

Abstract
Glutamate dehydrogenase 1 (GLUD1) is a mitochondrial enzyme expressed in all tissues, including brain. Although this enzyme is expressed in glutamatergic pathways, its function as a regulator of glutamate neurotransmitter levels is still not well defined. In order to gain an understanding of the role of GLUD1 in the control of glutamate levels and synaptic release in mammalian brain, we generated transgenic (Tg) mice that over-express this enzyme in neurons of the central nervous system. The Tg mice have increased activity of GLUD, as well as elevated levels and increased synaptic and depolarization-induced release of glutamate. These mice suffer age-associated losses of dendritic spines, nerve terminals, and neurons. The neuronal losses and dendrite structural changes occur in select regions of the brain. At the transcriptional level in the hippocampus, cells respond by increasing the expression of genes related to neurite growth and synapse formation, indications of adaptive or compensatory responses to the effects of increases in the release and action of glutamate at synapses. Because these Tg mice live to a relatively old age they are a good model of the effects of a “hyperglutamatergic” state on the aging process in the nervous system. The mice are also useful in defining the molecular pathways affected by the over-activation of GLUD in glutamatergic neurons of the brain and spinal cord.
Keywords: Glutamate dehydrogenase, Depolarization-induced glutamate release, Spine loss, Synapse loss, LTP, Aging, Transcriptomic changes
Introduction
Glutamate dehydrogenase1 (GLUD1) is a mitochondrial enzyme that is considered to be a “housekeeping” protein involved in providing a substrate, α-ketoglutarate, to the tricarboxylic acid cycle through oxidative deamination of glutamate. Liver GLUD has been studied extensively with regard to the structure of the protein and the kinetic properties and allosteric regulation of enzyme activity (Frieden 1959, 1965; Smith et al. 2001; Li et al. 2009). GLUD1 has been isolated from various tissues of different animal species, including the human brain (Hussain et al. 1989). In the human brain, there are two forms of the enzyme, GLUD1 and GLUD2, representing the products of two genes, one located on chromosome 10 (Glud1) and the other on the X chromosome (Glud2) (Shashidharan et al. 1994). It has been suggested that the presence and expression in brain and retina of the enzyme GLUD2, besides the “housekeeping” GLUD1, represents an evolutionary step that provides for greater neurotransmitter (L-glutamate) flux and enhanced mental function in humans and chimpanzees (Burki and Kaessmann 2004). The evolution of Glud2 is important in determining the overall activity of the enzyme in brain and mutations in this gene may lead to significant neurological diseases, as suggested by the fact that gain-of-function mutations in Glud2 are linked to some cases of Parkinson’s disease (Plaitakis et al. 2010). But, it may not only be GLUD2 that is of significance with regard to cognitive function in mammals as the gene for Glud1 has also been identified as a memory-related gene in the hippocampus (Cavallaro et al. 1997). Yet, the function of GLUD1 and GLUD2 in synaptic transmission and, overall, in brain metabolism and physiology, is still not well characterized.
The GLUD proteins are distributed in regions of high glutamatergic neurotransmitter activity in brain (Aoki et al. 1987; Kugler and Baier 1992; Bao et al. 2009a; Spanaki et al. 2010). Activation of the enzyme by the allosteric activator leucine, leads to increases in glutamate flux in synaptosomes (Erecinska and Nelson 1990); this observation is suggestive of a role for this enzyme in the control of glutamate levels at nerve terminals. The deamination product of GLUD1 activity, α-ketoglutarate, would be expected to be the predominant product of increased GLUD1 activity in neurons, and its transport out of mitochondria and into the cytoplasm by the keto-dicarboxylic acid carriers of the mitochondria would provide the substrate for transamination by aspartate aminotransferase and the formation of glutamate that is stored and released by neurons (Palaiologos et al. 1989). Nevertheless, despite these observations, the potential role of GLUD activity in neuronal synapses has remained poorly defined. In an effort to determine the function of GLUD in neurotransmission, we developed transgenic (Tg) mice that over-express the GLUD1 gene in central nervous system neurons only (Bao et al. 2009a). This was accomplished by introducing the transgene under the control of the promoter for neuron specific enolase (Nse). The results from such studies are outlined below.
Development and Characterization of Glud1 Tg mice
Enzyme expression and activity in brain
As reported previously (Bao et al. 2009a), the Glud1 Tg mice were generated by pronuclear microinjections of mouse oocytes with a construct containing the cDNA for the mouse Glud1 under the control of the Nse promoter. We identified four founder Tg mice that had the Glud1 transgene and the gene-copy number in these mice was either 6 or 8 copies. The founder mice were mated with C57BL/6 wild type mice for over 10 generations. Inheritance of the transgene has been that of a normal Mendelian transmission pattern (Bao et al. 2009a). In all studies that we have conducted, we have used only hemizygous mice and for every characteristic that we have examined, we have not observed substantial differences among the mice derived from the different founders.
The Nse gene is expressed only in neurons of brain and spinal cord (Peel et al. 1997) and in studies with mice that we had generated and which had the transgene for Egfp under the control of the Nse promoter, we found that EGFP fluorescence was highly enriched in neuronal cell bodies and not in myelinated axonal tracks (Fig. 1). Therefore, we anticipated that the Glud1 transgene expression would also be confined to neurons. Furthermore, as the transgene for Glud1 includes the mitochondrial targeting sequence of the enzyme protein, it was expected that the GLUD1 formed from the transgene would be localized in neuronal mitochondria.
Figure 1.
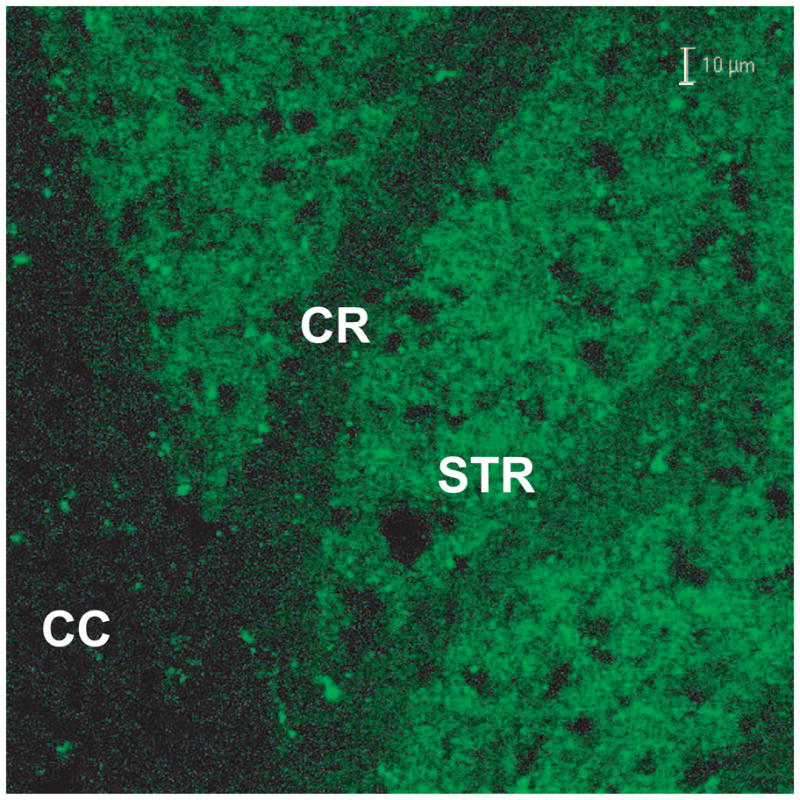
Expression pattern of EGFP in a transgenic mouse generated with the Egfp gene under the control of the Nse promoter. EGFP fluorescence in corpus callosum (CC) and striatum (Str) of a hemizygous Tg mouse is shown. Note the lack of, or low levels of, EGFP fluorescence in myelinated tracks, such as those in the CC and in part of corona radiata (CR). Line: 10 μm
The levels of GLUD1 protein in homogenates from the brains of the Tg mice are significantly higher (28.5 percent higher) than in littermate wt mice, as is also the GLUD enzyme activity (35–40% higher in Tg vs. wt) (Bao et al. 2009a). Importantly, the GLUD activity in the mitochondria and nerve-ending particle enriched P2 fraction isolated from brain homogenates of Tg mice, has an estimated Vmax for L-glutamate deamination that is significantly (65%) higher than that in wt P2. This enrichment in GLUD activity in the Tg mouse was even more pronounced in the purified synaptosome fraction in which the Vmax is 153% higher in Tg than wt mice. The P2 fraction contains mitochondria derived from neuronal and glial cell bodies, as well as from nerve endings, the higher Vmax in the Tg mice is indicative of a greater enrichment of GLUD1 activity in neuronal tissue, an assertion that is supported by the much higher Vmax in the isolated nerve ending particles (synaptosomes). The synaptosome fraction represents an enriched preparation of intraneuronal mitochondria. The KM values for GLUD activity do not differ significantly between Tg and wt mice for either glutamate or NAD+ (Bao et al. 2009a).
In situ histochemical detection of GLUD activity in brain also indicates that known glutamatergic regions of the brain, such as the pyramidal cell layer and dendritic fields of the stratum oriens (SO) and stratum radiatum (SR) of the hippocampus CA1 and CA3 subfields, and the molecular layer of the dentate gyrus, are more strongly labeled in Tg mice than the respective regions in wt mice (Bao et al. 2009a). The SO and SR fields of the hippocampus are known to be enriched with glutamatergic nerve terminals thus the histochemical labeling fits with the biochemical results of increased GLUD activity in isolated nerve ending particles. Finally, immunohistochemical labeling with anti-GLUD1 antibodies reveals that in the strata of hippocampal pyramidal neurons of both CA1 and CA3, GLUD1 immunoreactivity is significantly higher (53 percent higher) in Tg mice than wt mice. Significantly increased levels of GLUD1 immunoreactivity in Tg (52 percent higher) compared with wt mice was also detected in neurons of the cerebral cortex (Bao et al. 2009a). The biochemical, histochemical, and immunocytochemical data are indicative of overall increases in GLUD1 levels and activity throughout the brains of Tg mice and these endophenotypic characteristics of the Glud1 Tg mice, plus the characteristics that will be described below, are summarized in Table 1.
Table 1.
Differential Characteristics of Glud1 Tg Mice Compared with wt C57Bl/6 mice
| Biochemical and Cellular Characteristics | Brain Region/Subcellular Fraction | Tg vs. wt (↑-increase; ↓-decrease; →-no change) |
|---|---|---|
| GLUD Activity (Biochemical) | Whole brain homogenate | ↑ |
| Whole brain P2 | ↑↑ | |
| Whole brain synaptosomes | ↑↑↑ | |
| GLUD Activity (Histochemical) | Hippocampus | ↑↑ |
| GLUD Immunoreactivity | Hippocampus | ↑↑ |
| Cortex | ↑↑ | |
| Cerebellum | ↑↑ | |
| Striatum | ↑↑ | |
| Glutamate Levels | Striatum | ↑ |
| Hippocampus | ↑ | |
| Neuronal Loss | CA1 Hippocampus | ↓↓ |
| CA3 Hippocampus | → | |
| Dentate Gyrus | ↓↓ | |
| Motor/Somatosensory Cortex | ↓ | |
| Spinal Cord Motorneurons | ↓↓ | |
| MAP2A Dendrite Labeling | CA1 Hippocampus | ↓↓ |
| CA3 Hippocampus | → | |
| Motor/Somatosensory Cortex | ↓↓ | |
| Striatum | ↓↓ | |
| Depolarization-induced Glutamate Release | Striatum | ↑↑ |
| Synaptic Glutamate Release | CA1 Hippocampus | ↑↑↑ |
| Spine Density | CA1 Hippocampus | ↓↓ |
| Synaptophysin Labeling of Nerve Terminals | CA1 Hippocampus | ↓↓ |
| Transient and Sustained LTP | CA1 Hippocampus | ↓↓ |
Glutamate levels and glutamate release in brain
Two important issues with regard to the Glud1 Tg mice are: a) whether Glud1 expression in neurons leads to increases in brain concentrations of glutamate; and b) whether Glud1 over-expression causes increased synaptic and post-depolarization release of glutamate. To address the first issue, we determined whether Tg mice had higher levels of glutamate in brain by using magnetic resonance spectroscopy (Bao et al. 2009a). Since glutamate may function as the precursor for other amino acids in brain (e.g., GABA and glutamine), we also examined the concentrations of these amino acids in the hippocampus and striatum of Tg and wt living mice. The concentration of glutamate in both brain regions is significantly, though only modestly (9.76 μmol/mg in Tg vs. 8.99 μmol/mg in wt hippocampus; 8.87 μmol/mg in Tg vs. 8.01 μmol/mg in wt striatum), increased, the levels of GABA, aspartate, and glutamine are not, and those of alanine are significantly decreased (0.56 μmol/mg in Tg vs. 0.79 μmol/mg in wt hippocampus; 1.01 μmol/mg in Tg vs. 1.40 μmol/mg in wt striatum). Thus, the expression of Glud1 in neurons of the Tg mice causes modest, yet significant, increases in glutamate concentrations in brain without increasing GABA levels in inhibitory neurons.
The exact compartment in which the increases in glutamate levels occur, i.e., glial vs. neuronal, is not known. Glutamine levels in brain are primarily determined by glial cell metabolism and, as we pointed out previously, the lack of a change in glutamine levels might indicate that the increases in glutamate levels contribute a relatively small amount to glial glutamine synthesis (Bao et al. 2009a). We also surmise that there is an increased rate of conversion of neuronal glutamine to glutamate in neurons of the Tg mice thus maintaining glutamine levels within a narrow range of concentrations. If this is the case, then the elevated levels of NH4+ released as a result of increased glutamine to glutamate conversion in neurons, may lead to reductive amination of α-ketoglutarate to glutamate and thus elevations of neuronal glutamate (Bak et al. 2006). This may explain how increased levels of glutamate in neurons may occur in the Glud1 Tg mice.
The decreased alanine levels, we hypothesize, might be the result of increases in the transamination of excess α-ketoglutarate that is formed in mitochondria and transported to cytoplasm. Increases in the cytoplasmic levels of α-ketoglutarate may lead to enhanced transamination by alanine aminotransferase and the formation of glutamate and pyruvate (Bak et al. 2006). The glutamate formed may be stored in synaptic vesicles in neurons of the Tg mouse brain. These are complex metabolic pathways that have not yet been explored in the Tg mice. The measurements that were obtained were not designed to label and monitor the metabolic inter-conversions of glutamate and its metabolites through various metabolic pools but rather to estimate the steady state levels of these amino acids that are related to glutamate metabolism.
Increases in glutamate levels in the brains of Tg mice, even if they are occurring within neurons, may not, necessarily, reflect increases in pools of glutamate at synapses and which may be released following neuronal depolarization. For this reason, we undertook, first, the measurement of depolarization-induced release of glutamate. Using self-referencing microelectrode arrays, we measured glutamate release and clearance in vivo in the striatum of Tg and wt mice. Depolarization-induced release was evoked by local injections of nanoliter volumes of 70 mM KCl. The application of 70 mM KCl for 1–2 sec produced rapid and reproducible changes in extracellular glutamate concentrations and this depolarization-induced release of glutamate is consistently and significantly higher in Tg mice (Bao et al. 2009a). Almost identical volumes (135–140 μL) of KCl applications in Tg and wt mice produce a 57% higher level of glutamate release in Tg vs. wt mouse striata. It is important to note that the higher amplitude of glutamate release in Tg as compared with wt mice is not the result of any deficits in glutamate uptake by glia or neurons. The clearance rates of depolarization-induced glutamate release are almost identical in Tg and wt mice. In addition, nearly identical kinetics for glutamate clearance from the extracellular space are measured following local application of nanoliter volumes of 5 mM glutamate in the striatum of Tg and wt mice. These results are confirmatory of the assertion that the kinetics of glutamate transport into glia and neurons are as rapid and effective in Tg as in wt mice. Thus, changes in glutamate transport cannot explain the higher levels of released glutamate following a depolarizing stimulus.
To determine whether the increased release of glutamate following neuronal depolarization is due to excess release of neurotransmitter at known excitatory glutamatergic synapses, we measured both the amplitude and frequency of miniature excitatory currents (mEPSCs) in synapses of the CA1 subfield of the hippocampus. Based on studies in other species, such as Drosophila (Daniels et al. 2004), increased amounts of glutamate in synaptic vesicles, as might be occurring also in Glud1 Tg mice, would be expected to lead to increases in both mEPSC frequency and amplitude. The frequency of mEPSCs measured in CA1 excitatory synapses is increased by an estimated 500 percent in Tg vs. wt mice, and the amplitude by 100 percent (Bao et al. 2009a). Thus, the Glud1 Tg mice exhibit both increased spontaneous synaptic and depolarization-induced neuronal release of glutamate; but, because of normally functioning glutamate transport processes, the increases in extracellular glutamate levels are probably rapidly dissipating. Based on these observations, we conclude that the activity of the mitochondrial enzyme GLUD contributes to the maintenance of glutamate neurotransmitter pools at the synapses of glutamatergic neurons.
Glud1 Tg mice as models of age-associated neuronal degeneration
Neuronal structure in the brain of Tg mice
Assuming that the enhanced release of glutamate from neurons occurs in the Tg mice across the life-span of these animals, this characteristic makes these mice a useful model for studies focused on the effects of transient hyper-glutamatergic states in brain on neuronal structure and function during development as well as aging. In histological sections from the CA1 region of the hippocampus, the ventral dentate gyrus, and layers III to V of the primary motor/somatosensory cerebral cortex of 12–20 month old mice, we observed significant decreases in the number of neurons in Tg vs. wt mouse brains (Bao et al. 2009a). Besides the decreases in certain types of neurons in hippocampus and cerebral cortex, many neurons are shrunken and darkly stained (pyknotic). Such morphological changes have been described previously for neurons exposed to high levels of glutamate resulting from inhibition of glutamate transporters (Arias et al. 1997), thus linking the increased release of glutamate in the Glud1 Tg mice to the neuronal losses observed in select regions of the brain.
The losses of large pyramidal neurons observed in the cortex and hippocampus of Tg mice appear to be rather selective in nature as we did not observe losses of other large brain neurons, such as the Purkinje neurons of the cerebellar cortex. In addition to the selective loss of neurons in the brain of Tg mice, we also observed losses among the population of large motor neurons in the ventral horn of the cervical and lumbar spinal cord of Glud1 mice, 16 months old or older, when compared with wt mice of the same age (Fig. 2). Surviving motor neurons in the spinal cord appear to be shrunken and stain weakly with methylene blue. Unlike the ventral horn motor neurons, most neurons of the intermediate nucleus of the spinal cord appear to be surviving in Glud1 mice, although they stain less strongly than in wt mice.
Figure 2.

Large motor neuron loss in the ventral horn of the cervical spinal cord of a Glud1 Tg (B, D) as compared with a wt mouse (A,C). Both mice were of the same age (17 months). C, D: magnification of ventral horn from A and B.
In addition to the neuronal losses observed in select regions of the brain, neurons in hippocampus, cerebral cortex, and striatum of Glud1 mice exhibit an overall significant decrease in dendrite labeling by anti-microtubule-associated protein 2A (MAP2A) antibodies (Bao et al. 2009a). Decreases in dendrite labeling are pronounced in the CA1 subfield of the hippocampus but not in the CA3 subfield, a further indication of the selective nature of neuronal changes occurring in the brain of Glud1 mice. We noted that the decreases in anti-MAP2A labeling in dendrites of CA1 pyramidal neurons of Glud1 mice are similar to the decreases in MAP2A labeling in the hippocampus of rats following the injection of dihydrokainate, a glutamate transporter inhibitor (Zhang et al. 2007). In both the CA1 neurons of Glud1 Tg mice and those in the hippocampus of animals treated with an inhibitor of glutamate transporters, the cell bodies of the neurons are, generally, well-labeled by anti-MAP2A antibodies, i.e., there is continued synthesis of MAP2A in neuronal cells of Glud1 mice or in rats injected with dihydrokainate (Arias et al. 1997). These observations are strongly suggestive that a state of hyper-glutamate release in Glud1 Tg mice is the likely cause of the decreases in MAP2A labeling in dendrites of the CA1 field of the hippocampus.
Another observation made with regard to neuronal structure of the Glud1 Tg mice is that in areas of the hippocampus with significant loss of neurons and reduction of dendrite labeling by anti-MAP2A, there is also a significant decrease in axonal nerve terminal labeling with anti-synaptophysin antibodies (double immune labeling) when compared with the labeling observed in wt mice (Bao et al. 2009a). It is important to note, however, that the loss of nerve terminals in Tg mice is not a universal phenomenon in brain as losses of synaptophysin labeling are not apparent in the striatum of Tg mice.
We used a method of double-labeling of neurons with anti-MAP2A antibodies and the carbocyanine dye DiI, a cell membrane-labeling dye, in order to examine the integrity of neuronal dendrites and dendritic spines in susceptible regions of the Tg mouse brain, the CA1 and cortical regions. This double-labeling approach revealed that despite the discontinuities and decreases in dendrite labeling by anti-MAP2A antibodies in the Tg mouse brain, the labeling by DiI is indicative of intact membrane structure in the primary dendrites of these same neurons (Bao et al. 2009a). The DiI labeling, however, does reveal a significant loss of dendritic spines in CA1 neurons, especially of mushroom and stubby types of synapses. These types of synapses, especially the mushroom type, are important in the establishment and maintenance of long term potentiation of synaptic activity (Matsuzaki et al. 2004).
Aging, neuronal loss and changes in dendritic structure and function
It has been observed that synaptic and neuronal losses during the aging process, both in human and rodent brains, occur along pathways of glutamatergic neurotransmission (Terry et al. 1987; Masliah et al. 1993; Jacobs et al. 1997; Francis 2003; Morrison and Hof 2007). The brain regions where synaptic and neuronal losses have been observed include those of the hippocampus and cerebral cortex, regions that are also affected in the Glud1 Tg mice. The loss of synapses and neurons in these regions of the rodent brain may also correlate with the observed age-associated increases in extracellular glutamate resulting from diminished glutamate transport into neurons or glial cells during the aging process (Wheeler 1980; Freeman and Gibson 1987; Najlerahim et al. 1990; Massieu and Tapia 1997; Zoia et al. 2004; Nickell et al. 2005). In our studies, MAP2A labeling of cell bodies and dendrites of the CA1 region of the hippocampus was used to measure changes in the numbers of neurons and in dendritic structure with advancing age (Bao et al. 2009a). In Tg mice between the ages of 6 and 20 months, both the number of neurons and the labeling of dendrites is decreasing significantly with advancing age. In wt mice of identical ages, there is no significant trend of either neuronal loss or dendritic loss. A detectable decrease in MAP2A labeling of cell bodies and dendrites was observed in wt mice between the ages of 11 and 20 months. However, there is no statistically significant pattern of decreased labeling across the age span from 6 to 20 months for either neuronal cell body labeling or dendrite labeling in the wt mice. We did note, though, that the rates of decrease in MAP2A labeling between 11 and 20 months in both wt and Tg mouse CA1 are very nearly equal. Thus, even though the trends of age-associated decreases from 6 to 20 months are significant only in Tg mice, the decreases in MAP2A labeling between 11 and 20 months in wt mice might indicate that the over-expression of Glud1 and the resulting excess in glutamate release in Tg mice, accelerate a trend of age-related cell loss and altered dendrite structure in the CA1 region of the hippocampus.
We assume that these changes in the wt mice are occurring as a result of diminished glutamate transport and the increased accumulation of extracellular glutamate with advancing age, and that the effects of such glutamate hyperactivity are enhanced by the excess release of glutamate in the Tg mice. Such enhanced activity of glutamate in the extracellular space occurring even at young ages would explain the significant decreases in cell and dendrite labeling of Tg mice at 6, 11, and 20 months when compared with the wt mice. And, as described above, the age-associated effect of Glud1 over-expression on neuronal population numbers and dendrite structure is not manifested to any degree in the CA3 region of the hippocampus, a further indication of true selective vulnerability of neurons to the aging process and to the over-production and release of glutamate.
An important issue with regard to the losses in neurons and dendritic processes in Tg mice is the possible functional consequences of these structural changes. We have observed that the slope of field-evoked excitatory postsynaptic potentials (fEPSP) is significantly lower in hippocampal CA1 neurons from Tg vs. wt mice, and that the lower fEPSP slopes in Tg mice are evident during both the initial period as well as the sustained period of LTP (Bao et al. 2009a). The initial transient phase of LTP is apparently dependent on signals that enhance neurotransmitter release from pre-synaptic terminals over a prolonged period of time, whereas the sustained component of LTP is due to receptor changes at the post-synaptic sites (Volianskis and Jensen 2003). It is possible that in the Tg mice both components of LTP are altered because of decreases in the number of glutamatergic nerve terminals and of dendritic spines with which they make synapses. An alternative explanation might be that both phases of LTP are affected by an altered expression of certain genes and of the respective proteins, especially genes and proteins that have important functions in establishing LTP, such as the N-methyl-D-aspartate (NMDA) and the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) type of glutamate receptors, or the various regulators of presynaptic glutamate release during LTP, such as glycogen synthase kinase 3 beta (GSK3β).
Whole genome transcriptional activity in Glud1 Tg mice: Adaptive or compensatory responses to a chronic hyperglutamatergic state
In order to address the issue of differential expression of certain genes in Tg vs. wt mice, we performed an analysis of genome-wide expression in nine month-old Glud1 and wt mice (Wang et al. 2010). We selected the 9 month age because at that age the Tg mice exhibit moderate losses of neurons and dendritic processes, thus making it possible to probe for changes in gene expression in neurons and glia with minimized distortion of the results by excessive neuronal death. The microarray data from the Tg and wt mice fall into two distinct clusters, one corresponding to the group of Tg and the other to that of the wt mice. There are 707 probesets that are significantly more highly expressed in Tg than wt mice (up-regulated), and 311 that are significantly down-regulated in Tg vs. wt mice.
Gene Ontology (GO) analyses identified biological processes that are significantly enriched in up-regulated or down-regulated genes. The genes within these GO categories (Table 2 and also Fig. 3) fall into eight biological processes; four of the GO biological processes, those of Nervous system development, Intracellular signal transduction, Synaptic neurotransmission, and Ion homeostasis, are enriched in both up- and down-regulated genes, while four others, those of mRNA processing, Protein mis-folding correction and degradation, Cytoskeleton and focal adhesion, and Cognition and behavior, contain only significantly up-regulated genes in Tg mice. Pathway analysis of the gene expression data identified similar biological functions to those uncovered using GO analyses (Wang et al. 2010). However, some new pathways were also uncovered, such as those enriched with genes involved in oxidative stress responses and inflammation.
Table 2.
Biological Processes and Canonical Pathways identified by transcriptomic assays as being enriched with genes that are either up-regulated or down-regulated in Glud1 Tg mice when compared with wt littermates.
| GO Biological Processes | Canonical Pathways |
|---|---|
| Nervous system development | Axonal Guidance Signaling |
| Up-regulated genes (70); | Up-regulated genes (23) |
| Down-regulated (29) | Down-regulated genes (10) |
|
| |
| Intracellular signal transduction | Neurotrophin/TRK Signaling |
| Up-regulated (68) | Up-regulated genes (6) |
| Down-regulated genes (57) | Down-regulated genes (1) |
|
| |
| Synaptic neurotransmission | Synaptic Long Term |
| Up-regulated genes (21) | Potentiation |
| Down-regulated genes (10) | Up-regulated genes (9) |
| Down-regulated genes (2) | |
|
| |
| Ion transport & homeostasis | Ephrin Receptor Signaling |
| Up-regulated genes (23) | Up-regulated genes (14) |
| Down-regulated genes (4) | Down-regulated genes (6) |
|
| |
| mRNA processing | Integrin Signaling |
| Up-regulated genes (15) | Up-regulated genes (12) |
|
| |
| Protein mis-folding correction & degradation | Huntington’s Disease Signaling (Axoplasmic transport-related) |
| Up-regulated genes (9) | Up-regulated genes (17) |
| Down-regulated genes (5) | |
|
| |
| Cytoskeleton & focal adhesion | Neuregulin Signaling |
| Up-regulated genes (38) | Up-regulated genes (7) |
| Down-regulated genes (1) | |
|
| |
| Cognition & behavior | NRF2-mediated Oxidative |
| Up-regulated genes (8) | Stress Response |
| Up-regulated genes (11) | |
| Down-regulated genes (3) | |
|
| |
| Chemokine Signaling | |
| Up-regulated genes (9) | |
| Down-regulated genes (1) | |
|
| |
| Toll-like Receptor Signaling | |
| Up-regulated genes (5) | |
Figure 3.

A schematic depiction of the relationships of neurons, astrocytes, and microglial cells with respect to the major GO categories of genes that are differentially expressed in the Glud1 Tg vs. those in the wt mouse hippocampus. The GO categories are also summarized in Table 2.
Neuronal growth, differentiation and preservation of synaptic transmission are among the GO categories and pathways that are enriched with genes differentially expressed in Tg mice. The genes in these categories are mostly up-regulated in the hippocampus of Tg mice and are related to growth of nerve processes, differentiation of neurons, synapse formation, and synaptic transmission. Genes of importance with regard to neuro-transmission include two of the most up-regulated genes in Glud1 mice, those for the NMDA receptor subunits 2A and 2B (Grin2A and Grin2B). In addition to the up-regulation of expression of the NMDA receptor genes, the genes for three other glutamate receptors, Gria1 (Glutamate receptor ionotropic AMPA subunit 1), Gria3 (AMPA receptor subunit 3), and Grik2 (Glutamate receptor ionotropic kainate subunit 2), were also up-regulated, although not to the same level as the NMDA receptor genes. The differential level of expression of the glutamate receptor genes contributed to the identification of the pathway Synaptic long term potentiation as one that is significantly enriched with up-regulated genes (Table 2). Based on the gene expression patterns for glutamate receptors, some of which have been confirmed at the protein level by immunoassays (Wang et al. 2010), the decreases in LTP in the Tg mice are more likely to be related to nerve ending and spine losses than decreases in the levels of glutamate receptors.
The increased expression of NMDA receptor genes is, most likely, linked to growth of new synapses as a compensatory response in neurons of the Tg mouse hippocampus to the damage to dendrites and nerve terminals described above. Such an interpretation is supported by the observed increases in gene expression in Tg mouse hippocampus for the synaptic membrane scaffold proteins (Dlg1, Dlg2, and Dlg4), for a protein associated with the strengthening of synapse function (Arc, Activity-Regulated Cytoskeleton-Associated Protein), and for genes that determine axon, dendrite and synapse growth, including four genes for the ephrin receptors, the gene for brain-derived neurotrophic factor (BDNF), and those for two neurotrophic factor receptors, NTRK2 and NTRK3.
Increased expression in the hippocampus of Glud1 mice of another group of genes with implications for neurite elongation and synapse formation is that represented by the category of Cytoskeleton and focal adhesion. We constructed a schematic gene network for the genes that are involved in neurite growth and synapse formation and that are up-regulated in Tg mice (Fig. 4). In this network, we included proteins involved in focal adhesion of cell membranes to the extracellular matrix, such as the focal adhesion tyrosine kinase Ptk2b, as well as genes for the organizing elements of the cytoskeleton, such as the gene for the actin-organizing protein Arpc2 (Actin related protein 2/3 complex subunit 2). The gene-gene interactions displayed in this network are based on previous experimental evidence from the literature. The increases in expression of some of these genes have been confirmed by PCR (for mRNA) and immunochemical or immunohistochemical methods (for the respective proteins) (Wang et al. 2010). We are currently exploring the idea that changes in the expression of the genes described above are a compensatory response to loss of neurites and synapses throughout the lifespan of the Tg mice by examining the changes in gene expression in Tg and wt mice from 10 days to 20 months of age. Starting the examination of gene expression changes at a very young age (10 days), we may be able to distinguish changes in gene expression due to neuronal damage during development from those that represent compensatory or adaptive changes in neurons or glial cells during the life of the Tg mice.
Figure 4.

Scheme of a hypothesized network of genes involved in nerve process elongation and synapse formation, genes whose expression is up-regulated in Glud1 Tg mice. Depicted in this scheme is the tip of a hypothetical growth cone and the genes involved in nerve process elongation and synapse formation. The genes were grouped based on their function: b1 - growth factor and ephrin receptors; b2 - glutamate receptor and scaffolding genes; b3 - extracellular matrix interacting genes (integrin); b4 - actin-organizing genes; b5 - focal adhesion complex genes; and b6 - signal transduction genes. Interactions among the genes shown are portrayed by lines connecting individual genes. Such interactions were culled from previously published studies. Only genes that were up-regulated in Glud1 as compared with wt mouse hippocampus and that are related to neurite growth and synapse formation are shown in this scheme. The figure is reproduced from Wang et al. BMC Genomics 2010, 11:360.
A prominent example of differential gene expression in Tg mice that represents a compensatory response to a hyperglutamatergic state is the up-regulation of genes for the glutamate transporters, such as Slc1a2, the predominant transporter in the plasma membranes of glial cells (Lehre and Danbolt 1998), and Slc1a1, the neuronal glutamate transporter. The up-regulation of the latter transporter was confirmed by immunochemical assessment of the expression of the respective protein, and by measurements of neuronal glutamate uptake activity in vitro (comparative measurements of radioactive glutamate uptake by synaptosomal preparations of wt and Tg mice) (Wang et al. 2010). These measurements confirmed the fact that these gene and protein changes are linked to functional compensation for the higher levels of glutamate release from nerve terminals in Tg vs. wt mice.
Conclusions
The development of the Tg mouse that over-expresses the gene for Glud1 in neurons of the central nervous system was undertaken in order to determine the role of the enzyme GLUD1 in the maintenance of glutamate neurotransmitter levels in brain and, hopefully, to provide an animal model that exhibits lifelong, moderate excess formation and release of glutamate. Animal models of excess glutamate accumulation in the extracellular space have been generated previously through down-regulation or targeted disruption of glutamate transporters. These animal models include the Slc1a2 and Slc1a3 glutamate transporter null mutants, and the Tsc1 (Tuberous Sclerosis 1) knockout mice. All of these mice accumulate high levels of extracellular glutamate and this leads to the disruption of normal brain development, lethal spontaneous seizures, extensive neuronal death, and severely shortened lifespan (Rothstein et al. 1996; Tanaka et al. 1997; Matsugami et al. 2006; Zeng et al. 2007). In other words, these three types of mice, as the homozygous mutants, are not appropriate for the study of the effects of lifelong, pulsatile, moderately excessive glutamate release at neuronal synapses on nervous system structure and function. On the other hand, the heterozygous mice for the null mutation of Slc1a3 do survive to approximately the same age as wild type mice, have no morphological changes in the brain, and exhibit only modest differences in behavior, such as increased locomotor activity(Kiryk et al. 2008). But, to this date, there are no reports of changes in synaptic transmission in the heterozygous null mutant mice nor are there reports of age-dependent brain anomalies exhibited by these mice.
The over-expression of Glud1 in the Tg mice we created does result in moderate excess formation and synaptic release of glutamate and is compatible with a long life of the heterozygous Glud1 Tg mice. As described above, the Glud1 Tg mice suffer relatively confined structural and functional damage in their brain and spinal cord and the loss of neurons, dendritic spines, and nerve endings during the aging process. The latter changes seem to parallel some of the neuronal and synaptic losses observed in human brain during aging. The moderate and selective nature of neuronal damage in Glud1 mice probably accounts for the survival of these mice into early stages of aging. However, despite the observed changes in synaptic glutamate release, dendrite and spine morphology, and LTP formation, these Tg mice do not exhibit deficits in learning new behaviors (Bao et al. 2009b) or in memory formation of learnt behaviors (R. Pal and E. Michaelis, unpublished observations). These are unexpected results as the assumption has been that neuronal damage in the hippocampus and decreases in LTP formation would result in deficits in learning and memory. It is possible, however, that the relationship between glutamate neurotransmission in the hippocampus and learning and memory formation are not as tightly linked as has been thought. Mice that have genetic knockdown for the gene of phosphate-activated glutaminase (Gls1), have significantly decreased glutamate levels in the hippocampus and significantly lower levels of glutamate release at glutamatergic synapses in the hippocampus CA1 region, yet they exhibit normal spatial learning and memory retention of the learnt behaviors (Gaisler-Salomon et al. 2009).
Up until now, most of the ideas about glutamate hyperactivity have been based on results from acute exposure of neurons, either in vitro or in vivo, to sudden large excesses of extracellular glutamate or one of its congeners, such as NMDA (Buddle et al., 2003; Hoskison et al., 2007). Studies such as those cited here, and many others, have led to the expectation that all neurons in brain or spinal cord would suffer severe injury and cell death following exposure to increased extracellular glutamate concentrations. Such assertions have been supported by the previously developed animal models of excess extracellular glutamate, that is, those with down-regulation or targeted disruption of glutamate transporters described above. However, assumptions made on the basis of massively destructive effects due to large increases in extracellular glutamate may have to be revised upon better understanding of the more subtle effects on neuronal structure and physiology, as well as organismal biology and behavior, produced by moderate, pulsatile increases in synaptic glutamate release throughout the lifespan of an organism. For example, the selective synapse and neuronal losses observed in the Glud1 Tg mice, would not have been predicted if one focused only on models of glutamate-induced excitotoxicity brought about by large, excessive concentrations of extracellular glutamate. In support of the idea that excess glutamate release or accumulation in the extracellular space may lead to selective neuronal damage are the observation of a relative degree of selectivity in neuronal injury, such as that of neurons in layer II of the cortex and the CA1 region of the hippocampus, in null mutant Slc1a3 mice (Kiryk et al. 2008). Thus, the selective nature of neuronal and synaptic losses that are observed along select glutamatergic pathways in human brain during the aging process and in some neurodegenerative diseases, might indeed be explained as phenomena related to glutamate transmitter hyperactivity. And, we believe that the Tg mice we created are good models of such selective neuronal vulnerability to glutamate over-activity and may be used to uncover the molecular pathways that determine these selectively expressed changes in populations of neurons.
Another phenomenon that would have been difficult to predict on the basis of studies examining the toxic effects of large concentrations of glutamate would have been the type of up-regulation of gene transcription and protein levels of components of the neurite elongation and synapse formation pathways that we observed in the Glud1 Tg mice. Acute experimental manipulations leading to excitotoxicity do not provide an appropriate environment for the detection of compensatory or adaptive responses to glutamate-induced neuronal injury that may occur over a prolonged period of time. On the other hand, the 9 month old Glud1 Tg mice that have to cope with moderate increases in mitochondrial GLUD activity and synaptic glutamate release, do exhibit a significant up-regulation of gene and protein components that are part of signaling cascades and mechanical aspects of nerve process growth and synapse formation. Furthermore, some of the adaptive changes involved in synapse formation, such as the increased expression of various sub-types of glutamate receptors, would have been considered as unlikely processes to be up-regulated in a pure “excitotoxicity model” of glutamate hyperactivity in the brain (up-regulation of glutamate receptors would seem counterproductive as a response to excitotoxicity). In conclusion, the Glud1 mice offer a potentially useful animal model for the study of the role of the enzyme GLUD in the central nervous system, for exploring the direct and indirect effects of moderate excess of glutamate transmitter release in the nervous system, for examining the aging-related aspects of glutamate hyperactivity in brain, and for delving into the molecular and cellular pathways that lead to adaptive or compensatory neuronal responses to excess glutamate and which may be exploited in developing methods for suppression of neuronal damage or loss in neurodegenerative diseases.
Acknowledgments
This work was funded by grants from NIA, AG12993, NICHD, HD02528, NIAAA AA11419, AA04732, AA12276, NSF DBI-9987807, DBI-0352848, NIDA DA017186, NINDS NS39787, and NIMH MH58414, NIDA Training Grant DA022738, NIDA DA015088, Kansas Technology Enterprise Corporation, the Miller, Hedwig and Wilbur fund, and the University of Kansas Research Development Fund. The support of the Higuchi Biosciences Center is acknowledged.
Abbreviations
- DiI
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine
- fEPSPs
fast excitatory postsynaptic potentials
- GABA
γ-aminobutyric acid
- Glud1
glutamate dehydrogenase 1
- MAP2A
microtubule-associated protein 2A
- mEPSCs
miniature excitatory postsynaptic currents
- NMDA
N-methyl-D-Aspartate
- Nse
neuron-specific enolase
- PCR
polymerase chain reaction
- SO
stratum oriens
- SP
stratum pyramidale
- SR
stratum radiatum
- Tg
transgenic
- wt
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aoki C, Milner TA, Sheu KF, Blass JP, Pickel VM. Regional distribution of astrocytes with intense immunoreactivity for glutamate dehydrogenase in rat brain: implications for neuron-glia interactions in glutamate transmission. J Neurosci. 1987;7:2214–2231. doi: 10.1523/JNEUROSCI.07-07-02214.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias C, Arrieta I, Massieu L, Tapia R. Neuronal damage and MAP2 changes induced by the glutamate transport inhibitor dihydrokainate and by kainate in rat hippocampus in vivo. Exp Brain Res. 1997;116:467–476. doi: 10.1007/pl00005774. [DOI] [PubMed] [Google Scholar]
- Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. 2006;98:641–653. doi: 10.1111/j.1471-4159.2006.03913.x. [DOI] [PubMed] [Google Scholar]
- Bao X, Pal R, Hascup K, Wang Y, Wang W-t, Xu W, Hui D, agbas A, Wang X, Michaelis ML, Choi IY, Belousov AB, Gerhardt GA, Michaelis EK. Transgenic Expression of Glutamate Dehydrogenase 1 (Glud1) in Neurons: In Vivo Model of Enhanced Glutamate Release, Altered Synaptic Plasticity, and Selective Neuronal Vulnerability. Journal of Neuroscience. 2009a;29:13929–13944. doi: 10.1523/JNEUROSCI.4413-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Pal R, Hascup KN, Wang Y, Wang WT, Xu W, Hui D, Agbas A, Wang X, Michaelis ML, Choi IY, Belousov AB, Gerhardt GA, Michaelis EK. Transgenic expression of Glud1 (glutamate dehydrogenase 1) in neurons: in vivo model of enhanced glutamate release, altered synaptic plasticity, and selective neuronal vulnerability. J Neurosci. 2009b;29:13929–13944. doi: 10.1523/JNEUROSCI.4413-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burki F, Kaessmann H. Birth and adaptive evolution of a hominoid gene that supports high neurotransmitter flux. Nat Genet. 2004;36:1061–1063. doi: 10.1038/ng1431. [DOI] [PubMed] [Google Scholar]
- Cavallaro S, Meiri N, Yi CL, Musco S, Ma W, Goldberg J, Alkon DL. Late memory-related genes in the hippocampus revealed by RNA fingerprinting. Proc Natl Acad Sci U S A. 1997;94:9669–9673. doi: 10.1073/pnas.94.18.9669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased Expression of the Drosophila Vesicular Glutamate Transporter Leads to Excess Glutamate Release and a Compensatory Decrease in Quantal Content. J Neurosci. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erecinska M, Nelson D. Activation of glutamate dehydrogenase by leucine and its nonmetabolizable analogue in rat brain synaptosomes. J Neurochem. 1990;54:1335–1343. doi: 10.1111/j.1471-4159.1990.tb01967.x. [DOI] [PubMed] [Google Scholar]
- Francis PT. Glutamatergic systems in Alzheimer’s disease. Int J Geriatr Psychiatry. 2003;18:S15–21. doi: 10.1002/gps.934. [DOI] [PubMed] [Google Scholar]
- Freeman GB, Gibson GE. Selective alteration of mouse brain neurotransmitter release with age. Neurobiol Aging. 1987;8:147–152. doi: 10.1016/0197-4580(87)90024-8. [DOI] [PubMed] [Google Scholar]
- Frieden C. Glutamic dehydrogenase. I. The effect of coenzyme on the sedimentation velocity and kinetic behavior. J Biol Chem. 1959;234:809–814. [PubMed] [Google Scholar]
- Frieden C. Glutamate Dehydrogenase. Vi. Survey of Purine Nucleotide and Other Effects on the Enzyme from Various Sources. J Biol Chem. 1965;240:2028–2035. [PubMed] [Google Scholar]
- Gaisler-Salomon I, Miller GM, Chuhma N, Lee S, Zhang H, Ghoddoussi F, Lewandowski N, Fairhurst S, Wang Y, Conjard-Duplany A, Masson J, Balsam P, Hen R, Arancio O, Galloway MP, Moore HM, Small SA, Rayport S. Glutaminase-deficient mice display hippocampal hypoactivity, insensitivity to pro-psychotic drugs and potentiated latent inhibition: relevance to schizophrenia. Neuropsychopharmacology. 2009;34:2305–2322. doi: 10.1038/npp.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain MM, Zannis VI, Plaitakis A. Characterization of glutamate dehydrogenase isoproteins purified from the cerebellum of normal subjects and patients with degenerative neurological disorders, and from human neoplastic cell lines. J Biol Chem. 1989;264:20730–20735. [PubMed] [Google Scholar]
- Jacobs B, Driscoll L, Schall M. Life-span dendritic and spine changes in areas 10 and 18 of human cortex: a quantitative Golgi study. J Comp Neurol. 1997;386:661–680. [PubMed] [Google Scholar]
- Kiryk A, Aida T, Tanaka K, Banerjee P, Wilczynski GM, Meyza K, Knapska E, Filipkowski RK, Kaczmarek L, Danysz W. Behavioral characterization of GLT1 (+/−) mice as a model of mild glutamatergic hyperfunction. Neurotox Res. 2008;13:19–30. doi: 10.1007/BF03033364. [DOI] [PubMed] [Google Scholar]
- Kugler P, Baier G. Mitochondrial enzymes related to glutamate and GABA metabolism in the hippocampus of young and aged rats: a quantitative histochemical study. Neurochem Res. 1992;17:179–185. doi: 10.1007/BF00966797. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Smith CJ, Walker MT, Smith TJ. Novel inhibitors complexed with glutamate dehydrogenase: allosteric regulation by control of protein dynamics. J Biol Chem. 2009;284:22988–23000. doi: 10.1074/jbc.M109.020222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, DeTeresa R, Terry RD. Quantitative synaptic alterations in the human neocortex during normal aging. Neurology. 1993;43:192–197. doi: 10.1212/wnl.43.1_part_1.192. [DOI] [PubMed] [Google Scholar]
- Massieu L, Tapia R. Glutamate uptake impairment and neuronal damage in young and aged rats in vivo. J Neurochem. 1997;69:1151–1160. doi: 10.1046/j.1471-4159.1997.69031151.x. [DOI] [PubMed] [Google Scholar]
- Matsugami TR, Tanemura K, Mieda M, Nakatomi R, Yamada K, Kondo T, Ogawa M, Obata K, Watanabe M, Hashikawa T, Tanaka K. From the Cover: Indispensability of the glutamate transporters GLAST and GLT1 to brain development. Proc Natl Acad Sci U S A. 2006;103:12161–12166. doi: 10.1073/pnas.0509144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Hof PR. Life and death of neurons in the aging cerebral cortex. Int Rev Neurobiol. 2007;81:41–57. doi: 10.1016/S0074-7742(06)81004-4. [DOI] [PubMed] [Google Scholar]
- Najlerahim A, Francis PT, Bowen DM. Age-related alteration in excitatory amino acid neurotransmission in rat brain. Neurobiol Aging. 1990;11:155–158. doi: 10.1016/0197-4580(90)90049-6. [DOI] [PubMed] [Google Scholar]
- Nickell J, Pomerleau F, Allen J, Gerhardt GA. Age-related changes in the dynamics of potassium-evoked L-glutamate release in the striatum of Fischer 344 rats. J Neural Transm. 2005;112:87–96. doi: 10.1007/s00702-004-0151-x. [DOI] [PubMed] [Google Scholar]
- Palaiologos G, Hertz L, Schousboe A. Role of aspartate aminotransferase and mitochondrial dicarboxylate transport for release of endogenously and exogenously supplied neurotransmitter in glutamatergic neurons. Neurochem Res. 1989;14:359–366. doi: 10.1007/BF01000039. [DOI] [PubMed] [Google Scholar]
- Peel AL, Zolotukhin S, Schrimsher GW, Muzyczka N, Reier PJ. Efficient transduction of green fluorescent protein in spinal cord neurons using adeno-associated virus vectors containing cell type-specific promoters. Gene therapy. 1997;4:16–24. doi: 10.1038/sj.gt.3300358. [DOI] [PubMed] [Google Scholar]
- Plaitakis A, Latsoudis H, Kanavouras K, Ritz B, Bronstein JM, Skoula I, Mastorodemos V, Papapetropoulos S, Borompokas N, Zaganas I, Xiromerisiou G, Hadjigeorgiou GM, Spanaki C. Gain-of-function variant in GLUD2 glutamate dehydrogenase modifies Parkinson’s disease onset. Eur J Hum Genet. 2010;18:336–341. doi: 10.1038/ejhg.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Shashidharan P, Michaelidis TM, Robakis NK, Kresovali A, Papamatheakis J, Plaitakis A. Novel human glutamate dehydrogenase expressed in neural and testicular tissues and encoded by an X-linked intronless gene. J Biol Chem. 1994;269:16971–16976. [PubMed] [Google Scholar]
- Smith TJ, Peterson PE, Schmidt T, Fang J, Stanley CA. Structures of bovine glutamate dehydrogenase complexes elucidate the mechanism of purine regulation. J Mol Biol. 2001;307:707–720. doi: 10.1006/jmbi.2001.4499. [DOI] [PubMed] [Google Scholar]
- Spanaki C, Zaganas I, Kleopa KA, Plaitakis A. Human GLUD2 glutamate dehydrogenase is expressed in neural and testicular supporting cells. J Biol Chem. 2010;285:16748–16756. doi: 10.1074/jbc.M109.092999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Terry RD, DeTeresa R, Hansen LA. Neocortical cell counts in normal human adult aging. Ann Neurol. 1987;21:530–539. doi: 10.1002/ana.410210603. [DOI] [PubMed] [Google Scholar]
- Volianskis A, Jensen MS. Transient and sustained types of long-term potentiation in the CA1 area of the rat hippocampus. J Physiol. 2003;550:459–492. doi: 10.1113/jphysiol.2003.044214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bao X, Pal R, Agbas A, Michaelis EK. Transcriptomic responses in mouse brain exposed to chronic excess of the neurotransmitter glutamate. BMC Genomics. 2010;11:360. doi: 10.1186/1471-2164-11-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DD. Aging of membrane transport mechanisms in the central nervous system--high affinity glutamic acid transport in rat cortical synaptosomes. Exp Gerontol. 1980;15:269–284. doi: 10.1016/0531-5565(80)90031-5. [DOI] [PubMed] [Google Scholar]
- Zeng LH, Ouyang Y, Gazit V, Cirrito JR, Jansen LA, Ess KC, Yamada KA, Wozniak DF, Holtzman DM, Gutmann DH, Wong M. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2007;28:184–196. doi: 10.1016/j.nbd.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SX, Holmberg EG, Geddes JW. Artifactual dendritic beading in rat spinal cord induced by perfusion with cold saline and paraformaldehyde. Journal of Neuroscience Methods. 2007;163:38–43. doi: 10.1016/j.jneumeth.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Zoia C, Cogliati T, Tagliabue E, Cavaletti G, Sala G, Galimberti G, Rivolta I, Rossi V, Frattola L, Ferrarese C. Glutamate transporters in platelets: EAAT1 decrease in aging and in Alzheimer’s disease. Neurobiol Aging. 2004;25:149–157. doi: 10.1016/s0197-4580(03)00085-x. [DOI] [PubMed] [Google Scholar]