

Abstract
Aim
To evaluate a standardised MRI acquisition protocol and a new image rating scale for disease severity in patients with progressive supranuclear palsy (PSP) and multiple systems atrophy (MSA) in a large multicentre study.
Methods
The MRI protocol consisted of two-dimensional sagittal and axial T1, axial PD, and axial and coronal T2 weighted acquisitions. The 32 item ordinal scale evaluated abnormalities within the basal ganglia and posterior fossa, blind to diagnosis. Among 760 patients in the study population (PSP=362, MSA=398), 627 had per protocol images (PSP=297, MSA=330). Intra-rater (n=60) and inter-rater (n=555) reliability were assessed through Cohen's statistic, and scale structure through principal component analysis (PCA) (n=441). Internal consistency and reliability were checked. Discriminant and predictive validity of extracted factors and total scores were tested for disease severity as per clinical diagnosis.
Results
Intra-rater and inter-rater reliability were acceptable for 25 (78%) of the items scored (≥0.41). PCA revealed four meaningful clusters of covarying parameters (factor (F) F1: brainstem and cerebellum; F2: midbrain; F3: putamen; F4: other basal ganglia) with good to excellent internal consistency (Cronbach α 0.75–0.93) and moderate to excellent reliability (intraclass coefficient: F1: 0.92; F2: 0.79; F3: 0.71; F4: 0.49). The total score significantly discriminated for disease severity or diagnosis; factorial scores differentially discriminated for disease severity according to diagnosis (PSP: F1–F2; MSA: F2–F3). The total score was significantly related to survival in PSP (p<0.0007) or MSA (p<0.0005), indicating good predictive validity.
Conclusions
The scale is suitable for use in the context of multicentre studies and can reliably and consistently measure MRI abnormalities in PSP and MSA.
Clinical Trial Registration Number
The study protocol was filed in the open clinical trial registry (http://www.clinicaltrials.gov) with ID No NCT00211224.
Progressive supranuclear palsy (PSP) and multiple system atrophy (MSA) represent the two most common causes of progressive neurodegenerative akinetic rigid, multisystem syndromes (‘Parkinson's plus syndromes’; PPS) after idiopathic Parkinson's disease (IPD).1 2 In the early stages, it can be difficult to differentiate PSP and MSA from IPD. Symptoms of PSP include oculomotor abnormalities, early falls, pyramidal symptoms and frontal lobe dysfunction.3 Patients with MSA exhibit autonomic failure, cerebellar and pyramidal involvement.4 5 For the majority of patients with PSP and MSA, the course of the disease is one of relentless progression, increasing disability and death, with a median survival of 5–10 years from onset of symptoms.4 6 7
The disease processes in MSA and PSP involve many brain areas but particularly the basal ganglia, brainstem and cerebellum.8–10 Although a number of MRI abnormalities corresponding to underlying pathological changes have been described in PSP and MSA,11–16 these have not been subject to a systematic assessment. Furthermore, existing studies have used small samples, limiting the conclusions that can be drawn for routine practice.17–19 Several studies have examined the usefulness of quantitative measurements of atrophy taken in specific regions of interest.11 17–21 However, these restricted measurements do not capture the full extent of abnormalities seen on MRI. In order to provide a validated framework for a systematic and semiquantitative approach to assessment of MRI abnormalities in large multicentre studies of PPS, and to provide an outcome measure of disease progression in clinical trials, we incorporated a prospective standardised collection of MRIs as an ancillary component of the Neuroprotection and Natural History in Parkinson's Plus Syndromes (NNIPPS) study.22 NNIPPS was designed to investigate the natural history of Parkinson's plus syndromes—PSP and MSA—as part of a double blind, placebo controlled, randomised, multicentre (n=44) trial in France, Germany and the UK.
In this paper, we present the standardised MRI acquisition protocol and validation of the NNIPPS MRI rating scale which was intended to measure disease severity and progression in the context of large multicentre randomised clinical trials.
Methods
Subjects
From April 2000 to July 2002, subjects were included in the trial according to NNIPPS diagnostic criteria, and followed-up for 3 years or until death, whichever came first.22 Demographic information and clinical scales were collected at entry and during the course of the study (table 1). Detailed information on trial design and results, including accuracy of diagnostic criteria, and clinical assessments, has been reported previously.22 Members of the NNIPPS Study Group are listed in Appendix 1.
Table 1.
Comparisons between MSA and PSP patients with MRI (Student's t test or Pearson χ2)
| PSP (n=297) | MSA (n=330) | All (n=627) | p Value | |
| Gender (%F) | 42 | 45 | 44 | 0.49 |
| Mean (SD) age (years) (40–81) | 67 (7) | 62 (8) | 64 (8) | <0.001 |
| Mean (SD) age at onset (years) (35–79) | 64 (7) | 57 (8) | 60 (8) | <0.001 |
| Mean (SD) disease duration (years) (1–8) | 3.9 (1.9) | 4.3 (1.9) | 4.1 (1.9) | 0.002 |
| Clinical Global Impression of severity (1–6) | ||||
| Mean (SD) | 3.6 (1.0) | 3.6 (0.9) | 3.6 (1.0) | 0.73 |
| Borderline/moderately ill (0–2) (%) | 14 | 10 | 12 | |
| Markedly ill (3–4) (%) | 67 | 73 | 70 | |
| Severely/extremely ill (5–6) (%) | 19 | 17 | 18 | |
| Modified Hoehn and Yahr (0–5) (%) | ||||
| No sign to mild bilateral disease (0–2) | 15 | 24 | 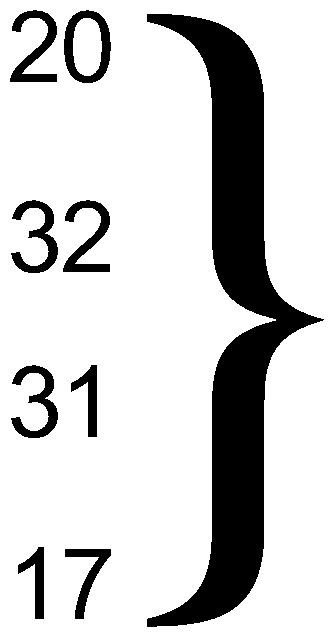 |
|
| Mild to moderate bilateral disease (3) | 36 | 29 | 0.02 | |
| Severe disability (4) | 30 | 32 | ||
| Wheelchair bound (5) | 19 | 15 | ||
| Mean (SD) Schwab and England activities of daily living scale (0–100%) | 50 (23) | 55 (24) | 53 (24) | 0.02 |
MSA, multiple systems atrophy; PSP, progressive supranuclear palsy.
Standardised MR image acquisition protocol
The main constraint in designing the acquisition protocol was to determine sequences that would accommodate the variability in scanner configuration according to centres, and that could be completed in 30 min, estimated as the maximum time these patients would tolerate the scanner. The Imaging Technical Committee determined, after initial testing and literature review, that the acquisitions would: (i) be done on >1 T magnets; (ii) include two-dimensional sagittal (at 5 mm slice thickness) and three-dimensional T1 acquisitions allowing reconstructions of axial images (at 5 mm slice thickness); and (iii) include axial PD as well as axial and coronal T2 (at 3 mm slice thickness). Axial slices were required to follow the bicallosal plane, while coronal acquisitions had to be orthogonal to that plane. The MRI protocol developed on a GE scanner (GE Medical, Milwaukee, USA) and adapted by site investigators for their particular configuration is described in table 2.
Table 2.
NNIPPS imaging protocol for Parkinson's plus syndromes
| Plane | Acquisition | Slice (mm) | Number slices | Film* | TR (ms) | TE (ms) | FOV (mm) | Matrix |
| Sagittal | FGE T1 weighted | 5 | 16 | 1 | 250–512 | 14–16 | 230–240 | 512×(224–256) |
| Axial bicallosal plane | FSE proton density | 3 | 40 | 2 | 5270–6000 | 12–20 | 230–240 | 256×(224–256) |
| Axial bicallosal plane | FSE T2 weighted | 3 | 40 | 2 | 5270–6000 | 75–110 | 230–240 | 256×(224–256) |
| Coronal orthogonal to the bicallosal plane | FSE T2 weighted | 3 | 40 | 2 | 4520–5200 | 96–110 | 230–240 | 512×(204–256) |
| Axial† | 3D IR T1 weighted | 0.9 | 160 | 2‡ | 2500 IT=500 | Minimum | 230–230 | 256×256 |
Printed films contain 20 images each.
The whole cerebrum, including the cerebellum and brainstem, should be included.
Reconstruction of 20 slices at 5 mm thickness in the bicallosal plane, centred on the basal ganglia.
3D, three-dimensional; FGE, fast gradient echo; FOV, field of view; FSE, fast spin echo; IR, inversion recovery; NNIPPS, Neuroprotection and Natural History in Parkinson's Plus Syndromes; TE, echo time; TR, repetition time.
At the time data acquisition began (2000), standardised DICOM format was not available in every centre and hence the use of printed films was the only practical option for centralised reading. The 160 images were printed by groups of 20 on 14×17 inch films, for a total of eight films per patient, with care taken to optimise contrast.
Image assessment
An image rating scale was developed in order to systematically and semiquantitatively evaluate MRI signs within the basal ganglia and posterior fossa (mesencephalon, pons and cerebellum), focussing on regions where both neuronal loss and gliosis have been well documented either in PSP8 10 or in MSA.23 24 Selection of items to be scored was based on a literature review of MRI abnormalities.11–16 In addition, based on neuropathology findings and background clinical and radiological experience, new undocumented items were added, including hyperintensity within the ventral area of the globus pallidus (table 3, items 12 and 29), and the area between the red nucleus and substantia nigra (table 3, item 15), as well as punctate upper mesencephalic hyperintensities (table 3, items 18 and 31).
Table 3.
NNIPPS MRI scale: inter-rater and intra-rater reliability
| Image | Measurement | Inter-rater (n=555) | Intra-rater (n=60) |
| Sagittal T1 | 1. Pontine atrophy | 0.59 | 0.80 |
| 2. Cerebellar atrophy | 0.56 | 0.78 | |
| 3. Fourth ventricle enlargement | 0.59 | 0.66 | |
| 4. Midbrain atrophy | 0.60 | 0.62 | |
| 5. Aqueduct of Sylvius enlargement | 0.53 | 0.63 | |
| Axial PD | 6. Ponto-cerebellar atrophy (Cross sign) | 0.70 | 0.88 |
| 7. Cerebellar peduncles hyperintensities | 0.64 | 0.94 | |
| Axial T2 | 8. Putamen marginal lateral rim | 0.52 | 0.67 |
| 9. Lateralisation of item 8 | 0.27* | 0.72* | |
| 10. Putamen marginal postero-medial rim | 0.29 | 0.68 | |
| 11. Hypointense posterior putamen | 0.42 | 0.41 | |
| 12. Hyperintense internal pallidum ventral area | 0.63 | 0.79 | |
| 13. Hypointense red nuclei | 0.37 | 0.61 | |
| 14. Hypointense substantia nigra | 0.30 | 0.55 | |
| 15. Hyperintensity between red nucleus and substantia nigra | 0.26 | 0.52 | |
| 16. Aqueduct of Sylvius enlargement | 0.51 | 0.60 | |
| 17. Periaqueductal hyperintensity | 0.45 | 0.41 | |
| 18. Punctate mesencephalic hyperintensities | 0.47 | 0.66 | |
| 19. Increased interpeduncular angle | 0.49 | 0.62 | |
| 20. Ponto-cerebellar atrophy (Cross sign) | 0.80 | 0.72 | |
| 21. Cerebellar peduncles hyperintensities | 0.65 | 0.81 | |
| 22. Middle cerebellar peduncles atrophy | 0.60 | 0.72 | |
| 23. Hypointense dentate nuclei | 0.63 | 0.72 | |
| 24. Fourth ventricle enlargement | 0.64 | 0.71 | |
| 25. Hyperintense base of the pons | 0.35 | 0.76 | |
| 26. Peripheral patches | 0.60 | 0.69 | |
| Coronal T2 | 27. Putamen marginal lateral rim | 0.45 | 0.75 |
| 28. Putamen marginal inferior rim | 0.17 | 0.38 | |
| 29. Hyperintense internal pallidum ventral area | 0.64 | 0.78 | |
| 30. Third ventricle enlargement | 0.58 | 0.69 | |
| 31. Punctate upper mesencephalic hyperintensities | 0.48 | 0.73 | |
| Axial T1 | 32. Putamen marginal lateral rim | 0.60 | 0.83 |
Values in cells are weighted kappa statistics except for (*) which is simple κ.
ICC, intraclass coefficient; NNIPPS, Neuroprotection and Natural History in Parkinson's Plus Syndromes; PD, proton density.
The semiquantitative scale was defined by expert consensus and included 32 parameters (table 3) with scores ranging from 0 (normal) to 3 (most severe) and one item for lateralisation with categorical rating (item 9: 1=R>L, 2=L>R). For all items, a score of 4 was given when the image was not interpretable and a score of 9 when the image was missing. For structures well seen in orthogonal planes (ie, pons, IVth ventricle, cerebellar peduncles, mesencephalon, aqueduct of Sylvius, putamen and internal globus pallidus), redundant reporting was achieved.
The scale was tested and standard operating procedures (SOPs) defined on an initial series of images from the first 72 patients included in France. The scale was thereafter presented to all raters in a training session, together with SOPs, and an MRI atlas was built for scoring guidance (see supplementary material available online only: NNIPPS-MRI atlas for scoring).
In each country, centralised double reading of each MRI scan was performed blind to clinical diagnosis, by independent experts. In case of disagreement between the two ratings on any item (ie, scoring difference greater than 1), images were re-evaluated by both raters until consensus was reached.
Statistical analysis
For each image series, ratings of the 32 items by individual raters were recorded. All statistical analyses were performed using SAS software V.11. Inter-rater reliability was assessed using a simple κ coefficient for the binary parameter and a linear weighted version of Cohen's κ statistics for ordinal data.25 26 Intra-rater reliability and training effect was assessed using a weighted version of Cohen's κ statistics to compare scale measures on the first 30 patients and the last 30 patients included in France, rated twice at 1 year intervals. Scale redundancy was checked using between item correlation with Spearman rank coefficient.
Extraction of principal components (PCA) with varimax rotation was performed on the scale using the consensus ratings and excluding the categorical item 9, or the highly correlated ones, to prevent overloading of signs, and using data from 441 patients with complete ratings for all parameters. Dimensional factorial scores were calculated by summing items correlated to the factor. Internal consistency of extracted components was explored with Cronbach's α coefficient and inter-rater reliability of factor scores with intraclass coefficient (ICC).27 Dimensional scores were calculated by summing items correlated to the factor. Discriminant validity was checked comparing factor scores and overall scores between (i) extreme groups of the Clinician Global Impression of disease severity (CGI-ds, score 1–2 (borderline–mild) vs score 5–6 (severe–extremely severe)) with two-way ANOVA, including interaction, and (ii) diagnostic strata by Student's t test. For each strata, sensitivity to change in disease severity from borderline–mild to severe–extremely severe was summarised through Cohen's d effect size coefficient (ES) for each factor and total score.28
Inter-rater and intra-rater reliability coefficients were interpreted according to proposed standards for strength of agreement as: ≤0=poor, 0.01–0.20=slight, 0.21–0.40=fair, 0.41–0.60=moderate, 0.61–0.80=substantial and 0.81–1.0=almost perfect.29 30 Individual item strength of agreement was considered as acceptable for >0.40 (moderate to almost perfect); for factorial score combining items, ICC threshold for acceptability was raised to 0.70. Internal consistency of the factorial scores were considered as acceptable for Cronbach >0.70. ES coefficients were interpreted as published: <0.5=small, 0.5–0.79=medium and ≥0.8=large.28
For predictive validity, relation between factorial or total MRI scale scores at inclusion and survival over the 3 year follow-up was evaluated using univariate and multivariate Cox model analysis.31
Results
Demographics and clinical tests
A total of 760 patients were included in the intent to treat analysis.22 MRI could not be performed in 133 patients. Within the study centres, images were obtained using MRI scanners from three manufacturers (GE Medical, Milwaukee, USA; Siemens Medical Systems, Erlangen, Germany; and Philips Medical, Best, The Netherlands) with field strengths between 1 and 1.5 T. Images as per protocol were collected at entry for 627 patients (83% of total), including 330 patients (53%) with MSA and 297 patients (47%) with PSP (see supplementary figure 1 available online only). Reasons for missing MRI included: contraindication to MRI scanning; technical difficulties in scanning patients due to advanced disease stage; lack of MRI facilities (three centres) or images not performed according to the NNIPPS acquisition protocol, as specified in table 2.
For the development of SOPs, the first 72 of the 296 French acquisitions were used which were subsequently found to include 33 (46%) PSP and 39 (54%) MSA cases. These scans were excluded from subsequent inter-rater reliability analyses performed on the remaining 555 scans (PSP=264, MSA=291).
Comparison of patients with MRI to those without showed that the sample with MRI as a whole was slightly less severely affected with a significant difference on Hoehn and Yahr staging (p=0.02). Within the sample with MRI (table 1), PSP patients were older at inclusion and at disease onset and had shorter duration of disease than MSA patients (p<0.002). Regarding the Hoehn and Yahr grade and Schwab and England activity scale, PSP patients were significantly more severe (p<0.04 and p<0.005, respectively) than MSA patients. Overall, 48% of the patient population with MRI were classified in the most severe grades (severe disability or wheelchair bound) of the Hoehn and Yahr staging.
Histogram results
All patients' MRIs displayed abnormalities that could be reliably assessed on the scale. Histogram plots for each scale measure showed that most measurements could be performed on all images, with <7% of the 627 patient images scored as not interpretable due to poor quality and 31 items with <2% missing images. One item, ‘putamen marginal lateral rim’ assessment in axial T1, could not be assessed because of missing images in 10% of cases. Significant signs (scores 2 and 3) with frequency >10% were present for most items, including known signs (eg, ‘cross sign’) and previously undocumented ones (eg, ‘punctate mesencephalic hyperintensities’) (figure 1).
Figure 1.

Distribution of scores (% of overall population) for selected a priori redundant measurements of known (A–B) and new signs (C–D). NA, not assessed due to poor quality of image. ND, not determined due to missing images. (A, B) Ponto-cerebellar atrophy (A) in axial (Ax) proton density (PD) (item 6) and (B) in Ax T2 (item 20), showing similar distribution although better sensitivity of the Ax PD sequence. (C, D) Punctate upper mesencephalic hyperintensities, (C) in Ax T2 (item 18) and (D) in coronal T2 (item 31), showing similar distribution and sensitivity.
Reliability analysis
Among the 32 items, 25 (78%) and 31 (97%) had acceptable inter-rater and intra-rater agreement, respectively (table 3). Intra-rater reliability was almost perfect for four items (≥0.81), substantial for 22 (0.61≤<0.81), moderate for five (0.41 ≤<0.61) and fair for one (=0.38). Inter-rater reliability was substantial for nine (0.61≤<0.81), moderate for 16 (0.41≤<0.61), fair for six (0.21≤<0.41) and slight for one (=0.17).
Item redundancy
Among the seven anatomical regions that were imaged on two separate planes and/or T1/T2 weighting, repeated scorings showed a high correlation (ρ>0.7) in four, indicating that these assessments were redundant (IVth ventricle, items 3 and 24; cerebellar peduncles, items 7 and 21; internal globus pallidus, items 12 and 29; mesencephalon, items 18 and 31) while three repeated scorings (pons, items 6 and 20; aqueduct of Sylvius, items 5 and 16; putamen, items 8 and 27) were only moderately correlated (0.5<ρ<0.7) (see supplementary table 1 available online only), indicating that each plane/weighting assessment of these regions might visualise separate abnormalities. In order to avoid bias from overemphasis of a particular sign (ie, to minimise overloading of signs in the scale score), redundant items 7, 12, 18 and 24 were deleted from subsequent analysis.
Principal component analysis
PCA (table 4; supplementary table 2 available online only) revealed four factors accounting for 50.5% of the total variance and corresponding to distinct anatomical regions: F1 related to the posterior fossa (see supplementary figure 2 available online only); F2 related to the midbrain and third ventricle (see supplementary figure 3 available online only); F3 related to the lateral putamen; and F4 related to the posterior putamen, substantia nigra and red nuclei (see supplementary figure 4 available online only). The remaining items were either not correlated to any factors (items 29 and 31) or clustered in a factor not found clinically or anatomically meaningful (F5: items 19, 25 and 26; 8.2% of the total variance).
Table 4.
Principal component analysis and reliability of factorial scores
| Factors (items in factor) | Anatomical dimension | Variance (% explained) | Consistency (Cronbach α) | Reliability (ICC) |
| F1 (1–3, 6, 20–23) | Brainstem and cerebellum | 21.0 | 0.93 | 0.92 |
| F2 (4–5,16–17, 30) | Midbrain | 10.2 | 0.75 | 0.79 |
| F3 (8,10, 27–28, 32) | Putamen | 9.9 | 0.75 | 0.71 |
| F4 (11, 13–15) | Basal ganglia (other) | 9.4 | 0.90 | 0.49 |
| F5 (19, 25–26) | Miscellaneous | 8.2 | 0.48 | 0.76 |
ICC, intraclass coefficient.
The first four meaningful factors had acceptable internal consistency (Cronbach α 0.75–0.93); the first three factors showed acceptable reliability (ICC 0.71–0.92) while the fourth was only moderate (ICC=0.49) (table 4).
Discriminant and predictive validity
In the overall population, extreme subgroups of disease severity (CGI-ds borderline–mild vs severe–extremely severe) showed significant differences on F2 (p<0.001) and the total score (p<0.01). A significant interaction was found for F3; borderline patients did not differ between MSA and PSP, while in the severe group, MSA patients displayed higher scores than PSP patients (figure 2). In the PSP group, MRI scale sensitivity to change in CGI-ds showed that F2 was the most discriminant score (ES=0.93) followed by the total score (ES=0.56), with the remaining scores having small ES values (ES 0.20–0.44). In the MSA group, F3 was the most discriminant score (ES=0.85) followed by the total score (ES=0.62), with the remaining scores having small ES values (ES 0.09–0.48).
Figure 2.

Comparison of factorial and total scores according to diagnosis at entry (A) and according to Clinician Global Impression (CGI) of disease severity (progressive supranuclear palsy (PSP)—(B); multiple systems atrophy (MSA)—(C)). Figures within bars are number of patients in each group. CGI disease severity score1–2=borderline–mild impairment, score 5–6=severe–extremely severe impairment. F1, brainstem and cerebellum; F2, midbrain; F3, putamen; F4, other basal ganglia—posterior putamen, substantia nigra, red nuclei. *p<0.05; **p<0.01; ****p<0.0001.
Overall comparisons between PSP and MSA on the factorial and total scores showed that all scores were significantly different, with three factors scoring significantly higher in the MSA group (F1, F3 and F4; p<0.0001) and one higher in the PSP group (F2; p<0.0001) (figure 2). ES for diagnosis ranked F1 as the most discriminant factor (ES=−1.02) followed by F2 (ES= 0.79), F4 (ES=−0.49) and F3 (ES=−0.46). On the total score, MSA patients rated significantly higher than PSP patients (ES=−0.78).
Among the 627 patients with usable MRI, 279 (44.5%) died during the 3 year follow-up (PSP 46%; MSA 43%). Predictive validity analysis using univariate Cox model analysis showed that the total score was significantly and linearly related to survival in the overall population (RR (95% CI) 1.036 (1.019 to 1.053), p<0.0001) and in PSP (RR (95% CI) 1.068 (1.028 to 1.108), p<0.0007) or MSA (RR (95% CI) 1.037 (1.016 to 1.059), p<0.0005).
Among the four-dimensional subscores, multivariate analysis in PSP showed F2 as the only predictive subscore (RR (95% CI) 1.154 (1.072 to 1.243), p<0.0002); in MSA, both F3 (RR (95% CI) 1.106 (1.045 to 1.171), p=0.0005) and F2 (RR (95% CI) 1.091 (1.008 to 1.181), p=0.031) were found to be significantly and independently related to survival.
Discussion
The main aims of this study were to establish the feasibility of acquiring standard imaging data in a large multicentre study of MSA and PSP; to show that standard imaging data can be summarised using a semiquantitative rating scale; and to assess the metric qualities of this scale in terms of construct validity and reliability, as mandatory preliminary steps for using the scale as an outcome measure in clinical trials.
In support of these goals, we have presented a standardised MRI acquisition protocol and a set of image rating criteria to evaluate brain lesions in MSA and PSP patients within the context of a large prospective multicentre study. The protocol was sufficiently universal to accommodate the heterogeneity of data from the many participating centres, with acquisition time compatible with routine clinical use, even in patients with advanced disease. Image assessments performed on all usable scans for reliability showed that 78% of the 32 items had acceptable agreement for intra-rater and inter-rater reliability. The scale was able to measure known abnormalities as well as other previously undocumented signs. PCA revealed that 22 out of the 32 criteria proposed could be grouped into four meaningful factors, excluding four items with high redundancy and five with unassigned significance.
Standard image rating scale
Assessment time was deemed acceptable by all raters, and analysis showed that signs were usable, measurable and reproducible. Abnormalities were noted on every image series. As expected, redundant signs were evident and to prevent overloading of signs, we deleted four highly correlated redundant items from the final scale. Other parameters rating the same signs in different sequences (ie, enlargement of the aqueduct, marginal putamen lateral and inferior rim, cerebellar atrophy, cross sign) were less correlated. Further analyses, including sensitivity to change, will help determine whether further scale reduction is appropriate. Neuropathology is still in progress and the findings will be used to confirm whether these signs assess separate abnormalities.
There were seven signs with low inter-rater reliability (κ values <0.4). These were based on signal intensity, taking a region of white matter as reference, and thus dependent on printing technique which is a potential source of variability. DICOM format, now routinely available, will allow improved standardisation and therefore reliability of such data in future studies. In addition, the signal intensity of midbrain nuclei in T2 weighted images may not have been optimal for assessing a small tilted midbrain structure such as the substantia nigra.32 The availability of devices at 3 T (or higher) have indicated that basal ganglia signs such as putaminal hyperintense rims on T2 weighted images can be influenced by field strength33 and that high field devices may be oversensitive in this context.34 It is certainly possible that some additional abnormalities may be detected by applying 3 T or higher field strengths but no reliable data yet exist indicating the degree of increased sensitivity or specificity for these purposes. Overall, improvements in reliability provided by the newer technologies should improve the performance of our scale.
Factorial clusters
The factorial clusters extracted by component analysis are consistent with the pathological literature.8–10 23 24 The first cluster (F1) consists of posterior fossa abnormalities (mainly pontine atrophy), enlargement of the fourth ventricle, hyperintensity within the cerebellar peduncles and cerebellar atrophy. These changes are consistent within image series (eg, enlargement of the fourth ventricle) and reflect degeneration of the ponto-cerebellar pathways.
The second cluster (F2) centres on mesencephalic atrophy and hypersignals associated with enlargement of the aqueduct and periaqueductal hypersignals. These coexist with enlargement of the third ventricle. These abnormalities are consistent with degenerative processes involving the dentatorubrothalamic pathway and the periaqueductal grey matter in both disorders, as indicated in figure 2. Further insights on the pathophysiological relevance of these findings will depend on analyses of the longitudinal imaging data, with detailed clinical and pathological correlations.
Cluster F3 is composed of marginal putamen hypersignals that are seen in both axial and coronal planes, while cluster F4 combines signs related to posterior hypointensities in the putamen, red nucleus and substantia nigra.
Overall, three of these four factors (F1–3) showed good reliability and internal consistency. F4 was highly consistent (Cronbach 0.90) but combined four items with only fair to moderate reliability, yielding to an overall moderate ICC, indicating a need for improved procedures, including acquisition, display media and/or readings.
It is important to note that we did not set out to test the diagnostic sensitivity and specificity of the NNIPPS MRI scale, so we cannot draw conclusions on the diagnostic usefulness of this scale. Thus the next step is to test the scale prospectively across a range of degenerative conditions, including IPD, PSP, MSA and other multisystem disorders. Furthermore, when the study was planned, MRI sequences such as fluid attenuated inversion recovery and diffusion weighted imaging, which might in theory contribute to diagnostic sensitivity, were not routinely available in the majority of centres. In addition, our protocol was designed to minimise factors (such as duration of scanning time) that might exclude more disabled patients and thus limit the general relevance of our results.
In the present study, the ES of the MRI scores for comparing severity stages were lower than those of standard clinical scales, such as the Schwab and England Activity of Daily Living or the Unified Parkinson's Disease rating Scale (data not shown). These results are not surprising given the high correlation between these clinical scales with the CGI-ds, all of which assess function. Analysis of sensitivity to change with time should provide a better and more relevant estimate of the MRI scale responsiveness. Nonetheless, our results support the fact that this MRI scale as it stands can be used to measure severity and progression in PSP in as much as in MSA, as shown by its good discrimination of severity stages within relevant brain structures, and significant prediction of survival.
Given the limited understanding of imaging changes that correlate with disease severity or progression, some bias is likely in our choice of items since we could not include items about which no information was available. Both clinical and histopathological analysis will help to refine the signs and clusters of signs that are most relevant for assessing disease severity in MSA and PSP, with the possibility that further redundant or non-discriminative signs can be removed. New pathological and imaging studies, including analysis of the NNIPPS longitudinal MRI data, will help to identify imaging changes of potential importance to inform future modifications and revisions of our scale.
With these limitations in mind, we believe that the MRI scale assessment of disease severity has important properties for randomised clinical trials that cannot be met by standard functional assessments since (i) it is a more robust end point with less liability to unblinding and (ii) quantification of neurodegeneration per se provides support for discriminating between symptomatic and neuroprotective therapies, an issue that confounds the interpretation of trials of putative disease modifying therapies in many neurodegenerative diseases.35
In summary, we have presented a standardised imaging protocol and image rating scale for quantifying neurodegeneration in patients with Parkinson's plus syndromes. We conclude that the NNIPPS MRI scale can reliably and consistently measure MRI abnormalities in PSP and MSA, within the context of a large multicentre trial.
Acknowledgments
The authors thank the patients and their families for their commitment and altruism, and the French and UK PSP Associations and the UK Parkinson's Disease Research Group for their help and support. The authors are grateful to the many colleagues who were not formally part of the NNIPPS consortium but whose support contributed to the success of the study.
Appendix 1. NNIPPS Study Group
Coordination
European and UK: PN Leigh (London, UK). France: G Bensimon (Paris, France). Germany: AC Ludolph (Ulm, Germany).
Steering Committee
Chair: PN Leigh (London, UK). Members: Y Agid, G Bensimon, M Dib, L Lacomblez, M Vidailhet (Paris, France), D Burn (Newcastle, UK), B Landwehrmeyer, AC Ludolph (Ulm, Germany).
Independent Data Monitoring and Safety Committee
Chair: B Asselain (Paris, France). Members: H Allain (Rennes, France), D Chadwick (Liverpool, UK), JE Perret (Grenoble, France), C Warlow (Glasgow, UK).
Technical committees
Clinical diagnostic criteria
Chair: D Burn (Newcastle, UK). Members: Y Ben-Shlomo (Bristol, UK), AM Bonnet, J Fermanian, C Payan, M Verny, M Vidailhet (Paris, France), P Moore (Liverpool, UK), C Tranchant (Strasbourg, France).
Motor function, quality of life and health service research
Chair: C Payan (Paris, France). Members: M Borg (Nice, France), P McCrone (London, UK), F Durif (Clermont-Ferrand, France), A Evans (London, UK), J Fermanian (Paris, France), F Viallet (Aix en Provence, France).
Neuroimaging
Chair: M Verin (Rennes, France). Members: N Deasy, J Jarosz (London, UK), TK Hauser (Tübingen, Germany), E Kraft (Ulm, Germany), E Broussolle (Lyon, France), D Dormont, C Marsault, A Tourbah, (Paris, France), L Defebvre, L Delmaire (Lille, France), Y Roland (Rennes, France).
Neuropathology
Chair: JJ Hauw (Paris, France). Members: C Duyckaerts, D Seilhean (Paris, France), S Al-Sarraj, T Revesz (London, UK), B Landwehrmeyer (Ulm, Germany), HA Kretzschmar (Munich, Germany).
Logistics, monitoring, data management and statistical analysis
Chair: G Bensimon (Paris, France). European Project Manager: M Graf (Paris, France). Data Manager: C Payan (Paris, France). Data entry: P Paillasseur (Theriamis-St Maur des Fossés, France). Senior Statistician: C Payan (Paris France). Assistant Statistician: HP Pham (Paris, France). Functional scales development: J Fermanian (Paris, France). Neuropsychology: R Brown (London, UK). Health economics: P McCrone (London, UK). Clinical research assistants: N Dedise, C Hermine, S Sagnes, B Poître, C Foucart (Paris, France), A Dougherty, C Murphy, H Mason (London, UK), T Hermann, K Klempp, A Niess, V Stange (Ulm, Germany). Regulatory affairs France: A Ouslimani (Paris, France).
Principal investigators (France/Germany/UK)
Y Agid (Paris), A Ludolph (Ulm), PN Leigh (London).
Investigators within countries
Centres, principal investigators, co-investigators (clinician/radiologist)
France. Aix en Provence: F Viallet, C Couratier, S Arguillère (clinicians), H Payan-Cassin, GM Vassault (radiologists); Angers: F Dubas, C Fressinaud (clinicians), JY Tanguy (radiologist); Besançon: L Rumbach, E Vidry (clinicians), J Kraehenbuhl, JF Bonneville (radiologists); Caen: F LeDoze, G Defer, F Viader (clinicians), H Huet (radiologist); Clermont-Ferrand: F Durif, B Debilly, Ph Derost, C Tilignac (clinicians), J Gabrillargues (radiologist); Grenoble: G Besson, C Mallaret (clinicians), S Grand (radiologist); Lille: A Destée, L Defebvre (clinicians), C Delmaire (radiologist); Limoges: P Couratier (clinician), MP Boncoeur-Martel (radiologist); Lyon: E Broussolle, H Mollion (clinicians), M Hermier (radiologist); Marseille: JP Azulay, T Witjas (clinicians) (radiologist cf Aix en Provence); Montpellier: W Camu, F Portet, J Khoris, N Pageot, G Garrigues (clinicians), B Viaud (radiologist); Nice: M Borg (clinician), S Chanalet (radiologist); Paris patient clinical selection: M Vidailhet, S Sangla (Hôpital St Antoine), D Ranoux (Hôpital St Anne), JP Brandel (Hôpital Leopold Belland), T De Broucker (Hôpital St Denis), Y Agid, B Dubois, V Meininger, M Verny (Hopital Pitié-Salpêtrière), P Cesaro (Hôpital Henri Mondor), G Fenelon (Hôpital Tenon); Paris CIC Pitié-Salpêtrière inclusion and follow-up: Y Agid, F Bloch, AM Bonnet, L Lacomblez, D Maltête, A Memin, FT Tornyi, ML Welter, J Worbe (clinicians), T Lalam, A Tourbah, C Marsault, Pr D Dormont (radiologists); Poitiers: R Gil, M Bailbé, S Venisse, H Moumy, V Mesnage, JL Houeto, F Petit (clinicians), P Vandermarcq (radiologist); Rennes: M Verin (clinician), Y Rolland (radiologist); Strasbourg: C Tranchant, G Steinmetz (clinicians), JL Dietemann (radiologist); Toulouse: O Rascol, M Galitzky, C Thalamas (clinicians), P Manelfe (radiologist); Tours: C Prunier, A Autret, P Corcsia (clinicians), P Cottier, S Gallas (radiologists).
Germany. Aachen: J Noth, C Kosinski, C Geyer, M Kronenbürger, C Schlangen (clinicians), M Doenges, S Kémeny, (radiologists); Berlin: K Einhaeupl, PDG Arnold, B Hauptmann, A Lipp (clinicians), A Villringer, A Lipp (radiologists); Bochum: H Przuntek, T Müller, G Gagel-Schweibold, M Siepmann, S Benz (clinicians), G Schmid (radiologist); Dresden: H Reichmann, B Herting (clinicians), R von Kummer, D Mucha (radiologists); Freiburg: CH Lücking, I Bötefür, S Braune, C Magerkurth, V Mylius (clinicians), M Schumacher, J Spreer, S Ziyeh (radiologists); Halle: S Zierz, M Kornhuber, T Mueller, S Neudecker, U Seifert (clinicians), C Behrmann, A Schlueter (radiologists); Hannover: R Dengler, A Hauswedell, H Kolbe, T Peschel, C Schrader, S Siggelkow, J Stewen, H-H Kapels, C Winkler (clinicians), H Heinze, G Kauffmann, M Rotte (radiologists); Magdeburg: CW Wallesch, C Bartels, M Fork (clinicians), S Reissberg (radiologist); München: T Brandt, F Asmus, M Bauer, T Gasser, S Maass, J Velden, A Viehöver, D Wassilowsky, K Bötzel (clinicians), T Youssri, T Wesemann, H Brückmann, R Brüning (radiologists); Regensburg: U Bogdahn, J Klucken, Z Kohl, M Lange, C Thun, J Winkler, B Winner (clinicians), G Schuierer (radiologist); Rostock: R Benecke, D Dressler, A Wolters, G Zegowitz (clinicians), G Grau (radiologist); Tübingen: J Dichgans, O Eberhardt, K Gröschel, TK Hauser, JB Schulz (clinicians), M Skalej, TK Hauser (radiologists); Ulm: AC Ludolph, D Ecker, A Jung, B Kramer, GB Landwehrmeyer, A Storch, SD Sussmuth (clinicians), E Kraft (radiologists).
UK. Belfast: M Gibson, R Forbes (clinicians), C Reynolds, CS McKinstry (radiologists); Birmingham—City Hospital: C Clarke (clinician), S Chavda (radiologist); Birmingham—Queen Elizabeth Hospital: H Pall, D Nicholls (clinicians), S Chavda (radiologist); Cambridge: J Hodges T Bak (clinicians), A Carpenter (radiologist); Liverpool: P Moore (clinician), T Dixon (radiologist); London IOP and GKT: PN Leigh, KR Chaudhuri, D Heaney, C Blain, S Azam, V Williams, J Isaacs, C Smallman, B Stanton (clinicians), J Jarosz, N Deasey (radiologists); London NHNN and Queen Square Hospital: A Lees, N Quinn, A Evans, T Scaravilli, N Russo, E Trikouli, D Paviour, L Massey (clinicians), C Andrews, J Stevens (radiologists); Middlesborough: P Newman, D Bathgate (clinicians), N Bradey (radiologist); Newcastle upon Tyne: D Burn, A Zermansky, N Warren (clinicians), P English, A Gholkar (radiologists); Stafford: B Summers (clinician), D Steventon (radiologist); Aberdeen: C Counsell (clinician), A Murray (radiologist).
Footnotes
Funding: NNIPPS was an academic led study with core funding from the European Union 5th Framework programme (QLG1-CT-2000-01262); support from the French Health Ministry, Programme Hospitalier de Recherche Clinique (AOM97073, AOM01125) and from Sanofi-Aventis affiliates in the UK, France and Germany, providing an unconditional research grant and drug supply throughout the study. Three academic institutions (Institute of Psychiatry, King's College London; Assistance Publique-Hôpitaux de Paris; and University of Ulm) were sponsors of the study in each country, and jointly own the data. The authors did not receive any financial contribution from these funding sources. The funding sources had no involvement in the study design or data collection, subsequent analysis and interpretation of data, writing of the report or in the decision to submit the paper for publication. SD received support from the Fonds pour la Recherche en Santé du Québec.
Competing interests: None.
Ethics approval: The protocol and amendments were reviewed and approved by the Comité de Protection des Personnes of Pitié-Salpêtrière Hospital (France), the UK Multicentre Research Ethics Committee (MREC) (UK), Ethikkommission of the University of Ulm (Germany) and by local institutional review boards (ethics committees) where appropriate (UK, Germany).
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.de Rijk MC, Tzourio C, Breteler MM, et al. Prevalence of parkinsonism and Parkinson's disease in Europe: the EUROPARKINSON Collaborative Study. European Community Concerted Action on the Epidemiology of Parkinson's disease. J Neurol Neurosurg Psychiatry 1997;62:10–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet 1999;354:1771–5 [DOI] [PubMed] [Google Scholar]
- 3.Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele–Richardson–Olszewski syndrome): report of the NINDS-SPSP International Workshop. Neurology 1996;47:1–9 [DOI] [PubMed] [Google Scholar]
- 4.Wenning GK, Ben SY, Magalhaes M, et al. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain 1994;117:835–45 [DOI] [PubMed] [Google Scholar]
- 5.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670s–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-Shlomo Y, Wenning GK, Tison F, et al. Survival of patients with pathologically proven multiple system atrophy: a meta-analysis. Neurology 1997;48:384–93 [DOI] [PubMed] [Google Scholar]
- 7.Golbe LI, Ohman-Strickland PA. A clinical rating scale for progressive supranuclear palsy. Brain 2007;130:1552–65 [DOI] [PubMed] [Google Scholar]
- 8.Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964;10:333–59 [DOI] [PubMed] [Google Scholar]
- 9.Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele–Richardson–Olszewski syndrome (progressive supranuclear palsy). Neurology 1994;44:2015–19 [DOI] [PubMed] [Google Scholar]
- 10.Jin C, Katayama S, Hiji M, et al. Relationship between neuronal loss and tangle formation in neurons and oligodendroglia in progressive supranuclear palsy. Neuropathology 2006;26:50–6 [DOI] [PubMed] [Google Scholar]
- 11.Quattrone A, Nicoletti G, Messina D, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology 2008;246:214–21 [DOI] [PubMed] [Google Scholar]
- 12.Savoiardo M, Girotti F, Strada L, et al. Magnetic resonance imaging in progressive supranuclear palsy and other parkinsonian disorders. J Neural Transm Suppl 1994;42:93–110 [DOI] [PubMed] [Google Scholar]
- 13.Schrag A, Good CD, Miszkiel K, et al. Differentiation of atypical parkinsonian syndromes with routine MRI. Neurology 2000;54:697–702 [DOI] [PubMed] [Google Scholar]
- 14.Stern MB, Braffman BH, Skolnick BE, et al. Magnetic resonance imaging in Parkinson's disease and parkinsonian syndromes. Neurology 1989;39:1524–6 [DOI] [PubMed] [Google Scholar]
- 15.Warmuth-Metz M, Naumann M, Csoti I, et al. Measurement of the midbrain diameter on routine magnetic resonance imaging: a simple and accurate method of differentiating between Parkinson disease and progressive supranuclear palsy. Arch Neurol 2001;58:1076–9 [DOI] [PubMed] [Google Scholar]
- 16.Yagishita A, Oda M. Progressive supranuclear palsy: MRI and pathological findings. Neuroradiology 1996;38:S60–6 [DOI] [PubMed] [Google Scholar]
- 17.Schocke MF, Seppi K, Esterhammer R, et al. Trace of diffusion tensor differentiates the Parkinson variant of multiple system atrophy and Parkinson's disease. Neuroimage 2004;21:1443–51 [DOI] [PubMed] [Google Scholar]
- 18.Seppi K, Schocke MF, Mair KJ, et al. Progression of putaminal degeneration in multiple system atrophy: a serial diffusion MR study. Neuroimage 2006;31:240–5 [DOI] [PubMed] [Google Scholar]
- 19.Shiga K, Yamada K, Yoshikawa K, et al. Local tissue anisotropy decreases in cerebellopetal fibers and pyramidal tract in multiple system atrophy. J Neurol 2005;252:589–96 [DOI] [PubMed] [Google Scholar]
- 20.Nicoletti G, Fera F, Condino F, et al. MR imaging of middle cerebellar peduncle width: differentiation of multiple system atrophy from Parkinson disease. Radiology 2006;239:825–30 [DOI] [PubMed] [Google Scholar]
- 21.Kato N, Arai K, Hattori T. Study of the rostral midbrain atrophy in progressive supranuclear palsy. J Neurol Sci 2003;210:57–60 [DOI] [PubMed] [Google Scholar]
- 22.Bensimon G, Ludolph A, Agid Y, et al. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: The NNIPPS Study. Brain 2009;132:156–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lantos PL, Papp MI. Cellular pathology of multiple system atrophy: a review. J Neurol Neurosurg Psychiatry 1994;57:129–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci 1989;94:79–100 [DOI] [PubMed] [Google Scholar]
- 25.Cohen J. A coefficient of agreement for nominal scales. Educ Psychol Meas 1960;20:37–46 [Google Scholar]
- 26.Cohen J. Weighted kappa: nominal scale agreement with provision for scaled disagreement for partial credit. Psychol Bul 1968;70:213–20 [DOI] [PubMed] [Google Scholar]
- 27.Shrout PE, Fleiss JL. Intraclass correlations: uses in assessing rater reliability. Psychol Bull 1979;86:420–8 [DOI] [PubMed] [Google Scholar]
- 28.Cohen J. Statistical power analysis for the behavioral sciences, 2nd Edn Hillsdale, NJ: Lawrence Earlbaum Associates, 1988 [Google Scholar]
- 29.Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics 1977;33:159–74 [PubMed] [Google Scholar]
- 30.Sim J, Wright CC. The kappa statistic in reliability studies: use, interpretation, and sample size requirements. Phys Ther 2005;85:257–68 [PubMed] [Google Scholar]
- 31.Cox DR. Regression models and life tables. J R Stat Soc 1972;B34:187–220 [Google Scholar]
- 32.Oikawa H, Sasaki M, Tamakawa Y, et al. The substantia nigra in Parkinson disease: proton density-weighted spin-echo and fast short inversion time inversion-recovery MR findings. Am J Neuroradiol 2002;23:1747–56 [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe H, Ito M, Fukatsu H, et al. Putaminal magnetic resonance imaging features at various magnetic field strengths in multiple system atrophy. Mov Disord 2010;25:1916–23 [DOI] [PubMed] [Google Scholar]
- 34.Fujii S, Matsusue E, Kinoshita T, et al. Hyperintense putaminal rim at 3T reflects fewer ferritin deposits in the lateral marginal area of the putamen. Am J Neuroradiol 2007;28:777–81 [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke CE. A “cure” for Parkinson's disease: can neuroprotection be proven with current trial designs? Mov Disord 2004;19:491–8 [DOI] [PubMed] [Google Scholar]