

Abstract
Many proteins play a dual role in embryonic development. Those that regulate cell fate determination in a specific tissue can also affect the development of a larger region of the embryo. This makes defining its role in a particular tissue difficult to analyze. For example, noggin overexpression in Xenopus laevis embryos causes the expansion of the entire anterior region, including the eye1,2. From this result, it is not known if Noggin plays a direct role in eye determination or that by causing an expansion of neural tissue, Noggin indirectly affects eye formation. Having this complex phenotype makes studying its eye-specific role in cell fate determination difficult to analyze. We have developed an assay that overcomes this problem. Taking advantage of the pluripotent nature of the Xenopus laevis animal cap 3, we have developed an assay to test the ability of gene product(s), like noggin or the eye field transcription factors (EFTFs), to transform caps into particular tissue or cell types by transplanting this tissue onto the side of the embryo 4. While we have found either Noggin protein treatment or a collection of transcription factors can determine retinal cell fate in animal caps, this procedure could be used to identify gene product(s) involved in specifying other tissues as well.
Protocol
Part I. Set-up for the ACT Assay
Generate capped RNA for injection as suggested by the manufacturer, using their phenol:chloroform purification method (e.g. mMessage mMachine Kit; Applied Biosystems/Ambion, Austin, TX). RNA encoding a fluorescent protein is used as a tracer. We transcribe either yellow fluorescent protein (YFP) or mCherry 5 cDNA, which is in a pCS2+ expression vector.
Pull borosilicate glass capillary tubes to form tapered glass tips using a micropipette puller. We use Sutter Micropipette Puller, P-97 (Sutter Instruments, Inc., Novato, CA), as previously described 6.
Make sterile solutions in Table 1 from a 10 X Marc's Modified Ringer's (MMR) solution stock (10 mM MgCl2 ; 20 mM KCl; 20 mM CaCl2 ; 50 mM HEPES; 1 M NaCl; adjusted to pH 7.5, autoclaved and stored at RT).
Isolate testes from a male frog and store the tissue at 4°C in a sterile solution of 1X MMR/gentamicin (50 μg/mL gentamicin sulfate; BioWhittaker, Cat # 17-518Z). This can be done up to a week before the start of the experiment but it is looses its efficacy over time.
Egg laying solution (1X: 150 mM NaCl, 2.62 mM KCl, 0.8 mM Na2HPO4, 20 mM Trizma base, 2.62 mM NaHCO3 and 2.75 mM MgSO4; adjusted to pH 7.6) can be made as an 8X stock, which can be stored at 4°C for up to two weeks. Note: watch out for contamination in this solution, as this will affect the quality of your eggs.
Generate injection plates. To do this, melt 1% agarose in 0.4X MMR in a microwave, add ~6 mL melted agarose solution into 60 mm plates, gently unroll an elastomer mold (Figure 1) on top of the solution to prevent bubbles and let cool for about 20 minutes at RT. To determine the number needed, consider the number of experimental conditions (e.g. 1, YFP; 2, YFP+Noggin) and the number of transplanted host embryos needed. For our experiments, we generate at least 20 transplanted host embryos for each experimental condition. At 18-19°C, embryos will be at stage 15 for about an hour. If it takes one hour to perform 20 transplants, then two fertilization time points are needed, assuming the host embryos accept all transplants. Therefore, four injection dishes will be needed (2 exp conditions x 2 fertilization time points = 4). Store the plates at 4°C for up to one week.
Generate cap isolation plates using 1% agarose + 0.7X MMR with molds as for the injection plate. Make the same number of cap isolation as injection plates plus one extra per fertilization time point (4 + 2 fertilization time points = 6).
Inject females with 500 units human chorionic gonadotropin late in the day on the day before microinjection. Store females at 16-18°C overnight.
Part II. RNA Microinjection of Xenopus laevis embryos
A. Preparation for microinjection
Remove hormonally induced female and place them in individual tanks containing 1X egg laying solution (ELS). Note: we don't use eggs that have been in the ELS more than two hours as the quality of eggs is reduced. Therefore, about an hour before in vitro fertilization, we discard all eggs from the tank so that only freshly laid eggs are collected.
Prepare the needle for microinjection by snipping off the tip to obtain a 25-35 μm diameter. Place the needle onto the picoinjector and calibrate it so that it dispenses 10 nL per injection.
Set an incubator to 14°C.
Once females have laid eggs for about an hour, remove the eggs from the tank using a transfer pipette and place them into 60 mm Petri dishes. Remove as much ELS from the eggs as possible and replace with 1X MMR. The eggs can be fertilized right away or stay in 1X MMR for up to an hour. For the transplant assay, it is best to fertilize two dishes, 80% full of a single layer of eggs in up to three separate batches at around 11 am, noon and 1 pm. After injection, they will be placed at 14°C so that they will grow to the desired stages for cap isolation (stage 8.5-9) and transplantation (stage 15; see Part I, #6 above).
To check the quality of the testes, we perform an early morning fertilization (~8 am), which allows us to extract testes from another male if the sperm from the first is not viable. Note: we homogenize ~0.25 testes in 1 mL 1X MMR and add approximately 5 to 7 drops per plate of oocytes.
In vitro fertilize the eggs with homogenized testes and, an hour later, remove the jelly coat on the eggs using de-jelly solution (164μl 1M DTT + 10mL 1M Tris, pH 8.8 up to 50 mL autoclaved nanopure water). Wait until they reach two-cell stage to begin sorting, which takes about 2 hours at 18°C or 1.5 hours at room temperature (22°C).
Using a transfer pipette, sort uniformly pigmented and evenly dividing embryos, as shown in the left animal cap in Figure 3, into the well of an injection dish filled with 0.4X MMR + 6% Ficoll.
B. Microinjection
Stretch Parafilm over the top of a 60 mm Petri dish lid. Pipette 2 μl of your first RNA sample onto the Parafilm and aspirate using the PLI-100 to fill your injection needle. Be sure to check that the needle is still dispensing 10 nL of sample before starting injection.
Inject 20-30 two-cell stage embryos (both blastomeres) with 500 pg YFP RNA in one dish and your gene of interest (e.g. 20 pg Noggin RNA) plus YFP RNA in another dish 7.
Place 100-200 non-injected embryos per time point in another injection dish with the Ficoll solution. Move them in and out of the incubator at the same time as the injected embryos. These are the hosts to be used in Part IV.
After injection, incubate all embryos at 14°C overnight. Repeat for all time points.
Part III. Isolate animal caps from injected embryos
Arrive early in the morning to stage the embryos. The earliest time point should be at stage 9. Remove those that did not survive. Proceed with the following steps working with each time point in batches.
Pour off the Ficoll solution from each plate and add back 0.7X MMR/gentamicin solution. To a cap isolation dish, add a generous amount of 0.7X MMR/gentamicin solution. Dislodge embryos from their injection dish wells using a transfer pipette and place in the freshly prepared cap isolation dish.
Isolate animal caps in 0.7X MMR + gentamicin solution, keeping 2 or 3 intact embryos for staging. Either the Gastromaster, fitted with a yellow tip bent to 400 μm, or a pair of sharp number 5 forceps can be used.
Put caps back in 14°C incubator until the next day. The host embryos should be removed from the incubator at the same time as the caps to keep them at the same developmental stage as the donor tissue.
Part IV. Transplantation of caps onto flank
Remove caps and embryos from the first time point from the 14°C incubator and stage the embryos. Sort for uniform yellow or red fluorescent-positive caps, depending on the tracer injected (Figure 2).
Count the number of caps and remove double the number of stage 12-13 embryos to a freshly prepared injection dish with 0.7X MMR/gentamicin solution. With sharp #5 forceps, remove the vitelline membrane from each. Transfer them to the plate with the caps, but be careful. Without their vitelline membranes, embryos can quickly dissociate at the water's surface.
Using the gastromaster (yellow tip, 200-250μm bent wire, highest setting) or #5 forceps, remove a 200 μm square on the side of the embryo at stage 14-15 (Figure 3). Then, cut a cap in half (with the gastromaster or the #5 forceps) and place the tissue in the hole made in the host embryo. Rotate the embryo against the well to allow healing. Do this for as many embryos as you can while they are at stage 15. Repeat for all time points.
The embryo will usually heal within 30 minutes. Remove the dead cells (white) with a gentle brushing motion with a #5 forceps. Grow them at 18°C overnight.
Under the dissecting fluorescence microscope, sort embryos with fluorescently-labeled, transplanted tissue. Put them back at 18°C to grow until stage 42-43.
Anesthetize the animals in tricaine. At this point, you can take pictures under a fluorescence dissecting microscope. After you are done, fix, section and immunostain or perform in situ hybridization using markers for your tissue of interest.
 Figure 1. Design of Plexiglass for casting the elastomer mold used for making embryo holding dishes. A 38 x 38 mm round-cornered square (~5 mm deep) has been routed out of a piece of Plexiglass that is 50 x 50 mm square and 25 mm thick. Circular, evenly spaced wells (1.5 mm diameter) have been drilled 1 mm deep into the plastic. To make the elastomer mold for the microinjection dishes, we poured Sylgard elastomer into the Plexiglass and allowed it to solidify, as per manufacturer instructions. This elastomer mold is used to make injection and cap isolation plates.
Figure 1. Design of Plexiglass for casting the elastomer mold used for making embryo holding dishes. A 38 x 38 mm round-cornered square (~5 mm deep) has been routed out of a piece of Plexiglass that is 50 x 50 mm square and 25 mm thick. Circular, evenly spaced wells (1.5 mm diameter) have been drilled 1 mm deep into the plastic. To make the elastomer mold for the microinjection dishes, we poured Sylgard elastomer into the Plexiglass and allowed it to solidify, as per manufacturer instructions. This elastomer mold is used to make injection and cap isolation plates.
 Figure 2. Animal cap sorting prior to transplantation is important, as expression can vary. Animal cap on the left expresses mCherry evenly throughout tissue while the explant on the right has spotty expression and will be discarded.
Figure 2. Animal cap sorting prior to transplantation is important, as expression can vary. Animal cap on the left expresses mCherry evenly throughout tissue while the explant on the right has spotty expression and will be discarded.
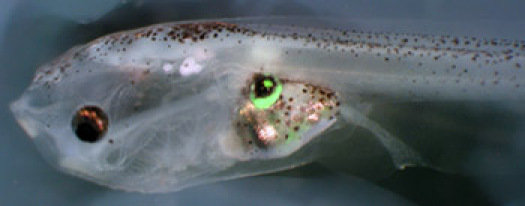 Figure 3. A host tadpole has developed an eye-like structure on its flank from transplanted animal cap cells. A fluorescent image has been overlayed on the brightfield image.
Figure 3. A host tadpole has developed an eye-like structure on its flank from transplanted animal cap cells. A fluorescent image has been overlayed on the brightfield image.
Discussion
In this video, we have demonstrated our version of classic techniques used by Xenopus biologists, which we rearranged to create the animal cap transplant assay. At room temperature, Xenopus embryos develop very quickly through stages 9 and 15. By placing them at 14°C, the development of the embryo is slowed down and many more transplants can be performed in one experiment. If it is necessary to grow the embryos to older stages (>42/43), then we recommend using transgenic venus YFP animals, since the injected fluorescent protein signal fades in older tadpoles. We used this assay to establish that animal cap tissue expressing EFTFs or Noggin is specified to form eye-like tissue. This assay could be used as a simple test to establish whether a gene(s) of interest is necessary for determining a particular cell fate.
Acknowledgments
This work was supported by grants from Research to Prevent Blindness (Career Development Awards to MEZ and ASV and an unrestricted grant to the Department of Ophthalmology), the E. Matilda Zeigler Foundation (MEZ and ASV) and the National Eye Institute/NIH (MEZ, grants R01EY015748 and R01EY017964).
References
- Smith WC, Harland RM. Expression cloning of noggin, a new dorsalizing factor localized to the Spemann organizer in Xenopus embryos. Cell. 1992;70:829–840. doi: 10.1016/0092-8674(92)90316-5. [DOI] [PubMed] [Google Scholar]
- Lamb TM. Neural induction by the secreted polypeptide noggin. Science. 1993;262:713–718. doi: 10.1126/science.8235591. [DOI] [PubMed] [Google Scholar]
- Green J. Methods in Developmental Biology. Towata, N.J: Humana Press; 1999. Molecular Methods in Developmental Biology: Xenopus & Zebrafish. [Google Scholar]
- Viczian AS, Solessio EC, Lyou Y, Zuber ME. Generation of functional eyes from pluripotent cells. PLoS Biol. 2009;7:e1000174–e1000174. doi: 10.1371/journal.pbio.1000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Brown AL, Johnson BE, Goodman MB. Making patch-pipettes and sharp electrodes with a programmable puller. J Vis Exp. 2008 doi: 10.3791/939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan L. Noggin Elicits Retinal Fate in Xenopus Animal Cap Embryonic Stem Cells. Stem Cells. 2009 doi: 10.1002/stem.167. [DOI] [PubMed] [Google Scholar]