

Lomaiviticin A (1) and lomaiviticin B (2) are novel C2-symmetric diazobenzofluorene glycoside marine natural products (Figure 1).[1] Lomaiviticin A (1) potently inhibits the growth of cultured cancer cell lines (GI50 values ranging from 0.007 to 72.0 nm), and both 1 and 2 exhibit impressive growth inhibition activity against Gram-positive bacteria. Compound 1 causes damage to DNA in vitro. It has been speculated that a reactive species derived from the diazobenzofluorenones of 1 and 2 causes damage to nucleic acids, leading to the cytotoxic activities of these molecules. Experimental studies have provided support for the formation of reactive species from diazofluorenone structures.[2] However, it remains to be determined how the full structures of 1 and 2 react under physiologically relevant conditions and what cellular components are perturbed by 1 and 2.
Figure 1.
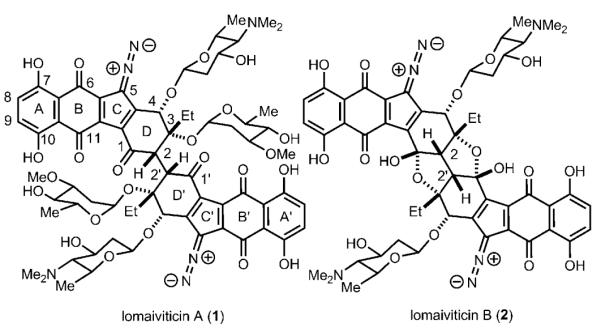
Structures of lomaiviticin A and B.
The structures of 1 and 2 are unprecedented and striking. The unusual structures of 1 and 2 pose interesting questions about how they are biosynthesized, especially how the C2–C2′ bond is made, and questions about how to achieve syntheses of these molecules. Although a synthesis of 1 or 2 has not yet been achieved, many approaches to these molecules have been reported,[3] including our own enantioselective synthesis of the central (C-D-D’-C’) ring system of 1.[3b] A synthesis of the full carbon skeleton of 1 and 2 has not yet been accomplished. To achieve syntheses of 1 and 2, there are significant challenges to overcome; the most difficult of which is likely formation of the C2–C2′ δ bond. The C2–C2′ bond links two highly functionalized “halves” of 1 and 2, which closely resemble the related natural product kinamycins.[4] The congested environment surrounding the C2–C2′ bond, stereochemical control during bond formation, and the potential lability of the D and D’ rings render the formation of this bond extremely challenging. Late-stage dimerization and formation of the C2–C2′ bond, in which double-processing is kept at a minimum, would be the most efficient means of synthesizing 1 and 2. Since the C2–C2′ bond in 1 and 2 is part of a 1,4-diketone (C1-C2-C2′-C1′), a late-stage oxidative enolate coupling would be the ideal reaction to construct this bond. However, the high potential for β-elimination of the C3 tertiary carbinol from a C1–C2 enolate, coupled with the likely poor stereoselectivity of such an oxidative enolate coupling reaction, renders this approach unattractive. To circumvent these obstacles, we developed a strategy utilizing the oxidative enolate coupling of 7-oxanorbornanones to achieve the first enantioselective synthesis of the central ring system of the aglycon of 1 (Scheme 1, see black structures in brackets). The 7-oxanorbornanone structure prevents β-elimination at C3 due to stereoelectronic effects, and it provides perfect diastereoselectivity during the formation of the C2–C2′ bond due to double diastereodifferentiation in the dimerization event. Herein we report stereo-selective coupling of the monomeric units of 1 and 2 leading to the first synthesis of the full carbon skeleton of the aglycons of 1 and 2. During the course of these studies, remarkable remote substituent effects during the key dimerization reaction have been observed, which may further complicate future syntheses of 1 and 2.
Scheme 1.

Synthesis plan.
As stated, our synthesis plan is to perform an oxidative dimerization of the enolate of 4 to afford 3 (Scheme 1). An annulation reaction[5] between enone 5 and a known cyanophthalide reagent[6] would provide access to 4 (see Scheme 3, 12). Enone 5 would be accessible from intermediate 6, a compound for which we had previously developed an enantioselective synthesis.[3b] Initially, an anionic annulation reaction on an enone similar to 5, but with C1 at the ketone oxidation state, was attempted. Unfortunately, the basic conditions of the annulation were not compatible with the C1 ketone. Therefore, we were forced to protect the C1 ketone of 6 in its reduced form by treatment with NaBH4 (Scheme 2). Surprisingly, 7 was obtained as the major product when the reduction was performed in allyl alcohol solvent. We speculate that transesterification of the oxazolidinone chiral auxiliary occurs from the boronate of allyl alcohol. Quenching the remaining borohydride with acetone allowed us to isolate 7 in 89 % yield. Protecting group manipulation of 7 afforded 8. The C5 carbinol of compound 8 was then oxidized under standard Swern conditions. Surprisingly, the product was obtained as the chlorinated β-ketoester 9.[7] The reductive dechlorination of 9 with Zn afforded β-ketoester 10, which underwent decarboxylative deallylation[8] upon exposure to [PdCl2(PPh3)2] and nBu3SnH, furnishing ketone 11 in 85 % yield. Finally, α-phenylselenation of the enolate of 11, followed by oxidation, delivered enone 5.[9]
Scheme 3.
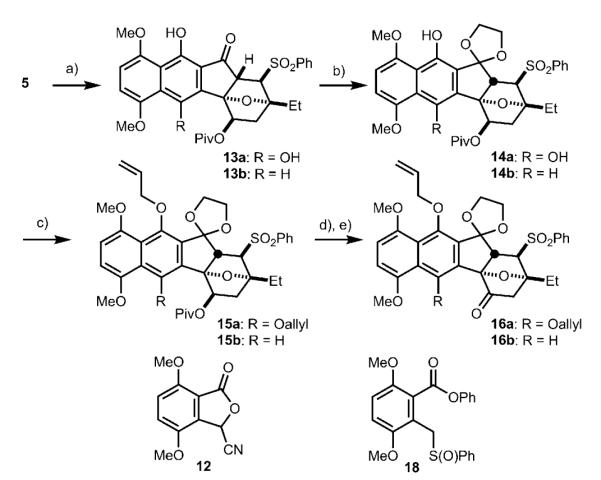
Synthesis of dimerization precursors 16 a and 16 b. a)For 13 a: 12, LHMDS, HMPA, THF, −78°C, 30 min; then 5, 50°C, 3 h, 85 %; for 13 b: 18, LHMDS, THF, −60°C, 6 h; then 5, −78°C, 10 min, 79 %; b) 1,2-bis(trimethylsilyloxy) ethane, TMSOTf, CH2Cl2, 23°C, 48 h, 76 % for 14 a, 72 % for 14 b; c) Cs2CO3, allyl bromide, DMF, 23°C, 2 h, 87 % for 15 a, 76 % for 15 b; d) NaBHEt3, THF, 0 °C, 4 h; e) TPAP, NMO, 4 Å M.S., CH2Cl2, 23°C, 2 h, 90 % for two steps for 16a, 88% for two steps for 16b. HMPA = hexamethylphosphoramide, DMF= dimethyl formamide, TMSOTf = trimethylsilyl trifluoromethanesulfonate, TPAP = tetrapropylammonium perruthenate, NMO = 4-methylmorpholine-N-oxide.
Scheme 2.
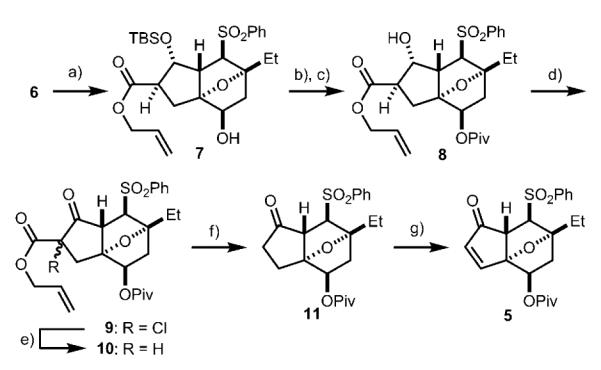
Synthesis of enone 5. a) NaBH4, allyl alcohol, −78°C, 5 min; then acetone, 23°C, 15 min, 89 %; b) PivCl, pyridine, CH2Cl2, 40°C, 24 h; c) TBAF, THF, 23°C, 16 h, 91 % over two steps; d) DMSO, (COCl)2, −78°C, 30 min; then Et3N, 23°C, 30 min; e) Zn dust, AcOH, 23°C, 2 h, 96 % over two steps; f) [PdCl2(PPh3)2], nBu3SnH, AcOH, toluene, 0°C, 30 min; then 110°C, 30 min, 85 %; g) LHMDS, THF, −78°C, 30 min; then PhSeBr, −78°C, 5 min; then H2O2,CH2Cl2/THF 1:1, 0°C, 4 h, 73 %. Piv = trimethylacetyl, TBAF = tetrabutylammonium fluoride, THF = tetrahydrofuran, DMSO = dimethyl sulfoxide, LHMDS = lithium bis(trimethylsilyl)amide.
For the key annulation reaction, we used the cyanophthalide method developed by Kraus[10] (Scheme 3). Addition of the anion of 12 to enone 5 afforded hydroquinone 13 a in 85 % yield. The C5 ketone of 13 a was protected as a dioxolane.[11] Protection of the two phenols of 14 a as their corresponding allyl ethers (compound 15 a), followed by reductive cleavage of the pivaloyl group, and subsequent oxidation at C1 afforded ketone 16 a, setting the stage for the key oxidative enolate coupling.
For synthesis of the C-D-D’-C’ central ring system of 1 and 2, we had developed conditions for the oxidative enolate coupling reaction involving deprotonation of the ketone with LHMDS at −78 °C, addition of [Cp2FePF6] and warming to −60 °C.[12] We then applied these conditions to the oxidative dimerization of ketone 16 a (Scheme 4). Unfortunately, none of the desired dimer (17 a) was formed under these conditions, and only starting material 16 a or decomposed material was isolated (Scheme 4).
Scheme 4.

Attempted oxidative dimerization of 16 a. a) LHMDS, HMPA, THF, −78°C, 2 h; then [Cp2FePF6], −60°C, 72 h, 0 %. LHMDS = lithium bis(trimethylsilyl)amide, [Cp2FePF6] = ferrocenium hexafluorophosphate.
In light of these disappointing results, we tried to rationalize why our C–D ring system published earlier underwent successful dimerization, but the A–D ring system here failed to dimerize. We speculated that nonbonded interactions may be developing in the transition state, which could be inhibiting dimerization. Since little is known about the mechanism of this reaction and the trajectory by which substrates approach each other, we attempted to understand possible developing nonbonded interactions in the transition state by studying the ground-state conformations of the desired product (compound 17 a).[13] We examined compound 17 a (Figure 2) and each of the three staggered conformations about the C2–C2′ bond, specifically H2–H2′ dihedral angles of 60, 180, and −60° (see Figure 2). The conformation with a H2–H2′ dihedral angle of 180° adopted an “n” conformation, the name of which is given because the shape of the conformer resembles that of the letter “n”. This conformer appeared to be very high in energy and unlikely to be a relevant conformer since the C3 and C3′ ethyl groups were suffering from severe nonbonded interactions. The 60° H2–H2′ dihedral angle conformation also exists in an “n” conformation, but it does not suffer from the destabilizing C3–C3′ ethyl interactions. However, we did notice potential nonbonded interactions between the C11 and C11′ allyloxy groups (see dotted arrow). Finally, the −60° H2–H2′ dihedral angle conformation exists in a “z” conformation, which was also given due to the resemblance to the letter “z” in shape, and in this conformation nonbonded interactions may develop between C11 (and C11′) allyloxy groups and the C3′ (and C3) ethyl groups. Since in both the “n” and “z” conformations, the C11 (and C11′) allyloxy groups may be preventing dimerization, we decided to synthesize the tetracyclic dimerization precursor where the C11 allyloxy group is replaced with a hydrogen atom (16 b, R = H). This forced us to devise a new annulation strategy to access this structure from enone 5.
Figure 2.
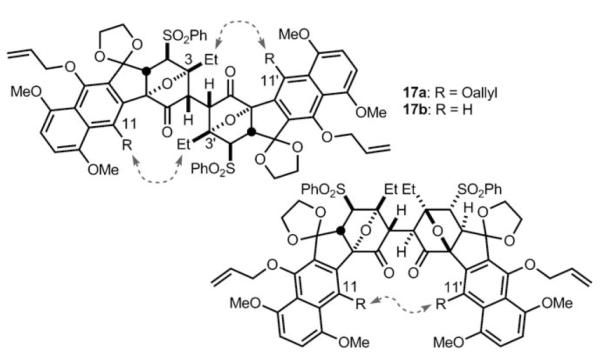
Postulated steric interactions in the dimeric products.
To construct 16 b, where the C11 substituent is a hydrogen atom, we synthesized Hauser-type sulfoxide annulation partner 18 (Scheme 3).[14] Deprotonation of 18 with LHMDS at −60 °C, followed by exposure to enone 5, delivered phenol 13 b in 79 % yield. It is worth noting that the use of the phenyl ester of 18, compared to the more commonly used methyl ester, dramatically enhanced the rate and yield of the annulation reaction-a finding that may be useful in other applications of this reaction.[15] Following the process we developed previously, 13 b was converted to dioxolane 14 b. Allyl protection of the phenol to afford 15 b, followed by cleavage of the C1-pivaloyl group, and subsequent oxidation, delivered ketone 16 b. With ketone 16 b in hand, we again attempted to perform oxidative enolate coupling under identical conditions (LHMDS, HMPA, [Cp2FePF6], −60 °C, THF). We were gratified to find that oxidative dimerization of 16 b afforded 17 b in 80 % yield as a single diastereomer (Scheme 5).
Scheme 5.

Oxidative dimerization of 16 b. a) LHMDS, HMPA, THF, 78°C, 2 h; then [Cp2FePF6], −60°C, 72 h, 80 %; b) [PdCl2(PPh 3)2], nBu3SnH, AcOH, CH2Cl2, 0°C, 30 min, 90 %.
To confirm the relative stereochemistry at C2 and C2′, and to determine the conformation of dimeric structure 17 b, we set out to obtain an X-ray crystal structure. Fortunately, we discovered that removal of the allyl groups of 17 b afforded 19 as a crystalline solid (Scheme 5), which was subjected to X-ray crystallographic analysis to provide the structure shown in Figure 3.[16] The crystal structure confirmed that the correct C2–C2′ stereochemistry had been established and also that the compound can exist in an “n” conformation.
Figure 3.
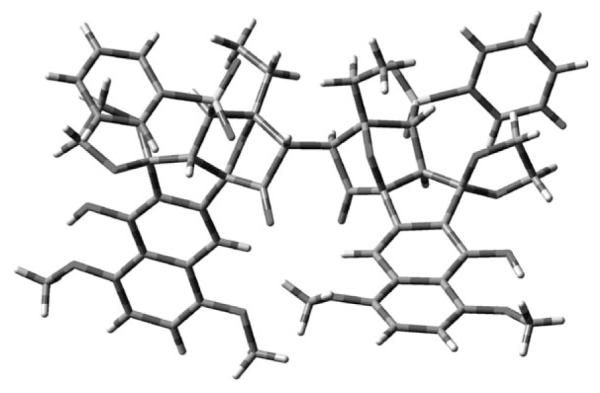
X-ray crystal structure of 19.
Closer inspection of the X-ray crystal structure revealed that the C11 hydrogen atom is in close proximity to the C1′ ketone oxygen atom (and conversely the C11′ hydrogen and C1 ketone oxygen; see Figure 4). In fact, the interatomic distance between H11 and O1′ (and H11′ and O1) is only 0.29 Å greater than the sum of their van der Waals radii. This close contact between the C11 substituent and O1′ (as well as C11′ and O1) may explain why the first attempted oxidative dimerization with a C11 allyloxy group failed. If, as in our first attempted dimerization, C11 bears an oxygen atom, its greater van der Waals radii of 1.52 Å would have suffered a nonbonded interaction with O1′. Interestingly, although our analysis of ground-state conformations led us to convert C11 from an oxygen substituent to hydrogen, we were surprised to find that it may be a steric clash between C11 substituent and O1′ that may be preventing dimerization.
Figure 4.
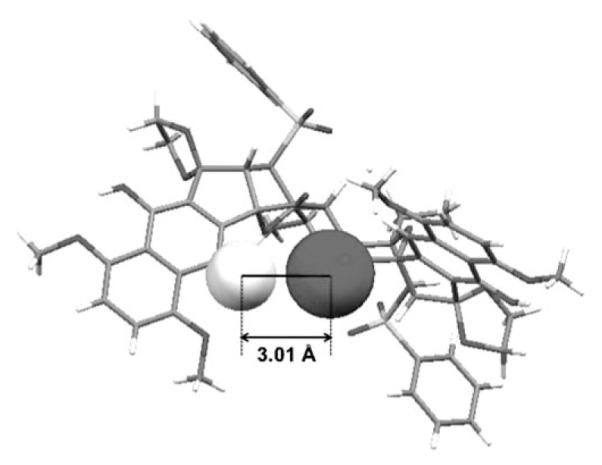
Interatomic distance of C11 hydrogen and C1′ oxygen.
Further examination of the X-ray crystal structure of 19 revealed unusual torsional strain in the C2–C2′ bond, with a C1-C2-C2′-C1′ dihedral angle of 29° (compared to a desired dihedral angle of 60°). The presence of C2–C2′ torsional strain in 19 and the potential for severe nonbonded interactions in the crystal structure suggested to us that there may be a measurable barrier to rotation about C2–C2′, resulting in atropisomerism. Unfortunately, heating to 90 or cooling to −80 °C did not result in a change in the 1H NMR spectrum of 19, preventing us from determining whether 19 exhibits atropisomerism.
In summary, we report the first synthesis of the full carbon skeleton of the lomaiviticin aglycon. An important aspect of this study was demonstrating that the oxidative enolate coupling strategy we reported earlier can be applied to the full monomeric units of the lomaiviticins to assemble the full carbon skeleton, again with complete stereochemical control in the formation of the C2–C2′ bond. During the course of these studies, we have discovered that remote oxygen substituents at C11 (and C11′), surprisingly, have dramatic effects on the success of the oxidative dimerization. It is interesting to speculate whether our observation that dimerization to form the lomaiviticin aglycon skeleton requires hydrogen atoms at C11 and C11′ extends to other dimerization strategies or even to the biosynthesis of 1 and 2. It may be that a biosynthetic dimerization also occurs with hydrogen at C11 and C11′ and that late stage oxidation leads to the lomaiviticins. An X-ray crystal structure of the lomaiviticin aglycon skeleton revealed that a nonbonded interaction between substituents at C11 and O1′ (and C11′–O1) may be suppressing oxidative dimerization when the C11 substituent is larger than hydrogen. In addition, the lomaiviticin aglycon skeleton exists (or at least crystallizes) in an unusual “n” conformation with significant torsional strain around the C2–C2′ δ bond. The lomaiviticins are challenging synthesis targets and the advances reported herein are important steps in eventually achieving their syntheses.
Experimental Section
Oxidative dimerization of 16 b
A two neck flask charged with a solid addition adaptor with ferrocenium hexafluorophosphate (2.32 g, 7.00 mmol, 5.00 equiv) in it was flame dried and charged with argon. Then the flask was charged with THF (8 mL), HMDS (0.502 mL, 2.38 mmol, 1.70 equiv), 2.29 m nBuLi solution in hexane (1.04 mL, 2.38 mmol, 1.70 equiv) and HMPA (0.487 mL, 2.80 mmol, 2.00 equiv) at −78°C, successively. 40 min later a solution of ketone 16 b (830 mg, 1.40 mmol, 1.00 equiv) in 10 mL of THF (plus 2 × mL rinse) was cannulated to the LHMDS/HMPA solution to give a pale yellow solution. 2 h later, ferrocenium hexafluorophosphate was added to the reaction system from the solid addition adaptor. The originally formed deep blue suspension turned yellowish green in 30 min. The reaction mixture was stirred at −60°C for 3 d. The reaction mixture was quenched with a saturated solution of NH4Cl and warmed to room temperature. The aqueous phase was extracted with dichloromethane (3 × 20 mL) and the combined organic phase was dried with anhydrous Na2SO4. The crude product was purified by flash column chromatography (EtOAc/hexane 1:1 to 1.5:1) to give dimer 17 b (665 mg, 1.12 mmol, 80 %) as a pale yellow solid.
Supplementary Material
Acknowledgements
Financial support for this project was provided by a grant from the NIH (CA125240). A.S.L. acknowledges financial support from an NDSEG fellowship. H.L. and J.A. acknowledge Sanofi-Aventis and Eli Lilly, respectively, for graduate fellowships. Shao-Liang Zheng is acknowledged for X-ray crystallographic analysis.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201002157.
References
- [1].He H, Ding W-D, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. J. Am. Chem. Soc. 2001;123:5362–5363. doi: 10.1021/ja010129o. [DOI] [PubMed] [Google Scholar]
- [2]a).Laufer RS, Dmitrienko GI. J. Am. Chem. Soc. 2002;124:1854–1855. doi: 10.1021/ja0167809. [DOI] [PubMed] [Google Scholar]; b) Feldman KS, Eastman KJ. J. Am. Chem. Soc. 2005;127:15344–15345. doi: 10.1021/ja053880w. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Feldman KS, Eastman KJ. J. Am. Chem. Soc. 2006;128:12562–12573. doi: 10.1021/ja0642616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3]a).Nicolaou KC, Denton RM, Lenzen A, Edmonds DJ, Li A, Milburn RR, Harrison ST. Angew. Chem. 2006;118:2130–2135. doi: 10.1002/anie.200504466. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:2076–2081. doi: 10.1002/anie.200504466. [DOI] [PubMed] [Google Scholar]; b) Krygowski ES, Murphy-Benenato K, Shair MD. Angew. Chem. 2008;120:1704–1708. doi: 10.1002/anie.200704830. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:1680–1684. doi: 10.1002/anie.200704830. [DOI] [PubMed] [Google Scholar]; c) Zhang W, Baranczak A, Sulikowski GA. Org. Lett. 2008;10:1939–1941. doi: 10.1021/ol800460a. [DOI] [PubMed] [Google Scholar]; d) Nicolaou KC, Nold AL, Li H. Angew. Chem. 2009;121:5974–5977. [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:5860–5863. doi: 10.1002/anie.200902509. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Gholap SL, Woo CM, Ravikumar PC, Herzon SB. Org. Lett. 2009;11:4322–4325. doi: 10.1021/ol901710b. [DOI] [PubMed] [Google Scholar]
- [4]a).Gould SJ, Tamayo N, Melville CR, Cone MC. J. Am. Chem. Soc. 1994;116:2207–2208.; Mithani S, Weeratunga G, Taylor NJ, Dmitrienko GI. J. Am. Chem. Soc. 1994;116:2209–2210.; Marco-Contelles J, Molina MT. Curr. Org. Chem. 2003;7:1433–1442.; for total syntheses, see:Lei X, Porco JA. J. Am. Chem. Soc. 2006;128:14790–14791. doi: 10.1021/ja066621v.; Nicolaou KC, Li H, Nold AL, Pappo D, Lenzen A. J. Am. Chem. Soc. 2007;129:10356–10357. doi: 10.1021/ja074297d.; Woo CM, Lu L, Gholap SL, Smith DR, Herzon SB. J. Am. Chem. Soc. 2010;132:2540–2541. doi: 10.1021/ja910769j.
- [5]a).Mal D, Pahari P. Chem. Rev. 2007;107:1892–1918. doi: 10.1021/cr068398q.; Rathwell K, Brimble MA. Synthesis. 2007:643–662.
- [6].Okazaki K, Nomura K, Yoshii E. Synth. Commun. 1987;17:1021–1027. [Google Scholar]
- [7].Smith AB, III, Leenay TL, Liu H-J, Nelson LAK, Ball RG. Tetrahedron Lett. 1988;29:49–52. [Google Scholar]
- [8].Tsuji J, Nisar M, Shimizu I. J. Org. Chem. 1985;50:3416–3417. [Google Scholar]
- [9].Reich HJ, Renga JM, Reich IL. J. Am. Chem. Soc. 1975;97:5434–5447. [Google Scholar]
- [10].Kraus GA, Sugimoto H. Tetrahedron Lett. 1978;19:2263–2266. [Google Scholar]
- [11].Tsunoda T, Suzuki M, Noyori R. Tetrahedron Lett. 1980;21:1357–1358. [Google Scholar]
- [12].It is crucial to keep the reaction temperature below −60°C to suppress undesired β-elimination.
- [13].The analyses were made by a simple hand-held plastic model. For more clear 3D representations of each staggered conformation, see the Supporting Information.
- [14]a).Hauser FM, Rhee RP. J. Org. Chem. 1978;43:178–180.; b) Sulfoxide 18 was prepared in six steps from commercially available 2,5-dimethoxy benzaldehyde. See the Supporting Information for details.
- [15].Sun C, Wang Q, Brubaker JD, Wright PM, Lerner CD, Noson K, Charest M, Siegel DR, Wang Y, Myers AG. J. Am. Chem. Soc. 2008;130:17913–17927. doi: 10.1021/ja806629e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].CCDC-777057 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.