFP-tagged eNOS constructs clearly suggest that NO is preferentially channeled to sites in proximity to those showing eNOS activity.
Additionally, we visualized cellular NO distribution generated by NO donors such as S-nitroso- glutathione (GSNO) and S-nitroso-N-acetylpenicillamine (SNAP) [4]. The dye-loaded COS-7 cells showed an increase in fluorescent intensity immediately after adding the NO donor. The fluorescent signal was diffusely distributed throughout the cells, suggesting that DAF is not concentrated on specific organelles in COS-7 cells. Collectively, these observations suggest that eNOS and NO donors provide different spatial NO concentration.
4. Protein S-nitrosylation is restricted to regions of the cell where eNOS is localized
We also demonstrated that S-nitrosylated proteins are formed preferentially at the primary site of eNOS localization[4]. Using an S-nitrosocysteine-specific antibody, we performed immunofluorescent analysis for protein S-nitrosylation in COS-7 cells transfected with RFP-tagged eNOS WT or RFP-tagged eNOS-NLS. If an NO pool is formed at the primary site of eNOS localization in the cells, then it is probable that S-nitrosylation of proteins may also be localized as suggested by other researchers [17, 18]. We found S-nitrosylated proteins concentrated at the primary site of eNOS localization, that is, the Golgi apparatus. In contrast, cells treated with the NO donor, SNAP, showed a markedly different pattern of S-nitrosylated protein immunoreactivity. Thus, the location of eNOS may determine the specificity of S-nitrosylation at that site by creating a high NO concentration. This would favor S-nitrosylation of specific proteins in endothelial cells because of their proximity to the eNOS site and, by corollary, to the site of high NO concentration. This would explain the different cellular responses induced by NO donors not at an eNOS site, i.e. not at a site of active endogenous NO synthesis[3]. This may reflect the concept that all sources of NO are not bioequivalent.
5. Endothelial NOS localization influences cellular processes
Localization of eNOS on the Golgi apparatus can enhance S-nitrosylation of Golgi proteins and may influence their functions[4, 19]. One such example is protein trafficking. We demonstrated that eNOS which is localized on the Golgi apparatus reduces the speed of protein transport from the endoplasmic reticulum (ER) to the plasma membrane[4]. We showed this using the transport of a temperature-sensitive vesicular stomatitis virus glycoprotein (VSVG) tagged with green fluorescent protein (GFP) (labeled as VSVG-GFP), as an indicator of protein transport from the ER to the cell surface. Cells were microinjected with VSVG-GFP plus a control plasmid, RFP-tagged eNOS WT or RFP-tagged eNOS-NLS. We then monitored movement of VSVG-GFP from the ER to the Golgi apparatus, and then to the cell surface in the presence of the control plasmid, RFP-tagged eNOS WT, or RFP-tagged eNOS-NLS. In the cells injected with RFP-tagged eNOS WT (mostly localized on the Golgi apparatus), there was a marked delay in targeting VSVG-GFP to the cell surface, whereas the cells injected with RFP-tagged eNOS-NLS (localized in the nucleus) exhibited similar rates of transport to those cells injected with the control plasmid. To further determine if the reduced rate of transport observed in the cells with RFP-tagged eNOS WT is dependent on NO, we treated those cells with a NOS inhibitor (L-NAME) to abrogate NO release and then monitored VSVG transport. The cells treated with the NOS inhibitor restored the rate of VSVG-GFP transport to that observed in the control or RFP-tagged eNOS-NLS-expressing cells, demonstrating the control of basal protein transport by NO, perhaps in this case via the S-nitrosylation of proteins important for protein traffic signaling, such as N-ethylmaleimide sensitive fusion (NSF) protein.
6. Cellular processes in endothelial cells regulated by protein S-nitrosylation
Protein trafficking is one example of the cellular processes that are influenced by NO via S-nitrosylation of specific proteins. Focusing on eNOS and endothelial cells, this section discusses recent findings that protein S-nitrosylation influences various cellular processes in endothelial cells.
6-1. Protein trafficking
NO regulation of vesicle/protein trafficking is an example of a molecular control mechanism that explains many endothelial cell functions and cardiovascular effects, and may be of broad physiological significance.
Exocytosis
NO inhibits the exocytosis of Weibel-Palade bodies, endothelial storage granules[6]. Weibel-Palade bodies contain von Willebrand factor (vWF), P-selectin, tissue plasminogen activator and CD63, and are rapidly released from endothelial cells, mediating vascular thrombosis and inflammation[20, 21]. NO decreases granule trafficking from the Golgi apparatus to the plasma membrane by targeting a key component of the exocytic machinery, n-ethylmaleimide sensitive fusion (NSF) protein, which facilitates disassembly of soluble NSF attachment protein receptor (SNARE) complexes[22, 23] (Figure 2). NO inhibits this process by S-nitrosylating critical cysteine residues of NSF[6]. Golgi localization of eNOS facilitates S-nitrosylation of NSF and decreases granule trafficking from the Golgi apparatus to the plasma membrane[4, 24]. Therefore, eNOS localization on the Golgi apparatus is an important factor that regulates protein trafficking and may influencing a wide range of physiological processes, vascular inflammation and thrombosis.
Figure 2. Nitric oxide prevents exocytosis of granules via S-nitrosylation of N-ethylmaleimide sensitive fusion protein (NSF).
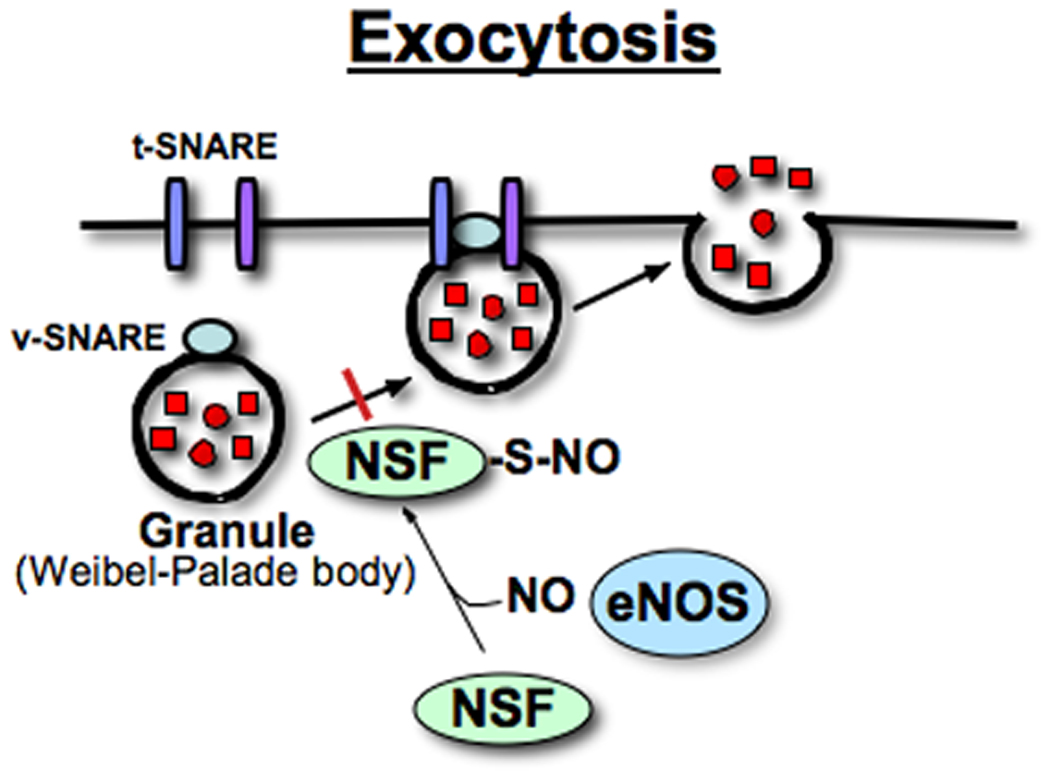
NO decreases granule trafficking from the Golgi apparatus to the plasma membrane by targeting a key component of the exocytic machinery, NSF, which facilitates disassembly of soluble NSF attachment protein receptor (SNARE) complexes[22, 23]. NO inhibits this process by S-nitrosylating NSF[6]. Weibel-Palade bodies are endothelial storage granules that contain von Willebrand factor, P-selectin, tissue plasminogen activator, and CD63 and are rapidly released from endothelial cells, mediating vascular thrombosis and inflammation[20, 21]. Thus, S-nitrosylation of NSF inhibits thrombosis, limiting vascular inflammation in endothelial cells. The diagram was adapted from Matsushita et al.[6] and a review paper by Lowenstein[76].
Endocytosis
While having the inhibitory effect on exocytosis mentioned above, NO also accelerates endocytosis by S-nitrosylating dynamin, a GTPase [5, 7] (Figure 3). There are three types of endocytosis: phagocytosis, macropinocytosis and receptor-mediated uptake[25]. S-nitrosylation of dynamin increases receptor-mediated endocytosis[5, 7], which involves surface receptors that interact and make a cluster with soluble ligands at clathrin-coated pits[25]. After a coated vesicle is formed at the cell surface, dynamin is assembled into a cluster forming a collar around the neck of the nascent vesicle and facilitates scission of the vesicle from the membrane[26]. S-nitrosylation of dynamin increases its ability to oligomerize with a subsequent increased rate of GTP hydrolysis. This oligomeric assembly of dynamin around a nascent vesicle drives the mechanism of cleavage of the vesicle from the plasma membrane. Interestingly, an interaction between dynamin and eNOS localizes NO production to the site of endocytosis[27]. Again, this suggests that an NO pool generated around eNOS facilitates S-nitrosylation of dynamin, leading to accelerated endocytosis.
Figure 3. Nitric oxide enhances endocytosis of vesicles via S-nitrosylation of dynamin.
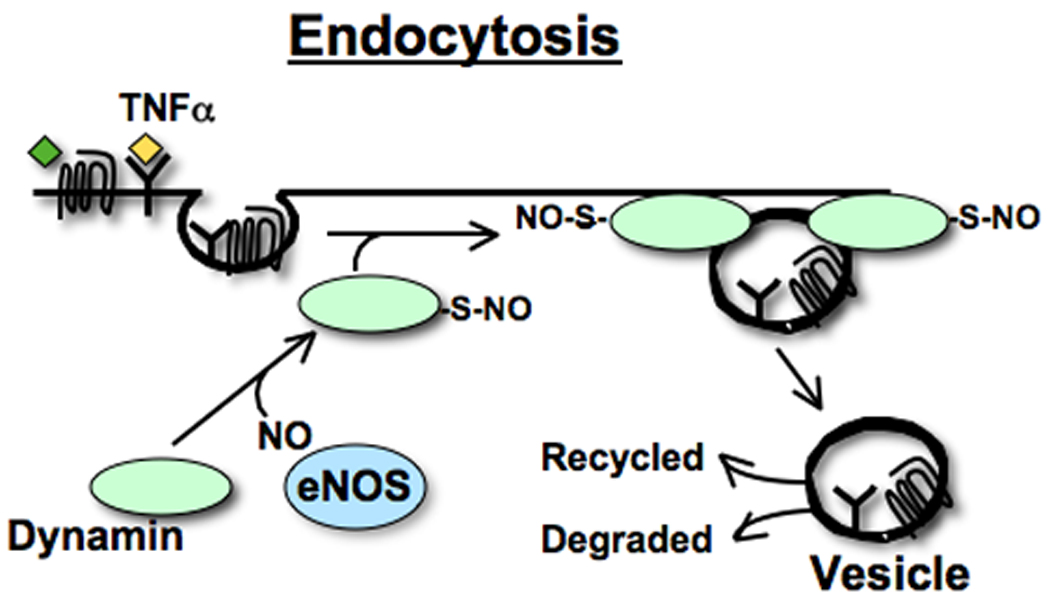
In endothelial cells, eNOS interacts with dynamin[27] through the production of NO which S-nitrosylates dynamin, enhancing its GTPase activity. This in turn facilitates receptor mediated endocytosis[5, 7]. After a coated vesicle is formed at the cell surface, dynamin assembles into multimers, forming a collar around the neck of the nascent vesicle, and facilitates scission of the vesicle from the membrane[26]. The diagram was adapted from Kang-Decker et al.[7], Wang et al.[5] and a review paper by Lowenstein[76].
Another example of NO-mediated receptor endocytosis is the regulation of β-adrenergic receptor (β-AR) internalization[28]. β-AR is a prototypic G-protein coupled receptor (GPCR) and plays an important role in cardiovascular[29] and pulmonary functions[30]. NO regulates β-AR internalization and desensitization via S-nitrosylation of three key players in these processes: β-arrestin2, GPCR kinase 2 (GRK2), and dynamin[28]. First, β-arrestin2 serves as a scaffold that functionally co-localizes β-AR and eNOS [31]. Upon ligand stimulation on β-AR, β-AR associates with β-arrestin2 and eNOS and also activates eNOS, which facilitates S-nitrosylation of β-arrestin2. This S-nitrosylation promotes dissociation of β-arrestin2 from eNOS and an association of β-arrestin2 with clathrin heavy chain/β-adaptin. These processes ultimately accelerate β-AR internalization through the clathrin-based endocytotic pathway. Once internalized, β-AR is either degraded or recycled. β-arrestin2 is subsequently de-nitrosylated and then forms a complex with eNOS. Second, S-nitrosylation of GRK2 decreases β-AR phosphorylation and subsequent recruitment of β-arrestin2 to the receptor, thereby decreasing β-AR internalization and desensitization[28]. Third, S-nitrosylation of dynamin facilitates clathrin-dependent endocytosis of membrane receptors, including β-AR, thereby leading to downregulation of β-AR signaling. Thus, NO can prevent the loss of β-AR signaling via S-nitrosylation of GRK2, while facilitating the loss of β-AR signaling via S-nitrosylation of β-arrestin2 and dynamin.
6-2. Redox regulation
Endothelial NADPH oxidase is a major source of the superoxide radical (O2−) in blood vessels and is implicated in the oxidative stress related to vascular diseases including atherosclerosis[32]. One study demonstrated that NO inhibits NADPH activity via S-nitrosylation, decreasing the generation of O2− in human microvascular endothelial cells, independent from peroxynitrite formation[33]. Thus, NO can act as an antioxidant agent through an inhibition of NADPH oxidase activity via S-nitrosylation in endothelial cells.
Thioredoxin 1 (TRX1) is an antioxidant enzyme that regulates the redox state in endothelial cells[34, 35]. S-nitrosylation of TRX1 at its Cys-69 residue is necessary for the redox regulatory activity of TRX1 and facilitates scavenging reactive oxygen species (ROSs)[35]. Statins have beneficial pleiotropic activities, not limited to their cholesterol-lowering activity, such as a vasoprotective effect by activating eNOS[34]. In addition, statins are found to promote S-nitrosylation of TRX1 subsequently stimulating TRX1 activity. As a consequence, statins reduce ROS production and thus protect endothelial cell integrity.
6-3. Anti-apoptosis
Endothelial cell-based angiogenesis are governed by a counterbalance of opposing endothelial cell survival signals and apoptosis signals. Nitric oxide signals are known to mediate cell survival and resistance to apoptosis, a key component of endothelial cell-based angiogenesis. Endothelial NOS can inhibit apoptosis of endothelial cells by S-nitrosylating several key players of apoptosis including dynamin-2[7], caspase-3[36], and thioredoxin[36].
Tumor necrosis factor-alpha (TNF-α) constitutes a prominent apoptotic stimulus for endothelial cells in pathological conditions[37]. S-nitrosylation of dynamin-2 stimulates its activity and enhances endocytosis in endothelial cells[7]. This biological consequence of dynamin-2 S-nitrosylation prevents TNFα-induced apoptosis in endothelial cells. In some pathological conditions, endothelial cell-based angiogenesis requires activation of survival signals against external apoptotic stimuli such as TNFα (Figure 3). Endothelial NOS S-nitrosylates dynamin-2, which then facilitates TNFα receptor internalization by enhancing endocytosis of the TNFα receptor. As a consequence, TNFα-induced apoptotic signals in endothelial cells are inhibited during angiogenesis in pathological conditions[7].
S-nitrosylation of an active cysteine residue on caspase-3 inhibits caspase activity [38–40] in endothelial cells[36]. Caspase-3 is known as a pro-apoptotic protease enzyme. Thus, eNOS-derived NO inhibits caspase-3 activity and subsequently inhibits apoptosis in endothelial cells.
S-nitrosylation of thioredoxin 1 (TRX1) protects endothelial cells from apoptosis[35]. Since S-nitrosylated TRX1 reduces ROS levels in endothelial cells and ROS are known to induce apoptosis[41], reduced ROS levels may contribute to the anti-apoptotic effect resulting from S-nitrosylation of TRX1.
Besides control of ROS levels in endothelial cells, TRX1 and TRX reductase 1(TRX1-TRXR1), collectively known as the TRX system, play a critical role in the regulation of S-nitrosylated protein levels by de-nitrosylating target proteins[42, 43]. One such protein is caspase-3[42]. When cells are depleted of TRX1-TRXR1 by underexpression using siRNAs, levels of S-nitrosylated-caspase-3 are significantly increased, while overexpression results in decreased S-nitrosylated caspase-3 levels. Currently, it has not been directly demonstrated whether the TRX system also controls S-nitrosylated caspase-3 levels in endothelial cells. However, since S-nitrosylation of caspase-3 plays a role in the promotion of endothelial cell integrity by inhibiting caspase-3 activity and decreasing apoptosis in endothelial cells, it is possible that the TRX system decreases endothelial cell apoptosis, not only by decreasing ROS levels in endothelial cells, but also by directly denitrosylating and decreasing caspase-3 activity.
6-4. Cell permeability
Increased permeability in endothelial cells is an initial event that induces VEGF-mediated angiogenesis. Junction proteins, such as VE-cadherin and β-catenin, control VEGF-induced permeability in endothelial cells[44, 45]. It is known that eNOS-derived NO is essential for VEGF-induced vascular permeability since eNOS−/− mice show markedly reduced VEGF-induced vascular permeability[46]. The mechanism by which eNOS and thus NO increase this permeability is related to S-nitrosylation of the Cys-619 residue on β-catenin[46]. S-nitrosylation of β-catenin facilitates its dissociation from the VE-Cadherin/β-catenin junction complex, leading to an increase in endothelial cell permeability. VEGF is unable to promote S-nitrosylation of β-catenin in the absence of eNOS and is thus unable to increase permeability in endothelial cells from eNOS−/− mice.
6-5. Cell migration
Stromal cell-derived factor-1α (SDF-1α) is one of the pivotal chemokines that facilitate angiogenesis by enhancing endothelial cell migration [47, 48]. SDF-1α facilitates endothelial cell migration by inducing a signaling cascade that involves eNOS activation, JNK, and MAPK phosphatase 7 (MKP7) [10]. SDF-1α activates eNOS via phosphorylation and thus increases NO production in endothelial cells. Further, SDF-1α activates JNK3, a critical activator of endothelial cell migration. The activation of eNOS and subsequent increase in NO production are required for JNK3 activation (i.e., phosphorylation of JNK3). However, phosphorylated and activated JNK3 can be inactivated by MAPK phosphatase 7 (MKP7) and it is here that eNOS plays an important role. Endothelial NOS S-nitrosylates MKP7 thus lowering its activity and thereby sustaining the phosphorylation and activation of JNK3. In brief, SDF-1α activates eNOS via phosphorylation. Subsequently eNOS-derived NO inactivates MKP7 via S-nitrosylation, promoting JNK3 activation and ultimately endothelial cell migration.
6-6. Cell cycle
Nitric oxide promotes cell cycle progression via S-nitrosylation of p21Ras and activation of the p21Ras-ERK1/2 MAP signaling pathway in endothelial cells[11]. A small G protein, p21Ras, is a guanine nucleotide-binding protein that cycles between the inactive GDP-bound and active GTP-bound states to regulate a diverse array of cellular processes, including cell growth, apoptosis and differentiation[49, 50]. Nitric oxide has been shown to promote guanine nucleotide exchanges on p21Ras and increase cellular Ras-GTP levels (the active form)[51–54]. In this process, NO S-nitrosylates p21Ras at Cys-118 and increases the active GTP-bound form of p21Ras[55]. Furthermore, S-nitrosylation of p21Ras triggers its downstream ERK1/2 MAP signaling pathway[51]. Oliveira and colleagues[11] demonstrated that endothelial cells stably expressing a p21Ras mutant at this S-nitrosylation site (C118S) inhibits the ERK1/2 MAP signaling pathway. Moreover, treatment with the NO donor, SNAP, enhances levels of Cyclin D1, Cdk4, Cdk6 and increases phosphorylation of RB protein, all of which are known to promote progression of the cell cycle. Collectively, these observations suggest that NO promotes endothelial cell cycle progression by p21Ras S-nitrosylation, which in turn facilitates the ERK1/2 MAP signaling pathway, and enhances expression/activation of factors that are associated with cell cycle progression.
6-7. Energy production
Nitric oxide decreases energy production by S-nitrosylating cytochrome C-oxidase (Complex IV) in the mitochondria of endothelial cells. Complex IV is a terminal enzyme of the mitochondrial electron transport chain and is an essential enzyme for regulating energy production. It has been shown that Complex IV is inactivated by S-nitrosylation at either of two cysteine residues at its active site[56]. Prolonged exposure of porcine pulmonary artery endothelial cells to 1 mM NOC-18 (a slow releasing NO donor, equivalent to 1– 5 µM NO) results in a gradual, persistent inhibition of Complex IV, which is concomitant with a reduction in the ratio of mitochondrial GSH to GSSG. Further, an overexpression of TRX1 in the mitochondria attenuates NO-induced loss of Complex IV activity in pulmonary artery endothelial cells. These findings provide a mechanism for NO toxicity in lung endothelial cells resulting from excessive levels of NO seen in inflammation, polluted air and tobacco smoke. Persistent inhibition of cytochrome-c oxidase by S-nitrosylation and the subsequent reduction in energy production contributes to enhanced oxidant production and programmed cell death or apoptosis of lung cells.
6-8. Nitric oxide production
Endothelial NOS itself is regulated by S-nitrosylation[57–59]. S-nitrosylation decreases eNOS activity. Endothelial NOS is active only as a homodimer[60]. Exogenous NO facilitates S-nitrosylation of eNOS, disrupts its dimer formation and decreases its activity[61]. Thus, TRX1 enhances eNOS activity by inhibiting its S-nitrosylation which prevents its monomerization. Endothelial NOS is S-nitrosylated at a basal level[57]. However, upon agonist stimulation, such as VEGF, eNOS is de-nitrosylated and phosphorylation at Ser-1179, a site associated with eNOS activation, is increased. Thus, S-nitrosylation and de-nitrosylation of eNOS regulate its activity and subsequent NO production in endothelial cells.
Endothelial NOS is indeed regulated by complex post-translational modifications and protein-protein interactions[1]. Heat shock protein 90 (Hsp90) interacts with eNOS, leading to eNOS activation[62]. This interaction between eNOS and Hsp90 is also regulated by S-nitrosylation at a cysteine residue (Cys-597) in the region of the C-terminal domain that interacts with eNOS[63]. S-nitrosylation of Hsp90 inhibits both its ATPase activity and its positive effects on eNOS activity, which may serve as a negative feedback mechanism of eNOS activation.
The enzyme dimethylarginine dimethylaminohydrolase (DDAH1) hydrolyses asymmetrically methylated arginine residues which are endogenously produced inhibitors of eNOS. Thus, DDAH1 positively regulates eNOS activity. Under certain conditions when NO generation is increased, DDAH1 is S-nitrosylated. S-nitrosylation of DDAH1 diminishes DDAH1 activity, which then increases an accumulation of asymmetric dimethylarginine and inhibits eNOS activity. This process has been proposed as a mechanism for the expression of iNOS, which causes a transient increase in NO levels, often leading to inhibition of the activity of constitutively expressed NOS isozymes, including eNOS[64].
6-9. Anti-thrombosis and cardio-protective effects
Tissue plasminogen activator (tPA) is a serine protease found on endothelial cells[65]. Tissue PA mediates the breakdown of blood clots by catalyzing the conversion of plasminogen to plasmin, the major enzyme responsible for clot breakdown. This beneficial effect of tPA is facilitated by S-nitrosylation of tPA. S-nitrosylation of tPA enhances the vasodilatory, anti-platelet and fibrinolytic properties of the endothelium[66]. Furthermore, S-nitrosylated tPA inhibits the adherence of neutrophils to the vascular endothelium and decreases P-selectin localization in the ischemic region[67]. Thus, NO by S-nitrosylation of tPA contributes to the maintenance of vascular health.
7. Endogenous stimuli that facilitate S-nitrosylation of proteins in endothelial cells
Given that eNOS-derived NO is an important source of S-nitrosylation of proteins in endothelial cells, any stimuli that enhance eNOS activity are expected to promote S-nitrosylation of proteins in endothelial cells. Those stimuli include VEGF[9], estrogen[68, 69], TNFα[36], oxidized LDL[36], hypoxia[70] and shear stress[71, 72].
Cellular levels of S-nitrosylated proteins are also regulated by the enzyme S-nitroso-glutathione reductase (GSNOR) which degrades S-nitroso-glutathione (GSNO) and lowers levels of S-nitrosylated proteins[73, 74]. It has been shown that genetic deletion of GSNOR (GSNOR−/−) increases levels of S-nitrosylated proteins, including SNO-Hb[74], GRK2[28], β-arrestin2[31] and HIF-1[75], all important molecules for maintenance of the cardiovascular system. This suggests a role for S-nitrosylated proteins in cardiovascular health.
8. Endothelial NOS-derived NO and S-nitrosylation in other cell types
Endothelial NOS-derived NO regulates T-cell receptor-dependent ERK activation by activating the p21Ras/ERK signaling pathway upon stimulation by antigen in antigen-presenting cells[19]. This process is mediated by S-nitrosylation of the N-form of p21Ras at Cys-118, a known S-nitrosylation site of this protein[55]. The S-nitrosylation is specific for the N- but not K-form of p21Ras. Furthermore, N-p21Ras is S-nitrosylated on the Golgi apparatus by WT eNOS, not by the G2A mutant of eNOS which localizes in the cytosol. This suggests that compartmentalization of eNOS with N-p21Ras on the surface of the Golgi apparatus is necessary for this process. Furthermore, N-p21Ras S-nitrosylation increases T-cell-dependent apoptosis, suggesting that it contributes to activation-induced T-cell death.
9. Summary and future direction
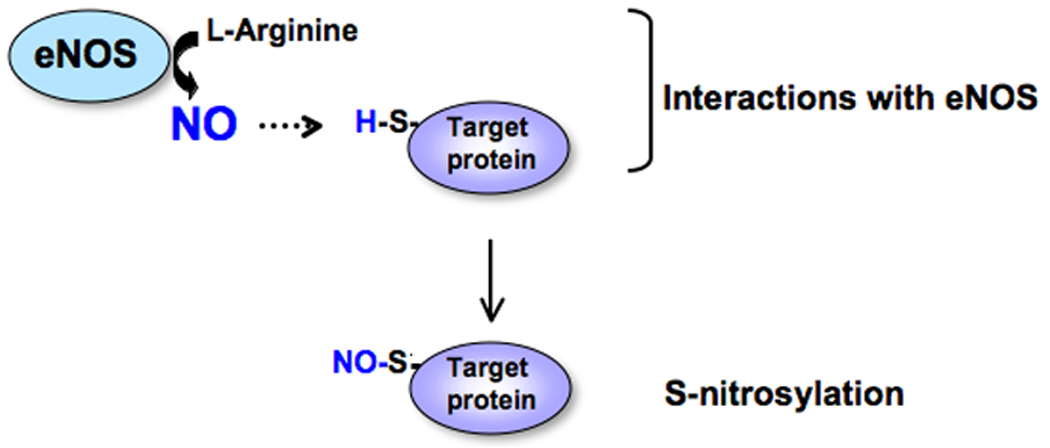
The interaction between eNOS and proteins that are to be targets for S-nitrosylation is a key determinant of the specificity of NO signaling[3] (Figure 4). Endothelial NOS-derived NO inhibits or stimulates activities of target proteins by S-nitrosylation. This goes on to influence a variety of cellular processes in endothelial cells and other cells such as T-cells. Localization of eNOS on the Golgi apparatus can enhance S-nitrosylation of Golgi proteins overall and may influence their function. Since the Golgi apparatus is constantly being remodeled during the cell cycle and is a site of several important post-translational modifications, local regulation of protein function via their S-nitrosylation may be the unique property of eNOS localization on the Golgi apparatus and is an important area to explore.
Figure 4. A key determinant of the specificity of NO signaling is the location of the interaction between eNOS and proteins targeted for S-nitrosylation.
ACKNOWLEDGEMENTS
This work was supported by NIH grants K01DK067933 and R01DK082600, and the Yale Center for Clinical Investigation Scholar award (ULRR024139).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Sessa WC. eNOS at a glance. J Cell Sci. 2004;117:2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- 2.Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3':5'-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci U S A. 1977;74:3203–3207. doi: 10.1073/pnas.74.8.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 4.Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, Groszmann RJ, Shah VH, Sessa WC. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci U S A. 2006;103:19777–19782. doi: 10.1073/pnas.0605907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang G, Moniri NH, Ozawa K, Stamler JS, Daaka Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc Natl Acad Sci U S A. 2006;103:1295–1300. doi: 10.1073/pnas.0508354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, Hara MR, Quick RA, Cao W, O'Rourke B, Lowenstein JM, Pevsner J, Wagner DD, Lowenstein CJ. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 2003;115:139–150. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J Cell Sci. 2007;120:492–501. doi: 10.1242/jcs.03361. [DOI] [PubMed] [Google Scholar]
- 8.Lee SJ, Kim KM, Namkoong S, Kim CK, Kang YC, Lee H, Ha KS, Han JA, Chung HT, Kwon YG, Kim YM. Nitric oxide inhibition of homocysteine-induced human endothelial cell apoptosis by down-regulation of p53-dependent Noxa expression through the formation of S-nitrosohomocysteine. J Biol Chem. 2005;280:5781–5788. doi: 10.1074/jbc.M411224200. [DOI] [PubMed] [Google Scholar]
- 9.Thibeault S, Rautureau Y, Oubaha M, Faubert D, Wilkes BC, Delisle C, Gratton JP. S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol Cell. 2010;39:468–476. doi: 10.1016/j.molcel.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Pi X, Wu Y, Ferguson JE, 3rd, Portbury AL, Patterson C. SDF-1alpha stimulates JNK3 activity via eNOS-dependent nitrosylation of MKP7 to enhance endothelial migration. Proc Natl Acad Sci U S A. 2009;106:5675–5680. doi: 10.1073/pnas.0809568106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oliveira CJ, Curcio MF, Moraes MS, Tsujita M, Travassos LR, Stern A, Monteiro HP. The low molecular weight S-nitrosothiol, S-nitroso-N-acetylpenicillamine, promotes cell cycle progression in rabbit aortic endothelial cells. Nitric Oxide. 2008;18:241–255. doi: 10.1016/j.niox.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6448–6453. doi: 10.1073/pnas.93.13.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Q, Church JE, Jagnandan D, Catravas JD, Sessa WC, Fulton D. Functional Relevance of Golgi- and Plasma Membrane-Localized Endothelial NO Synthase in Reconstituted Endothelial Cells. Arterioscler Thromb Vasc Biol. 2006;26:1015–1021. doi: 10.1161/01.ATV.0000216044.49494.c4. [DOI] [PubMed] [Google Scholar]
- 14.Jagnandan D, Sessa WC, Fulton D. Intracellular location regulates calcium-calmodulin dependent activation of organelle-restricted eNOS. Am J Physiol Cell Physiol. 2005;289:C1024–C1033. doi: 10.1152/ajpcell.00162.2005. [DOI] [PubMed] [Google Scholar]
- 15.Fulton D, Babbitt R, Zoellner S, Fontana J, Acevedo L, McCabe TJ, Iwakiri Y, Sessa WC. Targeting of endothelial nitric-oxide synthase to the cytoplasmic face of the Golgi complex or plasma membrane regulates Akt- versus calcium-dependent mechanisms for nitric oxide release. J Biol Chem. 2004;279:30349–30357. doi: 10.1074/jbc.M402155200. [DOI] [PubMed] [Google Scholar]
- 16.Fulton D, Fontana J, Sowa G, Gratton JP, Lin M, Li KX, Michell B, Kemp BE, Rodman D, Sessa WC. Localization of endothelial nitric-oxide synthase phosphorylated on serine 1179 and nitric oxide in Golgi and plasma membrane defines the existence of two pools of active enzyme. J Biol Chem. 2002;277:4277–4284. doi: 10.1074/jbc.M106302200. [DOI] [PubMed] [Google Scholar]
- 17.Gow AJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, Stamler JS. Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J Biol Chem. 2002;277:9637–9640. doi: 10.1074/jbc.C100746200. [DOI] [PubMed] [Google Scholar]
- 18.Gow AJ, Ischiropoulos H. Nitric oxide chemistry and cellular signaling. J Cell Physiol. 2001;187:277–282. doi: 10.1002/jcp.1085. [DOI] [PubMed] [Google Scholar]
- 19.Ibiza S, Perez-Rodriguez A, Ortega A, Martinez-Ruiz A, Barreiro O, Garcia-Dominguez CA, Victor VM, Esplugues JV, Rojas JM, Sanchez-Madrid F, Serrador JM. Endothelial nitric oxide synthase regulates N-Ras activation on the Golgi complex of antigen-stimulated T cells. Proc Natl Acad Sci U S A. 2008;105:10507–10512. doi: 10.1073/pnas.0711062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wagner DD. The Weibel-Palade body: the storage granule for von Willebrand factor and P-selectin. Thromb Haemost. 1993;70:105–110. [PubMed] [Google Scholar]
- 21.Weibel ER, Palade GE. New Cytoplasmic Components in Arterial Endothelia. J Cell Biol. 1964;23:101–112. doi: 10.1083/jcb.23.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Block MR, Glick BS, Wilcox CA, Wieland FT, Rothman JE. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc Natl Acad Sci U S A. 1988;85:7852–7856. doi: 10.1073/pnas.85.21.7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burgoyne RD, Morgan A. Analysis of regulated exocytosis in adrenal chromaffin cells: insights into NSF/SNAP/SNARE function. Bioessays. 1998;20:328–335. doi: 10.1002/(SICI)1521-1878(199804)20:4<328::AID-BIES9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 24.Qian J, Zhang Q, Church JE, Stepp DW, Rudic RD, Fulton DJ. Role of local production of endothelium-derived nitric oxide on cGMP signaling and S-nitrosylation. Am J Physiol Heart Circ Physiol. 2010;298:H112–H118. doi: 10.1152/ajpheart.00614.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mellman I, Warren G. The road taken: past and future foundations of membrane traffic. Cell. 2000;100:99–112. doi: 10.1016/s0092-8674(00)81687-6. [DOI] [PubMed] [Google Scholar]
- 26.Huang Y, Man HY, Sekine-Aizawa Y, Han Y, Juluri K, Luo H, Cheah J, Lowenstein C, Huganir RL, Snyder SH. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–540. doi: 10.1016/j.neuron.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 27.Cao S, Yao J, McCabe TJ, Yao Q, Katusic ZS, Sessa WC, Shah VV. Direct interaction between endothelial nitric oxide synthase and dynamin-2: Implications for nitric oxide synthase function. J Biol Chem. 2001;18:18. doi: 10.1074/jbc.M006258200. [DOI] [PubMed] [Google Scholar]
- 28.Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, Koch WJ, Daaka Y, Lefkowitz RJ, Stamler JS. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 29.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 30.Johnson M. The beta-adrenoceptor. Am J Respir Crit Care Med. 1998;158:S146–S153. doi: 10.1164/ajrccm.158.supplement_2.13tac110. [DOI] [PubMed] [Google Scholar]
- 31.Ozawa K, Whalen EJ, Nelson CD, Mu Y, Hess DT, Lefkowitz RJ, Stamler JS. S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol Cell. 2008;31:395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li JM, Shah AM. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem. 2002;277:19952–19960. doi: 10.1074/jbc.M110073200. [DOI] [PubMed] [Google Scholar]
- 33.Selemidis S, Dusting GJ, Peshavariya H, Kemp-Harper BK, Drummond GR. Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc Res. 2007;75:349–358. doi: 10.1016/j.cardiores.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 34.Haendeler J, Hoffmann J, Zeiher AM, Dimmeler S. Antioxidant effects of statins via S-nitrosylation and activation of thioredoxin in endothelial cells: a novel vasculoprotective function of statins. Circulation. 2004;110:856–861. doi: 10.1161/01.CIR.0000138743.09012.93. [DOI] [PubMed] [Google Scholar]
- 35.Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat Cell Biol. 2002;4:743–749. doi: 10.1038/ncb851. [DOI] [PubMed] [Google Scholar]
- 36.Hoffmann J, Haendeler J, Zeiher AM, Dimmeler S. TNFalpha and oxLDL reduce protein S-nitrosylation in endothelial cells. J Biol Chem. 2001;276:41383–41387. doi: 10.1074/jbc.M107566200. [DOI] [PubMed] [Google Scholar]
- 37.Zhang W, Zheng S, Storz P, Min W. Protein kinase D specifically mediates apoptosis signal-regulating kinase 1-JNK signaling induced by H2O2 but not tumor necrosis factor. J Biol Chem. 2005;280:19036–19044. doi: 10.1074/jbc.M414674200. [DOI] [PubMed] [Google Scholar]
- 38.Dimmeler S, Haendeler J, Galle J, Zeiher AM. Oxidized low-density lipoprotein induces apoptosis of human endothelial cells by activation of CPP32-like proteases. A mechanistic clue to the 'response to injury' hypothesis. Circulation. 1997;95:1760–1763. doi: 10.1161/01.cir.95.7.1760. [DOI] [PubMed] [Google Scholar]
- 39.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 40.Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, Stamler JS. Fas-induced caspase denitrosylation. Science. 1999;284:651–654. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- 41.Abello PA, Fidler SA, Buchman TG. Thiol reducing agents modulate induced apoptosis in porcine endothelial cells. Shock. 1994;2:79–83. doi: 10.1097/00024382-199408000-00001. [DOI] [PubMed] [Google Scholar]
- 42.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10:721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 44.Wallez Y, Vilgrain I, Huber P. Angiogenesis: the VE-cadherin switch. Trends Cardiovasc Med. 2006;16:55–59. doi: 10.1016/j.tcm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 45.Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–4380. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- 46.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL, Jain RK. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A. 2001;98:2604–2609. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mirshahi F, Pourtau J, Li H, Muraine M, Trochon V, Legrand E, Vannier J, Soria J, Vasse M, Soria C. SDF-1 activity on microvascular endothelial cells: consequences on angiogenesis in in vitro and in vivo models. Thromb Res. 2000;99:587–594. doi: 10.1016/s0049-3848(00)00292-9. [DOI] [PubMed] [Google Scholar]
- 48.Askari AT, Unzek S, Popovic ZB, Goldman CK, Forudi F, Kiedrowski M, Rovner A, Ellis SG, Thomas JD, DiCorleto PE, Topol EJ, Penn MS. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet. 2003;362:697–703. doi: 10.1016/S0140-6736(03)14232-8. [DOI] [PubMed] [Google Scholar]
- 49.Nagata K, Suzuki T, Shibagaki Y, Mizumoto K, Okano Y, Kaziro Y, Nozawa Y. Characterization and site-directed mutagenesis of a low M(r) GTP-binding protein, ram p25, expressed in Escherichia coli. J Biol Chem. 1992;267:19600–19606. [PubMed] [Google Scholar]
- 50.Satoh T, Kaziro Y. Ras in signal transduction. Semin Cancer Biol. 1992;3:169–177. [PubMed] [Google Scholar]
- 51.Lander HM, Hajjar DP, Hempstead BL, Mirza UA, Chait BT, Campbell S, Quilliam LA. A molecular redox switch on p21(ras). Structural basis for the nitric oxide-p21(ras) interaction. J Biol Chem. 1997;272:4323–4326. doi: 10.1074/jbc.272.7.4323. [DOI] [PubMed] [Google Scholar]
- 52.Lander HM, Milbank AJ, Tauras JM, Hajjar DP, Hempstead BL, Schwartz GD, Kraemer RT, Mirza UA, Chait BT, Burk SC, Quilliam LA. Redox regulation of cell signalling. Nature. 1996;381:380–381. doi: 10.1038/381380a0. [DOI] [PubMed] [Google Scholar]
- 53.Lander HM, Ogiste JS, Pearce SF, Levi R, Novogrodsky A. Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J Biol Chem. 1995;270:7017–7020. doi: 10.1074/jbc.270.13.7017. [DOI] [PubMed] [Google Scholar]
- 54.Lander HM, Sehajpal PK, Novogrodsky A. Nitric oxide signaling: a possible role for G proteins. J Immunol. 1993;151:7182–7187. [PubMed] [Google Scholar]
- 55.Yun HY, Gonzalez-Zulueta M, Dawson VL, Dawson TM. Nitric oxide mediates N-methyl-D-aspartate receptor-induced activation of p21ras. Proc Natl Acad Sci U S A. 1998;95:5773–5778. doi: 10.1073/pnas.95.10.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang J, Jin B, Li L, Block ER, Patel JM. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am J Physiol Cell Physiol. 2005;288:C840–C849. doi: 10.1152/ajpcell.00325.2004. [DOI] [PubMed] [Google Scholar]
- 57.Erwin PA, Lin AJ, Golan DE, Michel T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2005;280:19888–19894. doi: 10.1074/jbc.M413058200. [DOI] [PubMed] [Google Scholar]
- 58.Erwin PA, Mitchell DA, Sartoretto J, Marletta MA, Michel T. Subcellular targeting and differential S-nitrosylation of endothelial nitric-oxide synthase. J Biol Chem. 2006;281:151–157. doi: 10.1074/jbc.M510421200. [DOI] [PubMed] [Google Scholar]
- 59.Ravi K, Brennan LA, Levic S, Ross PA, Black SM. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proc Natl Acad Sci U S A. 2004;101:2619–2624. doi: 10.1073/pnas.0300464101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodriguez-Crespo I, Gerber NC, Ortiz de Montellano PR. Endothelial nitric-oxide synthase. Expression in Escherichia coli, spectroscopic characterization, and role of tetrahydrobiopterin in dimer formation. J Biol Chem. 1996;271:11462–11467. doi: 10.1074/jbc.271.19.11462. [DOI] [PubMed] [Google Scholar]
- 61.Black SM, Heidersbach RS, McMullan DM, Bekker JM, Johengen MJ, Fineman JR. Inhaled nitric oxide inhibits NOS activity in lambs: potential mechanism for rebound pulmonary hypertension. Am J Physiol. 1999;277:H1849–H1856. doi: 10.1152/ajpheart.1999.277.5.H1849. [DOI] [PubMed] [Google Scholar]
- 62.Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- 63.Martinez-Ruiz A, Lamas S. Detection and proteomic identification of S-nitrosylated proteins in endothelial cells. Arch Biochem Biophys. 2004;423:192–199. doi: 10.1016/j.abb.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 64.Leiper J, Murray-Rust J, McDonald N, Vallance P. S-nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc Natl Acad Sci U S A. 2002;99:13527–13532. doi: 10.1073/pnas.212269799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Loscalzo J, Braunwald E. Tissue plasminogen activator. N Engl J Med. 1988;319:925–931. doi: 10.1056/NEJM198810063191407. [DOI] [PubMed] [Google Scholar]
- 66.Stamler JS, Simon DI, Jaraki O, Osborne JA, Francis S, Mullins M, Singel D, Loscalzo J. S-nitrosylation of tissue-type plasminogen activator confers vasodilatory and antiplatelet properties on the enzyme. Proc Natl Acad Sci U S A. 1992;89:8087–8091. doi: 10.1073/pnas.89.17.8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delyani JA, Nossuli TO, Scalia R, Thomas G, Garvey DS, Lefer AM. S-nitrosylated tissue-type plasminogen activator protects against myocardial ischemia/reperfusion injury in cats: role of the endothelium. J Pharmacol Exp Ther. 1996;279:1174–1180. [PubMed] [Google Scholar]
- 68.Zhang HH, Feng L, Livnat I, Hoh JK, Shim JY, Liao WX, Chen DB. Estradiol-17beta stimulates specific receptor and endogenous nitric oxide-dependent dynamic endothelial protein S-nitrosylation: analysis of endothelial nitrosyl-proteome. Endocrinology. 151:3874–3887. doi: 10.1210/en.2009-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chakrabarti S, Lekontseva O, Peters A, Davidge ST. 17beta-Estradiol induces protein S-nitrosylation in the endothelium. Cardiovasc Res. 85:796–805. doi: 10.1093/cvr/cvp368. [DOI] [PubMed] [Google Scholar]
- 70.Chen SC, Huang B, Liu YC, Shyu KG, Lin PY, Wang DL. Acute hypoxia enhances proteins' S-nitrosylation in endothelial cells. Biochem Biophys Res Commun. 2008;377:1274–1278. doi: 10.1016/j.bbrc.2008.10.144. [DOI] [PubMed] [Google Scholar]
- 71.Huang B, Chen SC, Wang DL. Shear flow increases S-nitrosylation of proteins in endothelial cells. Cardiovasc Res. 2009;83:536–546. doi: 10.1093/cvr/cvp154. [DOI] [PubMed] [Google Scholar]
- 72.Hoffmann J, Dimmeler S, Haendeler J. Shear stress increases the amount of S-nitrosylated molecules in endothelial cells: important role for signal transduction. FEBS Lett. 2003;551:153–158. doi: 10.1016/s0014-5793(03)00917-7. [DOI] [PubMed] [Google Scholar]
- 73.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 74.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 75.Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, Messina E, Bowles D, Kontos CD, Hare JM, Stamler JS, Rockman HA. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A. 2009;106:6297–6302. doi: 10.1073/pnas.0901043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lowenstein CJ. Nitric oxide regulation of protein trafficking in the cardiovascular system. Cardiovasc Res. 2007;75:240–246. doi: 10.1016/j.cardiores.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]