

Abstract
The adenovirus E4 11k protein, product of E4 ORF3, is required in infection for processes including normal accumulation of viral late mRNAs. 11k restructures both the nucleus and cytoplasm of infected cells by relocalizing specific host cell target proteins, most strikingly components of nuclear PML oncogenic domains. It is likely that in many cases relocalization inactivates target proteins to produce 11k’s effects, although the mechanism and targets for stimulation of late mRNA accumulation is unknown. We have identified a new set of proteins relocalized by 11k: at least five protein components of cytoplasmic mRNA processing bodies (p-bodies) are found in 11k-induced cytoplasmic aggresomes, sites where proteins are inactivated or destroyed. One of these p-body proteins, RNA helicase Ddx6, binds 11k, suggesting a mechanism for relocalization. Because p-bodies are sites for mRNA degradation, their modification by 11k may provide an explanation for the role of 11k in viral late mRNA accumulation.
Keywords: Adenovirus E4 11k, Adenovirus E4 ORF3, P-body, aggresome, Ddx6, RCK, relocalization, reorganization, mRNA degradation, late gene expression, host-virus interaction
INTRODUCTION
Adenovirus early proteins function in a variety of processes in infected cells with the common goal of establishing a host cell environment favorable for viral replication (Tauber and Dobner, 2001). Viral early proteins can act both to commandeer host cell machinery for use in viral replication and to inhibit host cell pathways that might otherwise interfere with the virus’ life cycle. E4 11k, product of E4 open reading frame 3 [ORF 3], is one such protein. E4 11k stimulates late mRNA splicing and accumulation and viral DNA replication, participates in shutoff of host protein synthesis, and inhibits DNA damage sensing and prevents viral genome concatenation (Boyer et al., 1999; Bridge and Ketner, 1989; Halbert et al., 1985; Huang and Hearing, 1989; Medghalchi et al., 1997; Ohman et al., 1993; Stracker et al., 2002; Weiden and Ginsberg, 1994). At least a subset of the activities of E4 11k are mediated by redistribution of target host cell proteins in infected cells, which can result in localization near or away from sites of viral replication. Most dramatically, E4 11k is necessary and sufficient to redistribute components of nuclear structures containing the promyelocytic leukemia protein (PML), known as PML oncogenic domains or Nuclear Domain 10 (PODs/ND10), from punctate foci into elongated tracks that eventually surround viral replication centers (Carvalho et al., 1995; Doucas et al., 1996). PODs/ND10 have been implicated in diverse cellular processes including innate antiviral responses, and adenovirus mutants that fail to relocalize POD/ND10 proteins are sensitive to both interferon-induced and interferon-independent inhibition of replication (Ullman and Hearing, 2008; Ullman et al., 2007) and to growth inhibition mediated by the Mre11-Rad50-Nbs1 (MRN) complex (Ullman et al., 2007; Weitzman and Ornelles, 2005). E4 11k is selective in its rearrangement of proteins associated with PODs/ND10. PML, Sp100, Daxx, SUMO-1, and TIF1α (Doucas et al., 1996; Stracker et al., 2002; Stracker et al., 2005; Ullman et al., 2007; Yondola and Hearing, 2007). The basis for this specificity is not known.
Among its known targets, the activity of E4 11k on the Mre11-Rad50-Nbs1 (MRN) DNA damage sensing complex is unique. While early in infection MRN is reorganized into characteristic track-like structures as are POD/ND10 components, E4 11k ultimately targets MRN to cytoplasmic aggresomes (Araujo et al., 2005). Aggresomes are microtubule-dependent cytoplasmic inclusion bodies located adjacent to the nucleus, frequently in an indentation of the nuclear envelope, which colocalize with centrosome markers including γ–tubulin and are often described as surrounded by a vimentin cage (Kopito, 2000). Aggresomes are believed to contain aggregates of misfolded proteins (Garcia-Mata et al., 2002) and are enriched in components of the ubiquitin-proteasome pathway, suggesting that they function as sites for rapid degradation of unwanted proteins. Aggresomes are rare in normal cells but prominent in cells that have been stressed by factors that include viral infection, heat shock, and chemical treatments (Garcia-Mata et al., 2002; Kopito, 2000; Song et al., 2008; Wileman, 2007). In adenovirus-infected cells, expression either of E4 11k or another viral early protein, E1b 55k, can individually induce aggresome formation (Araujo et al., 2005; Liu et al., 2005; Stracker et al., 2002). Both E4 11k and E1b 55k relocate MRN to aggresomes, and E1b 55k also relocalizes p53 (Zantema et al., 1985). Both of these proteins have antiviral activity: MRN inhibits adenovirus growth by activation of the DNA damage response and is required for the viral genome concatenation seen in E4 mutant infections, and p53, which is induced by E1a during adenovirus infection, is potently pro-apoptotic. Relocalization of at least MRN to E1b 55k aggresomes interferes with its function and accelerates proteasomal degradation, (Carson et al., 2009; Liu et al., 2005) and it is likely that aggresomal p53, without access to its sites of action in the nucleus, is also nonfunctional. Thus, aggresome induction and host cell protein relocalization to aggresomes are among the mechanisms exploited by adenoviruses to abrogate inherent antiviral defenses and optimize the host cell environment for viral growth.
Here, we show that several proteins that are components of cytoplasmic RNA processing bodies (p-bodies) are relocalized to aggresomes induced by E4 11k expression. Most also appear in aggresomes induced by chemical stress. However, one of the p-body proteins relocalized to E4 11k aggresomes, Ddx6, appears specifically in E4 11k-induced aggresomes and is absent from aggresomes induced by chemical stress. Ddx6 coimmunoprecipitates with E4 11k, indicating a physical association between the two proteins, suggesting a mechanism for 11k-induced relocalization. P-bodies are sites of mRNA degradation (Eulalio et al., 2007). E4 11k stimulates viral late gene expression by promoting the accumulation of viral late mRNAs which are rapidly degraded in E4 mutant infections. The activity of E4 11k on p-body components therefore may reveal a mechanism for stimulation of late gene expression by E4 11k. The mechanisms of aggresome formation and virus-induced relocalization to aggresomes are not well understood. The interaction described here between E4 11k and Ddx6 therefore also provides a tool that will be useful in investigating these processes.
RESULTS
E4 11k targets several p-body components to aggresomes
In the course of a study to determine whether E4 proteins affected the integrity of mRNA processing bodies (p-bodies; Figure 1A), we noticed that plasmid-driven expression of Ad5 E4 11k (product of E4 ORF 3) in HeLa cells relocalized a portion of the p-body protein Lsm1 to E4 11k-containing foci located near the nucleus (not shown). Similar foci containing both Lsm1 and E4 11k were also found in HeLa cells infected by Ad5 (Figure 1B) and in cells infected with a defective adenovirus vector expressing an HA epitope-tagged Ad5 E4 11k (E1::11k; Figure 1C). E4 11k induces the formation of aggresomes, microtubule-dependent cytoplasmic inclusion bodies characterized by juxtanuclear location and colocalization with centrosome markers including γ–tubulin (Araujo et al., 2005; Johnston et al., 1998). The E4 11k-LSM1 foci that we observed resembled aggresomes. Therefore, we infected HeLa cells with the HA-tagged 11k vector and co-stained the infected cells for Lsm-1, HA-tagged E4 11k, and γ–tubulin as an aggresome marker. Approximately half of the infected cells contained perinuclear foci that stained for E4 11k. All of these foci also contained Lsm1 and all co-stained for γ–tubulin, confirming that they are bona fide aggresomes (Figure 1C). The distribution of Lsm1 within aggresomes was frequently irregular (Figure 1B and C). This was noted previously for members of the MRN complex, which are also relocalized to aggresomes by E4 11k (Araujo et al., 2005), and similar uneven distributions are seen for the other p-body components relocated to aggresomes by E4 11k (below). Irregularity may reflect the history of the aggresomes, which are thought to arise by coalescence of smaller cytoplasmic aggregates, or internal functional differentiation.
Figure 1. Lsm1 colocalizes with E4 11k in aggresomes.
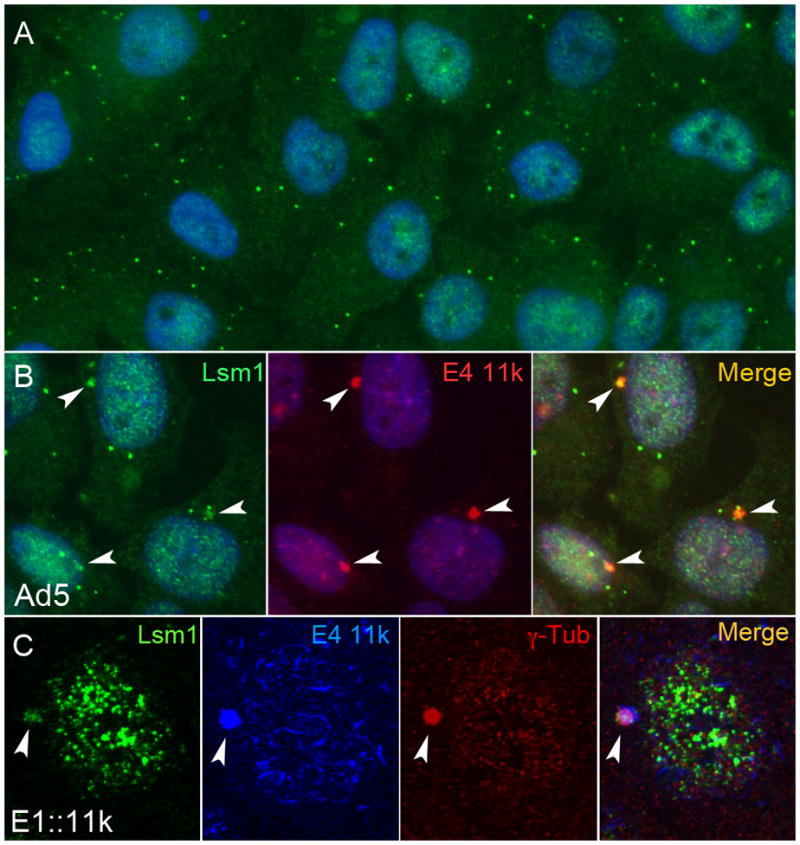
A. Mock-infected HeLa cells were immunostained for Lsm1 (green) and stained for DNA with DAPI (blue) and photographed under ultraviolet illumination. P-bodies, discrete cytoplasmic foci that contain Lsm1, are visible in each cell. B. HeLa cells were stained for Lsm1 (green), E4 11k (red) and DNA (blue) 36h after infection with Ad5 at a multiplicity of infection of 3000 particles per cell. E4 11k and Lsm1 colocalze in foci that lie near the nucleus (arrows). C. Cells infected with a defective adenovirus vector carrying an influenza hemagglutinin (HA) epitope-tagged E4 11k gene in place of E1 (E1::11k) were stained for Lsm1 (green), E4 11k (HA, blue) and aggresomal marker γ–tubulin (red). The juxtanuclear E4 11k/Lsm1 foci (arrows) co-stain for γ–tubulin, indicating that they are aggresomes. The cell shown here was one of 20 in the micrograph; of those, nine contained E4 11k-positive foci all of which costained for Lsm1 and γ-tubulin.
P-bodies are dynamic accumulations of a large number of proteins involved in mRNA metabolism. To determine whether E4 11k can direct p-body components other than Lsm1 to aggresomes, we evaluated localization of additional p-body components including RNA helicase Ddx6, scaffolding protein Ge-1, RNAi endonuclease Ago2, and 5′-3′ exonuclease Xrn1 in cells containing E4 11k expressed from Ad5 (Ddx6) or from the defective adenovirus vector (all four proteins). Each of these proteins was found in most E4 11k aggresomes (Figure 2; see the legend for Table 1, below). Thus, multiple p-body components are relocalized to aggresomes induced by E4 11k expression.
Figure 2. Other p-body proteins colocalize with E4 11k aggresomes.

A – D. HeLa cells infected with the E1::11k vector were immunostained for p-body proteins Ddx6 (red), Ge-1 (green). Xrn1 (red,), Argonaute 2 (red) and E4 11k [HA] (green in A, C, and D or red in B). Nuclei were stained with DAPI (blue). In infected cells, each of the p-body proteins is found in juxtanuclear foci that co-stain with E4 11k (arrows). E. P-body proteins are not found in comparable juxtanuclear foci in uninfected cells, consistent with the absence of aggresomes from most uninfected cells.
Table 1. P-bodies in cells with and without aggresomes.
Cells infected with the Ad5 E4 11k vector were stained for E4 11k and for Ddx6 and Lsm1, or for E4 11k and for Ge-1. The number of Ddx6, Lsm1, or Ge-1 p-bodies visible in each of 100 well-defined cells was determined in each experiment. Cells were then scored for aggresomes by examining E4 11k staining. Significance was determined using Student’s T test (2-tailed, equal variance). * Ddx6 and Lsm1 p-bodies were counted in the same experiment in cells triply stained for Ddx6, Lsm1, and E4 11k.
In this experiment, Lsm1 and Ddx6 were each present in 58 of 60 E4 11k aggresomes examined, while two aggresomes were negative for both proteins. Ge-1 was present in 61 of 67 aggresomes examined.
| p-body stain | Aggresome status | p-bodies per cell (average) | p | Number of cells |
|---|---|---|---|---|
| Ddx6* | − | 18.5 | 0.014 | 40 |
| + | 13.9 | 60 | ||
|
| ||||
| Lsm-1* | − | 9.4 | 0.0016 | 40 |
| + | 6.3 | 60 | ||
|
| ||||
| Ge-1 | − | 12.9 | 0.68 | 33 |
| + | 13.6 | 67 | ||
P-bodies persist in the cytoplasm of most cells that contain E4 11k aggresomes (Figure 1B). We investigated whether the numbers of p-bodies are reduced as p-body proteins are relocalized to aggresomes by counting p-bodies in E4 11k vector-infected cells co-stained for E4 11k and for Ddx6, Lsm1, or Ge-1. Ddx6, Lsm1, or Ge-1 foci were counted in 100 cells, which were then scored for the presence of an aggresome by E4 11k fluorescence (Table 1). Cells with aggresomes contained significantly fewer p-bodies that stain for either Ddx6 or Lsm-1 than did cells without aggresomes, consistent with the hypothesis that relocalization of Lsm-1 and Ddx6 to E4 11k aggresomes depletes p-bodies of those proteins. In contrast, in neither of two experiments was a significant reduction in Ge-1 foci observed in aggresome-containing cells. This may indicate that an insufficient fraction of the Ge-1 present in p-bodies is relocated to affect the number of Ge-1 foci detected by immunofluorescence, or that Ge-1 appearing in E4 11k aggresomes does not originate in p-bodies.
The E4 34k/E1b 55k E3 ubiquitin ligase shares some targets with E4 11k, such as Mre11. To determine whether p-body proteins were targets for virus-mediated degradation, particularly late in infection, we examined steady-state levels of Ddx6, Lsm-1 and a number of other p-body proteins in infected cells. None of the proteins examined, including Lsm1 and Ddx6, were noticeably degraded under conditions where DNA Ligase IV was completely ablated (Suplementary Figure 1 and data not shown) and the proteins examined therefore are not targets of the E4 34k/E1b 55k E3 ubiquitin ligase.
E4 11k aggresome formation is specific to Ad5
Proteins reorganized by E4 11k include PML, the members of MRN complex and transcription factor TIF1α While TIFlα reorganization is conserved across adenovirus serotypes, ability to reorganize MRN is restricted to Ad5 E4 11k, indicating that E4 11k-induced reorganization of TIF1α and MRN are separable functions (Stracker et al., 2005; Yondola and Hearing, 2007). To determine whether ability to form aggresomes and/or relocalize p-body components is conserved, we examined E4 11k, Lsm1, and Ddx6 distribution in HeLa cells infected with vectors expressing HA-tagged E4 11k from Ad3, Ad4, Ad9 and Ad12, which together with Ad5 represent five of the six subgroups of human adenoviruses. Expression of E4 11k proteins from these vectors was similar, as assessed by immunoblotting for the HA tag (not shown) and E4 11k from all serotypes formed the early nuclear tracks characteristic of E4 11k (Doucas et al., 1996; Figure 3A). Ad5 E4 11k progressed to form aggresomes as defined by location and co-staining with γ–tubulin (Figure 3A). Ad9 E4 11k formed rare perinuclear accumulations (Figure 3A and B) and both Ad4 and Ad9 E4 11k formed elongated cytoplasmic structures in about 25% of infected cells (Figure 3C and D). However, none of the structures formed by either Ad4 or Ad9 E4 11k co-stained for γ–tubulin (Figure 3C and D). In cells stained for E4 11k and for Lsm1 or Ddx6, 80–95% of the cytoplasmic Ad4 and Ad9 E4 11k accumulations contained Lsm1 (n=27 and 29 for Ad4 and Ad9 respectively) or Ddx6 (n=10 and 5). We conclude that classical aggresome formation by E4 11k is Ad5 specific, but that other serotypes relocalize at least Lsm1 and Ddx6 to discrete cytoplasmic structures which may have functions analogous to those of aggresomes in Ad5 infection.
Figure 3. Serotype specificity and effects of E4 11k mutations on aggresome formation.
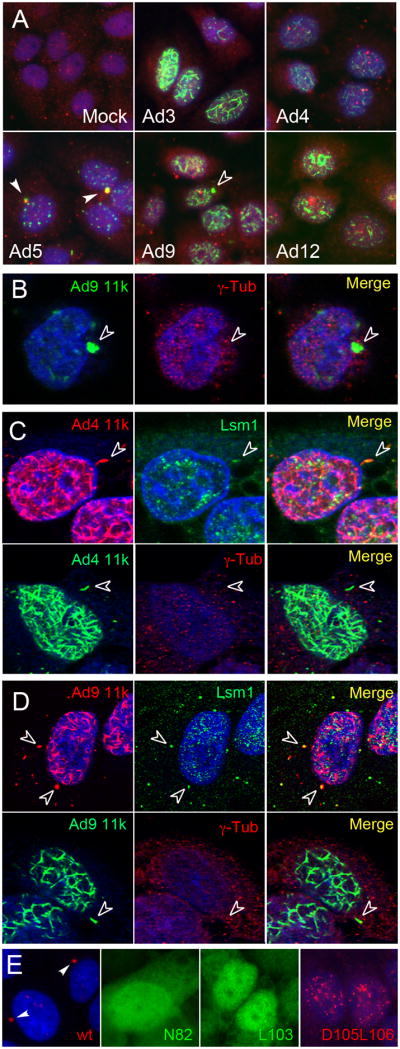
HeLa cells were infected with vectors expressing HA-tagged E4 11k from Ad3, Ad4, Ad5, Ad9 or Ad12. A, B. Cells were immunostained for E4 11k (HA, green), for the aggresomal marker γ–tubulin (red), and for DNA (blue). Ad5 E4 11k and γ–tubulin co-localize (solid arrows). Juxtanuclear foci containing Ad9 E4 11k do not contain γ–tubulin (open arrows), indicating that they are not aggresomes. Lsm1 is also absent from these foci (data not shown). C, D. Top rows: HeLa cells were infected with vectors expressing HA tagged-E4 11k from Ad4 or Ad9 and were immunostained for Lsm1 (green) and E4 11k [HA] (red). DNA is stained with DAPI (blue). E4 11k from both Ad4 and Ad9 can be found in elongated cytoplasmic accumulations that also contain Lsm1 but that are distinguishable from Ad5 aggresomes by morphology and location (open arrows). Bottom rows: Cytoplasmic structures containing Ad 4 or Ad9 E4 11k do not co-stain for γ–tubulin. E. Wild type Ad5 E4 11k forms aggresomes (arrows, first panel). The substitution mutant proteins N82A, and L103A show diffuse cytoplasmic and nuclear localization but do not appear in aggresomes. D105A/L106A forms early track-like nuclear structures, but not aggresomes. Stained for E4 11k (red or green); first and fourth panels also stained with DAPI.
A series of Ad5 E4 11k mutants have been evaluated for their ability to reorganize the previously identified E4 11k targets PML, MRN and TIF1α (Evans 2005; Yondola 2007). Single amino acid substitution mutants N82A and L103A fail to form track-like structures and do not reorganize PML, MRN or TIF1a. The double point mutant D105A/L106A can relocalize PML to early track-like structures, but cannot reorganize MRN or TIF1α We examined aggresome formation by these mutants by infecting HeLa cells with adenovirus E1-replacement vectors expressing HA-tagged versions of N82A, L103A, and D105A/L106A and co-stained the cells for HA (E4 11k) and Ddx6 or Lsm1. None of the mutants formed aggresomes and none displayed co-localization with Ddx6 or Lsm1 (Figure 3E and not shown).
P-body proteins in aggresomes induced by chemical stress
At least two mechanisms might account for the relocalization of p-body components to E4 11k-induced aggresomes. E4 11k might interact with one or more p-body components and actively mediate their movement to aggresomes. Alternatively, the presence of aggresomes, once induced by 11k, could promote relocalization of p-body components without further participation of 11k itself. To differentiate these hypotheses, we examined aggresomes formed without involvement of 11k for the presence of p-body components. Treatment with cadmium chloride (CdCl2) induces aggresome formation in a variety of cells (Song et al., 2008), and we evaluated the localization of Lsm1, Ge-1, and Ddx6 in HeLa cells treated with CdCl2 (Figure 4). Aggresomes were identified in cells after 6h of treatment with CdCl2 by staining for γ–tubulin and by juxtanuclear location. Lsm1 and Ge-1 were detected, respectively, in 21 out of 23 and 7 out of 9 of γ–tubulin positive CsCl2 aggresomes examined (Figure 4A and B), indicating that interactions with E4 11k are not necessary for relocalization of these proteins to aggresomes. In contrast, Ddx6 was detected in none of the 12 cadmium-induced aggresomes examined (Figure 4C). This suggests the possibility that Ddx6 is specifically relocated to aggresomes via an interaction with E4 11k.
Figure 4. P-body proteins in cadmium-induced aggresomes.
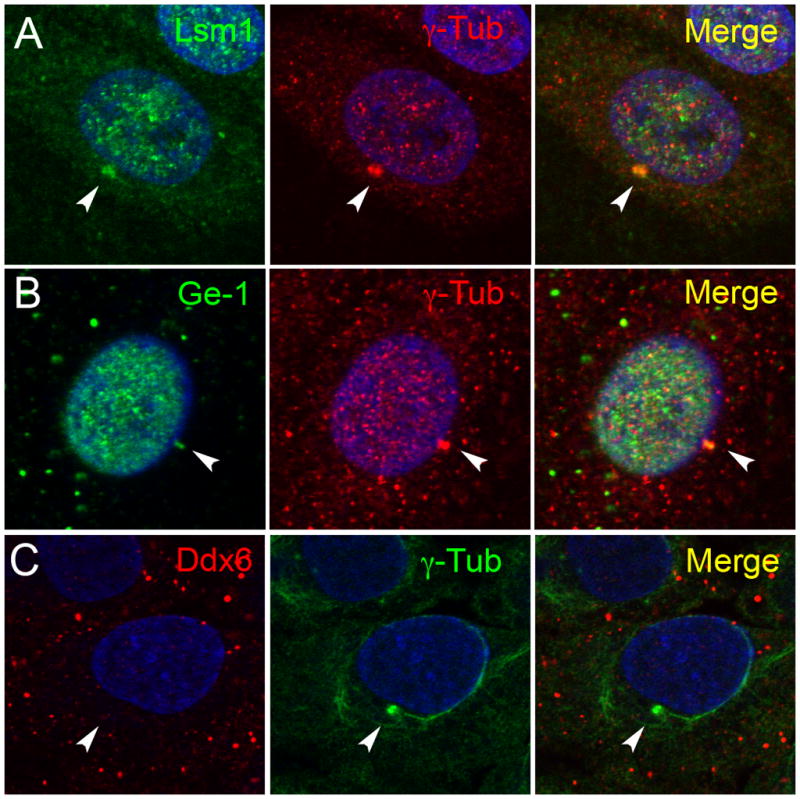
HeLa cells were treated with CdCl2 for 6 hours. Treated cells were immunostained for Lsm1 (green, A), Ge-1 (green, B) or Ddx6 (red, C) and the aggresomal marker γ–tubulin (red, A and B or green, C). DNA was stained with DAPI (blue). Lsm1 and Ge-1 are relocalized to cadmium-induced aggresomes, but Ddx6 is not. Arrows indicate aggresomes in each panel.
E4 11k and Ddx6 physically interact
The specificity of association between Ddx6 and E4 11k aggresomes suggests that E4 11k participates actively in the relocalization of Ddx6, perhaps through a physical interaction. To test the hypothesis that E4 11k and Ddx6 physically associate, we immunoprecipitated Ddx6 from mock-infected HeLa cells and from HeLa cells infected with vectors expressing HA-tagged wild type E4 11k, HA-tagged E4 11k mutant N82A, which neither reorganizes nuclear E4 11k targets nor forms aggresomes (Evans 2005; Yondola 2007 and data not shown), or GFP as a control. Immunoprecipitates were examined by immunoblotting for HA-tagged E4 11k and Ddx6 (to confirm successful immunoprecipitation) (Figure 5A). E4 11k was readily detected in Ddx6 precipitates from cells expressing the wild type protein, but not in those from cells expressing the N82A mutant despite substantial overexpression of the mutant protein. No HA-reactive material was detected in immunoprecipitates from mock-infected cells or cells infected with the GFP vector. Thus, E4 11k associates either directly or indirectly with Ddx6, and this association is abrogated by a mutation that interferes with normal E4 11k functions including aggresome formation. Similar experiments were performed in which immunoprecipitates made with anti-HA antibody, which precipitates HA-tagged E4 11k, were examined for the presence of Ddx6 (Figure 5B). In those experiments, Ddx6 was found at low levels in all precipitates, including those made from mock- and lacZ vector-infected cells. However, the signal was reproducibly more intense in wild type- but not in N82A-infected cells, consistent with the conclusion that Ddx6 and E4 11k interact, and that the interaction is disrupted by the N82A mutation. The E4 11k immunoprecipates were also examined for the presence of Lsm1 but none was detected (data not shown), suggesting that Lsm1 and E4 11k do not interact physically.
Figure 5. E4 11k and Ddx6 co-immunoprecipitate.

HeLa cells were infected with vectors expressing HA-tagged wild type or mutant N82A E4 11k, GFP or lacZ. 16 hours post-infection, cells were lysed in RIPA buffer and lysates were incubated with anti-Ddx6 (A) or anti-HA [E4 11k] (B) antibodies coupled to magnetic beads. Immune complexes were recovered, fractionated on 10–20% Tricine gels, and analyzed by immunoblotting for Ddx6 or E4 11k (HA). 1% of the input material blotted for Ddx6 and E4 11k is shown in the top rows of each panel and immunoprecipitates are shown in the bottom rows. Ddx6 immunoprecipitates specifically contain wild type HA-E4 11k, but not the mutant N82A. E4 11k immunoprecipitates from cells infected with a vector expressing wild type E4 11k are enriched in Ddx6 compared to those from cells infected with an N82A or lacZ vector control, or from mock-infected cells.
DISCUSSION
E4 11k and E4 34k, the products of E4 ORFs 3 and 6, respectively, each satisfy the requirement for E4 in processes in infected cells that include viral late mRNA accumulation and inhibition of DNA damage response and repair pathways (Bridge and Ketner, 1989; Huang and Hearing, 1989; Stracker et al., 2002). Despite their functional redundancy, the E4 34k and E4 11k proteins act by distinct mechanisms. E4 34k, with the 55kDa product of E1b, constitutes the substrate-binding component of an E3 ubiquitin ligase that targets specific host cell proteins for proteasomal destruction, while E4 11k acts by inducing relocalization of host cell proteins which, in at least one case, results in inactivation of its target (Carson et al., 2009; Liu et al., 2005; Querido et al., 2001). Both proteins thus are capable of interfering with the functions of host cell proteins, presumably inhibiting processes or pathways that otherwise would be inhibitory to viral growth. Some targets of E4 34k and E4 11k are held in common, such as MRN, while others such as DNA ligase IV, are not. In the latter cases, it is likely that the redundancy in the function of the E4 proteins reflects their inhibition of common pathways via effects on different specific targets.
The work reported here identifies a set of new proteins relocalized by E4 11k. The new targets are mRNA processing body (p-body) components Lsm1, Ge-1, Ago2, Xrn1 and Ddx6. While most previously identified E4 11k targets are nuclear proteins and are relocalized to structures within the nucleus, the targets identified here are principally cytoplasmic and E4 11k expression results in relocalization to cytoplasmic aggresomes. At least Lsm1 and Ge-1 are also present in aggresomes induced by cadmium treatment. This indicates that relocalization of these p-body proteins to aggresomes does not require a specific interaction with E4 11k. In contrast, Ddx6 is absent from cadmium-induced aggresomes. We have also shown that Ddx6 co-immunoprecipitates with wild type E4 11k from extracts of vector-infected cells, but not with non-functional mutant E4 11k. This strongly suggests a mechanism for relocalization of Ddx6 to aggresomes mediated by a physical interaction with E4 11k.
Aggresome formation occurs by coalescence of smaller aggregates of misfolded proteins directed to the centrosome by microtubules. P-bodies are also anchored to microtubules, display spatially confined motion dependent on microtubule motion, and stationary p-bodies can be identified at the centrosome (Aizer et al., 2008). Aggresome formation accompanies gross microtubule rearrangement within cells and it may not be surprising that as a result, p-bodies tethered to microtubules would move to aggresomes along with other material destined for aggresomal localization. This is consistent with our observation that most of the p-body proteins we have evaluated appear to be relocalized to aggresomes induced by E4 11k or cadmium. It is perplexing however, if p-bodies are trafficked en masse to aggresomes as a result of microtubule rearrangements, why Ddx6 is found only in E4 11k aggresomes and not in aggresomes induced by cadmium. Since Ddx6 remains associated with surviving p-bodies in cells that harbor aggresomes, its absence from cadmium-induced aggresomes must be a consequence of the structure or mechanism of formation of aggresomes, rather than the result of dissociation of Ddx6 from p-bodies prior to aggresome formation. For example, protein-protein interactions that occur in aggresomes may interfere with interactions that bind Ddx6 to other components of cytoplasmic p-bodies. Whatever explains the failure of Ddx6 to appear in aggresomes induced by other agents, the existence of a mechanism to specifically incorporate Ddx6 into aggresomes in infected cells reinforces the hypothesis that aggresomal localization and likely inactivation of Ddx6 is critical for viral growth.
Aggresomes are believed to arise when, under conditions of stress, misfolded proteins accumulate to levels beyond the capacity of constitutive protein quality-control mechanisms to eliminate them, and it is generally assumed that aggresomes are sites for emergency sequestration and inactivation of potentially harmful proteins. Adenoviruses exploit aggresomes for a similar purpose: MRN, when incorporated into aggresomes is inactive as assessed by its inability to form radiation-induced foci on chromosomal DNA or initiate DNA damage signaling (Carson et al., 2009; Liu et al., 2005), and aggresome formation thus contributes to the inactivation of MRN required for efficient viral growth. It is likely that aggresomal relocalization of p-body proteins inhibits their activity similarly. While classical aggresome formation is observed only for Ad5 E4 11k, the E4 11k proteins of at least two other serotypes, Ad4 and Ad9, formed γ-tubulin-negative cytoplasmic structures that contained Lsm-1 and Ddx6. These may be functionally analogous to the aggresomes induced by Ad5 E4 11k and inactivate similar pathways.
E4 11k and the E4 34k/E1b 55k complex redundantly promote efficient adenovirus late gene expression (Bridge and Ketner, 1989; Huang and Hearing, 1989). In their absence, late messages are transcribed at a normal rate, but do not accumulate and must therefore be degraded (Sandler and Ketner, 1989). P-bodies are sites of mRNA decay and contain proteins that participate in 5′ to 3′ mRNA decay, ARE-mediated degradation (AMD), mRNA surveillance by the nonsense mediated decay (NMD) pathway, and mRNA translational repression by the microRNA (miRNA)-mediated silencing pathway (Anderson and Kedersha, 2009; Eulalio et al., 2007). While the implications of the relocalization of Ddx6 and other p-body components on adenovirus growth are not yet certain, the late mRNA phenotype of E4 mutants and the connection of p-bodies to mRNA degradation suggests the possibility that effects on p-body function contributes to the effects of E4 11k on viral late mRNA accumulation.
Ddx6 is a member of the SF2 DEAD-box RNA helicase family (Weston and Sommerville, 2006) and a defining component of p-bodies in cells ranging from yeast to humans. The helicase activity of Ddx6 is essential for p-body formation (Minshall et al., 2009) and Ddx6 has been implicated in aspects of mRNA metabolism that include transcription, splicing, translation, decapping enhancement and degradation (Cordin et al., 2006; Linder, 2006; Rocak and Linder, 2004). Ddx6 has been shown to physically interact with several other p-body components including decapping enhancers Pat1, Lsm1–7, EDC3, Dcp1, Dcp2 and Ge-1 (Coller and Parker, 2005; Coller et al., 2001; Decker et al., 2007). Ddx6 also binds Ago1 and Ago2, and is essential for miRNA-mediated gene silencing (Chu and Rana, 2006). Disruption of any of these associations might contribute E4 11k-stimulated late mRNA accumulation. The C-terminal domain of Ddx6 is required for interactions with protein partners and localization to p-bodies (Minshall et al., 2009) and conserved RecA homology domain required for RNA binding is also located in the C-terminus (Cordin et al., 2006). It will be of interest to determine whether the interaction of E4 11k and Ddx6 is mediated by the C-terminus of Ddx6, and whether it is mutually exclusive with interactions with cellular protein partners.
P-bodies that contain Ddx6 persist in reduced numbers in cells expressing amounts of E4 11k sufficient to promote late gene expression. This indicates that quantitative removal of Ddx6 from p-bodies cannot be essential for E4 11k function. Modest reductions in Ddx6 content of p-bodies or in the number of p-bodies may suffice to accomplish the required functions of E4 11k. Alternatively, perhaps a critical population of Ddx6 either in p-bodies or elsewhere in cells is relocated to aggresomes and inactivated and the Ddx6 that remains in p-bodies is not relevant to E4 11k action. It is also possible that despite retention of some Ddx6, the p-bodies visible in cells containing E4 11k aggresomes are inactive due to E4 11k function. Finally, although without obvious precedent, relocation of Ddx6 to aggresomes may confer some activity upon Ddx6 that is required for viral growth, a hypothesis untroubled by residual p-body-associated Ddx6.
The implications of the interaction of adenovirus E4 11k with Ddx6 and other p-body components is not yet clear. We suggest that relocalization of p-body proteins, and Ddx6 in particular, interferes with mRNA decay functions that occur in p-bodies and that that interference is reflected in viral mRNA stabilization by E4 11k. Experiments intended to test that hypothesis should provide insight into an important unresolved issue in adenovirus biology - the mechanism of E4 stimulation of late gene expression - and may also clarify our view of p-body function in normal human cells.
MATERIALS AND METHODS
Cell culture
HeLa cells were propagated in Dulbecco modified Eagle’s medium (11965092, Invitrogen) supplemented with 10% FBS, penicillin and streptomycin.
Viruses and infections
E1-replacement vectors expressing hemagglutinin (HA)-tagged wild-type E4 11k proteins of Ad5, Ad3, Ad4, Ad9 and Ad12 and mutants of Ad5 E4 11k (N82A, L103A, D105A/L106A) have been described (Evans and Hearing, 2003, 2005). These recombinants carry deletions of E4 ORFs 1–3, and thus produce E4 11k only from the transgene located in E1. Virus particles were purified by cesium chloride density gradient centrifugation and were stored frozen in phosphate buffered saline containing 5% sucrose, 0.5mM CaCl2, 0.9mM magnesium acetate. Particle concentrations were determined from A260 (Challberg and Ketner, 1981; Mittereder et al., 1996). All infections were performed at an MOI of 3000 particles/cell. Virus was adsorbed in 10–20% normal media volume for 2 hours and then replaced with fresh media.
Antisera
Antibodies used for immunofluorescence experiments were purchased from the following companies and used at the indicated dilutions: HA (16B12, Covance), 1:1000; Lsm-1 (GW22100F, Sigma) 1:500; Ddx6 (A300-461A, Bethyl Laboratories) 1:500; Ge-1 [p70 S6 kinase (Stoecklin et al., 2006)] (sc-8416, Santa Cruz) 1:500; Ago2 (ab32381, Abcam) 1:50; Xrn1 (A300-443A, Bethyl Laboratories) 1:50; γ–tubulin (T3559, Sigma) 1:500; γ–tubulin (T6557, Sigma) 1:2000; E4 11k (rabbit polyclonal antiserum raised to a carboxy-terminal peptide (Boyer et al., 1999)) 1:1000. Alexa Fluor-conjugated secondary antibodies were purchased from Invitrogen and used at a dilution of 1:200.
HA and Ddx6 antibodies were used for immunoblots at dilutions of 1:1000. Secondary antibodies for immunoblotting experiments were purchased from GE Healthcare.
Immunoprecipitations and immunoblotting
Beads were prepared by covalently coupling 7mg anti-HA (Covance) or anti-Ddx6 (Bethyl Laboratories) per 1mg of Dynabeads (Invitrogen) according to the manufacturer’s protocol. Antibody bound-Dynabeads were then blocked in 5% BSA for 30 minutes at room temperature with rotation. Infected HeLa cells (2.5×107 cells per 14cm dish) were harvested 16 hours post-infection in 1mL radioimmunoprecipitation assay buffer (RIPA; 50mM Tris, pH 8.0; 150mM NaCl; 1.0% NP-40; 0.5% deoxycholic acid; 0.1% sodium dodecyl sulfate [SDS]) supplemented with a protease inhibitor cocktail (Sigma, P8340). Lysates were clarified by centrifugation at 13000 × g for 10 minutes at 4°C. 2.5mg total protein was incubated with 2mg of pre-blocked antibody bound-Dynabeads per reaction in the presence of 1% BSA for 2 hours at 4°C with rotation. Beads were washed three times with 0.5mL RIPA, and bound proteins were eluted by incubation with 50mL elution buffer [EB] (provided with the Dynabead kit) for 5 minutes with rotation.
Whole cell extracts and immunoprecipitation eluates were prepared in Tricine Sample Buffer (BioRad) boiled 5 minutes and separated on Novex 10–20% Tricine gels (Invitrogen). Proteins were electrophoretically transferred to nitrocellulose membranes and immunoblotted as described (Baker et al., 2007)
Immunofluorescence Microscopy
Cells were seeded on glass coverslips and infected or treated as indicated. Cells were fixed for 30 minutes in a solution of 2% formaldehyde in phosphate buffered saline [PBS], washed in PBS, and permeabilized in ice-cold acetone for 3 minutes. Primary and secondary antibodies were diluted in 2% BSA; 0.02% NaN3 in PBS. Cells were incubated with primary antibodies for 1 hour, washed three times in PBS and incubated with secondary antibodies for 30min. Cells were washed three times more in PBS and mounted on slides with mounting solution (100mM Tris, pH 8.8; 50% glycerol; 2.5% DABCO; and in some cases 0.2mg/mL DAPI). All manipulations were carried out at room temperature, and secondary antibody incubation and mounting were performed in the dark. Slides were viewed and images were acquired on a Zeiss 510 Meta confocal microscope using LSM Image software, or on Nikon E800 or 90i microscopes.
Cadmium Treatment
Cadmium chloride (C-2544, Sigma) was dissolved in distilled water. Tissue culture media was supplemented with cadmium chloride at a final concentration of 20mM. Cells were incubated for 6 hours under normal tissue culture conditions, and then prepared for immunofluorescence as described above.
Supplementary Material
Steady-state levels of Ddx6 (top panel), Lsm1 (bottom panel), and DNA ligase IV (both panels) were assessed by immunoblotting in cells infected by H5dl1013 (E4 11k−, E4 34k+) or Ad5. Actin served as a loading control. DNA ligase IV, a substrate of the adenovirus E4 34k/E1b 55k E3 ubiquitin ligase, is efficiently degraded in cells infected by either virus, as expected (Baker et al., 2007). Neither Ddx6 nor Lsm1 is reduced in amount at times where DNA ligase IV degradation is complete, indicating that neither is a substrate of the adenovirus-induces E3 ligase. For clarity, the Lsm1 panel was dodged in Photoshop to remove a shadow that partially obscured the 12 and 24h bands.
Acknowledgments
We thank Timra Gilson, Ph.D., for valuable discussions during the conduct of this work, Kasey Karen, Ph.D. for critical comments on the manuscript, Barbara Smith of the Johns Hopkins School of Medicine Department of Cell Biology Microscope Facility and Julia Romano, Ph.D for assistance with microscopy, and Cameron Ward for excellent technical assistance. Supported by NIH grants 5T32AI007417-17 and 5R01AI079132-02.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aizer A, Brody Y, Ler LW, Sonenberg N, Singer RH, Shav-Tal Y. The dynamics of mammalian P body transport, assembly, and disassembly in vivo. Mol Biol Cell. 2008;19:4154–4166. doi: 10.1091/mbc.E08-05-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol. 2009;10:430–436. doi: 10.1038/nrm2694. [DOI] [PubMed] [Google Scholar]
- Araujo FD, Stracker TH, Carson CT, Lee DV, Weitzman MD. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J Virol. 2005;79:11382–11391. doi: 10.1128/JVI.79.17.11382-11391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A, Rohleder KJ, Hanakahi LA, Ketner G. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J Virol. 2007;81:7034–7040. doi: 10.1128/JVI.00029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer J, Rohleder K, Ketner G. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology. 1999;263:307–312. doi: 10.1006/viro.1999.9866. [DOI] [PubMed] [Google Scholar]
- Bridge E, Ketner G. Redundant control of adenovirus late gene expression by early region 4. J Virol. 1989;63:631–638. doi: 10.1128/jvi.63.2.631-638.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson CT, Orazio NI, Lee DV, Suh J, Bekker-Jensen S, Araujo FD, Lakdawala SS, Lilley CE, Bartek J, Lukas J, Weitzman MD. Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J. 2009;28:652–662. doi: 10.1038/emboj.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho T, Seeler JS, Ohman K, Jordan P, Pettersson U, Akusjarvi G, Carmo-Fonseca M, Dejean A. Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J Cell Biol. 1995;131:45–56. doi: 10.1083/jcb.131.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challberg SS, Ketner G. Deletion mutants of adenovirus 2: isolation and initial characterization of virus carrying mutations near the right end of the viral genome. Virology. 1981;114:196–209. doi: 10.1016/0042-6822(81)90265-8. [DOI] [PubMed] [Google Scholar]
- Chu CY, Rana TM. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol. 2006;4:e210. doi: 10.1371/journal.pbio.0040210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller J, Parker R. General translational repression by activators of mRNA decapping. Cell. 2005;122:875–886. doi: 10.1016/j.cell.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller JM, Tucker M, Sheth U, Valencia-Sanchez MA, Parker R. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA. 2001;7:1717–1727. doi: 10.1017/s135583820101994x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordin O, Banroques J, Tanner NK, Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Decker CJ, Teixeira D, Parker R. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J Cell Biol. 2007;179:437–449. doi: 10.1083/jcb.200704147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucas V, Ishov AM, Romo A, Juguilon H, Weitzman MD, Evans RM, Maul GG. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 1996;10:196–207. doi: 10.1101/gad.10.2.196. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Behm-Ansmant I, Izaurralde E. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol. 2007;8:9–22. doi: 10.1038/nrm2080. [DOI] [PubMed] [Google Scholar]
- Evans JD, Hearing P. Distinct roles of the Adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J Virol. 2003;77:5295–5304. doi: 10.1128/JVI.77.9.5295-5304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JD, Hearing P. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J Virol. 2005;79:6207–6215. doi: 10.1128/JVI.79.10.6207-6215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata R, Gao YS, Sztul E. Hassles with taking out the garbage: aggravating aggresomes. Traffic. 2002;3:388–396. doi: 10.1034/j.1600-0854.2002.30602.x. [DOI] [PubMed] [Google Scholar]
- Halbert DN, Cutt JR, Shenk T. Adenovirus early region 4 encodes functions required for efficient DNA replication, late gene expression, and host cell shutoff. J Virol. 1985;56:250–257. doi: 10.1128/jvi.56.1.250-257.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MM, Hearing P. Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J Virol. 1989;63:2605–2615. doi: 10.1128/jvi.63.6.2605-2615.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Linder P. Dead-box proteins: a family affair--active and passive players in RNP-remodeling. Nucleic Acids Res. 2006;34:4168–4180. doi: 10.1093/nar/gkl468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Shevchenko A, Berk AJ. Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J Virol. 2005;79:14004–14016. doi: 10.1128/JVI.79.22.14004-14016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medghalchi S, Padmanabhan R, Ketner G. Early region 4 modulates adenovirus DNA replication by two genetically separable mechanisms. Virology. 1997;236:8–17. doi: 10.1006/viro.1997.8737. [DOI] [PubMed] [Google Scholar]
- Minshall N, Kress M, Weil D, Standart N. Role of p54 RNA helicase activity and its C-terminal domain in translational repression, P-body localization and assembly. Mol Biol Cell. 2009;20:2464–2472. doi: 10.1091/mbc.E09-01-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittereder N, March KL, Trapnell BC. Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. J Virol. 1996;70:7498–7509. doi: 10.1128/jvi.70.11.7498-7509.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohman K, Nordqvist K, Akusjarvi G. Two adenovirus proteins with redundant activities in virus growth facilitates tripartite leader mRNA accumulation. Virology. 1993;194:50–58. doi: 10.1006/viro.1993.1234. [DOI] [PubMed] [Google Scholar]
- Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001;15:3104–3117. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocak S, Linder P. DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol. 2004;5:232–241. doi: 10.1038/nrm1335. [DOI] [PubMed] [Google Scholar]
- Sandler AB, Ketner G. Adenovirus early region 4 is essential for normal stability of late nuclear RNAs. J Virol. 1989;63:624–630. doi: 10.1128/jvi.63.2.624-630.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Xiao Z, Nagashima K, Li CC, Lockett SJ, Dai RM, Cho EH, Conrads TP, Veenstra TD, Colburn NH, Wang Q, Wang JM. The heavy metal cadmium induces valosin-containing protein (VCP)-mediated aggresome formation. Toxicol Appl Pharmacol. 2008;228:351–363. doi: 10.1016/j.taap.2007.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Mayo T, Anderson P. ARE-mRNA degradation requires the 5′-3′ decay pathway. EMBO Rep. 2006;7:72–77. doi: 10.1038/sj.embor.7400572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- Stracker TH, Lee DV, Carson CT, Araujo FD, Ornelles DA, Weitzman MD. Serotype-specific reorganization of the Mre11 complex by adenoviral E4orf3 proteins. J Virol. 2005;79:6664–6673. doi: 10.1128/JVI.79.11.6664-6673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauber B, Dobner T. Molecular regulation and biological function of adenovirus early genes: the E4 ORFs. Gene. 2001;278:1–23. doi: 10.1016/s0378-1119(01)00722-3. [DOI] [PubMed] [Google Scholar]
- Ullman AJ, Hearing P. Cellular proteins PML and Daxx mediate an innate antiviral defense antagonized by the adenovirus E4 ORF3 protein. J Virol. 2008;82:7325–7335. doi: 10.1128/JVI.00723-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman AJ, Reich NC, Hearing P. Adenovirus E4 ORF3 protein inhibits the interferon-mediated antiviral response. J Virol. 2007;81:4744–4752. doi: 10.1128/JVI.02385-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiden MD, Ginsberg HS. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc Natl Acad Sci U S A. 1994;91:153–157. doi: 10.1073/pnas.91.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman MD, Ornelles DA. Inactivating intracellular antiviral responses during adenovirus infection. Oncogene. 2005;24:7686–7696. doi: 10.1038/sj.onc.1209063. [DOI] [PubMed] [Google Scholar]
- Weston A, Sommerville J. Xp54 and related (DDX6-like) RNA helicases: roles in messenger RNP assembly, translation regulation and RNA degradation. Nucleic Acids Res. 2006;34:3082–3094. doi: 10.1093/nar/gkl409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wileman T. Aggresomes and pericentriolar sites of virus assembly: cellular defense or viral design? Annu Rev Microbiol. 2007;61:149–167. doi: 10.1146/annurev.micro.57.030502.090836. [DOI] [PubMed] [Google Scholar]
- Yondola MA, Hearing P. The adenovirus E4 ORF3 protein binds and reorganizes the TRIM family member transcriptional intermediary factor 1 alpha. J Virol. 2007;81:4264–4271. doi: 10.1128/JVI.02629-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zantema A, Fransen JA, Davis-Olivier A, Ramaekers FC, Vooijs GP, DeLeys B, Van der Eb AJ. Localization of the E1B proteins of adenovirus 5 in transformed cells, as revealed by interaction with monoclonal antibodies. Virology. 1985;142:44–58. doi: 10.1016/0042-6822(85)90421-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Steady-state levels of Ddx6 (top panel), Lsm1 (bottom panel), and DNA ligase IV (both panels) were assessed by immunoblotting in cells infected by H5dl1013 (E4 11k−, E4 34k+) or Ad5. Actin served as a loading control. DNA ligase IV, a substrate of the adenovirus E4 34k/E1b 55k E3 ubiquitin ligase, is efficiently degraded in cells infected by either virus, as expected (Baker et al., 2007). Neither Ddx6 nor Lsm1 is reduced in amount at times where DNA ligase IV degradation is complete, indicating that neither is a substrate of the adenovirus-induces E3 ligase. For clarity, the Lsm1 panel was dodged in Photoshop to remove a shadow that partially obscured the 12 and 24h bands.