

Abstract
Exposure to airborne particles is associated with increased cardiovascular morbidity and mortality. During the combustion of chlorine-containing hazardous materials and fuels, chlorinated hydrocarbons chemisorb to the surface of transition metal-oxide-containing particles, reduce the metal, and form an organic free radical. These radical-particle systems can survive in the environment for days and are called environmentally persistent free radicals (EPFRs). This study determined whether EPFRs could decrease left ventricular function before and after ischemia and reperfusion (I/R) in vivo. Male Brown Norway rats were dosed (8 mg/kg, i.t.) 24 hr prior to testing with particles containing the EPFR of 1, 2-dichlorobenzene (DCB230). DCB230 treatment decreased systolic and diastolic function. DCB230 also produced pulmonary and cardiac inflammation. After ischemia, systolic, but not diastolic function was significantly decreased in DCB230-treated rats. Ventricular function was not affected by I/R in control rats. There was greater oxidative stress in the heart and increased 8-isoprostane (biomarker of oxidative stress) in the plasma of treated vs control rats after I/R. These data demonstrate for the first time that DCB230 can produce inflammation and significantly decrease cardiac function at baseline and after I/R in vivo. Furthermore, these data suggest that EPFRs may be a risk factor for cardiac toxicity in healthy individuals and individuals with ischemic heart disease. Potential mechanisms involving cytokines/chemokines and/or oxidative stress are discussed.
Keywords: inflammation, cytokines, chemokines, pressure-volume, particles, oxidative stress
Introduction
The public health impacts of airborne particles are well documented with studies consistently demonstrating correlations between environmental exposure and cardiovascular disease (1–4). In addition to the epidemiological data, animal studies show that fine and ultrafine particulates produce cardiovascular toxicity. For example, exposure to fine diesel exhaust particles increases the size and rate of atherosclerotic plaque formation in apoE knockout mice (5, 6). These lesions are accompanied by increased oxidative stress and inflammation in the blood vessels. Particles also decrease heart rate variability (7, 8), produce cardiac arrhythmias (9–11), increase thrombus formation (12) and alter vascular reactivity (5, 13). In mice, prior exposure to diesel exhaust particles significantly increases infarct size after coronary artery ligation, as well as increase inflammation and oxidative stress in the heart (13). While the specific pathways and mechanisms mediating particulate-associated cardiovascular toxicity are not well defined, systemic inflammation and oxidative stress are widely believed to play important contributory roles (see (14)). Although controversial, several studies show that fine and ultrafine particles can transit the lung and enter the systemic circulation where they may produce inflammation and/or oxidative stress in a number of tissues and organs (12, 15, 16).
The majority of airborne fine and ultrafine particles (mean diameter of < 2.5 μm and < 0.1 μm, respectively) are generated in combustion processes or result from secondary reactions involving combustion by-products (17, 18). Our group has shown that during combustion of chlorine-containing materials (e.g. hazardous waste incineration), chlorinated hydrocarbons are formed, which chemisorb to the surface of transition metal-oxide-containing particles, reduce the metal, and form an organic free radical (19–21). Chemisorption, by definition, is the formation of a chemical bond between the particle and pollutant to form a new particle-pollutant system, which persists until a subsequent chemical reaction separates them. Electron transfer from the chemisorbed organic to the surface of the particle reduces the transition metal and concomitantly forms the organic radical. The association of the free radical with the surface of the metal-containing particle stabilizes the radical and reduces its reactivity with atmospheric oxygen (19). This combination of stability and non-reactivity is referred to as environmental persistence and we refer to this radical-particle system as an environmentally persistent free radical (EPFR). EPFRs can persist for hours to days in air and are capable of redox cycling, resulting in the continuous release of reactive oxygen species. We have recently documented their existence in samples of airborne fine particles (22).
The ability of EPFRs to undergo prolonged redox cycling, producing large numbers of free radicals, raises the possibility that EPFRS may confer a level of biological activity/toxicity that is greater than that of either the particle or the pollutant alone. While studies examining the biological activity of EFPRs have only just begun, it was recently shown that EPFRs produce oxidative stress in human lung epithelial cells in vitro (23) and produce pulmonary inflammation in neonatal rats (24). Given the relationship between particulate exposure and cardiovascular disease, we hypothesized that inhaled EPFRs decrease left ventricular function under basal conditions and after cardiac ischemia followed by reperfusion.
To understand the chemistry and biological actions of EPFRs, we generated EPFRs which mimic those formed during combustion of chlorinated hydrocarbons and chlorine-containing fuels. These EPFRs are oxygen-centered semiquinone-type radicals similar to those found in cigarette smoke and airborne environmental samples (21, 22). The EPFRs used in this study were composed of the EPFR of 1, 2 dichlorobenzene formed by thermal reaction at 230°C (DCB230) with silica particles (~200 nm dia.) containing 3% CuO. A unique advantage of our approach is that we can reproducibly manufacture well-characterized EPFRs with environmentally-relevant particles of known size, composition, and quantity.
This study was designed to determine whether intratracheal (i.t.) instillation of DCB230 alters baseline cardiac function in vivo and exacerbates left ventricular dysfunction after a brief period of ischemia followed by reperfusion. We also sought to determine whether pulmonary and/or cardiac inflammatory infiltration had a role in DCB230-induced cardiotoxicity in vivo.
Materials and Methods
Synthesis of ultrafine DCB230 particles
The DCB230 used in this study was created using established protocols (21) and consisted of an ortho-chlorophenoxyl EPFR produced from the thermal reaction of 1, 2-dichlorobenzene with silica particles containing 3% CuO at 230°C (DCB230). Particle size was verified by transmission electron microscopy and flow cytometry (23). Electron paramagnetic resonance was used to confirm the presence of oxygen-centered EPFRs in our DCB230 samples (21).
In vivo studies
Animals
Male Brown Norway rats (250 to 320 g; Harlan, Indianapolis, IN) were housed in a temperature- and humidity-controlled room with a 12-hour light/dark cycle. Standard rat chow and water were available ad libitum. All procedures were performed in accordance with National Institutes of Health Guidelines for the Care and Use of Experimental Animals and were approved by the Institutional Animal Care and Use Committee at Louisiana State University Health Sciences Center.
Instillation of DCB230
DCB230 was suspended in isotonic saline with 0.02% Tween 80 and sonicated for 1 minute. The rats were anesthetized in an induction chamber with isoflurane (4%) and were intubated with an 18 ga. catheter. DCB230 (8 mg/kg) or vehicle (isotonic saline with 0.02% Tween 80) was instilled (1 μl/g body weight) down the intubation tube as a bolus dose 24 hours before conducting experiments. This dose of DCB230 was chosen because it is within the range of particle doses that have been shown to produce cardiac toxicity after a single i.t. instillation in rats and mice (13, 25, 26). Although the intratracheal route of administration is limited because it does not adequately model real world inhalation exposures, this method is a reliable and reproducible way to acutely administer known quantities of particles to the lungs. The 24 hour time point was chosen based on the report by Cozzi and colleagues (13) who showed increased infarct size, inflammation and oxidative stress 24 hours after particle instillation.
Left ventricular pressure-volume relationships
Left ventricular performance was assessed in DCB230 (n=6) and vehicle-treated rats (n=6) in vivo using Millar pressure-volume conductance catheters (SPR-838) (27). Rats were anesthetized with isoflurane (4%), intubated and ventilated (isoflurane 2% and 100% oxygen 3 l/min). Tidal volume (3–5 ml) and respiratory rate (60–70 breaths/min) were adjusted to maintain arterial blood pH between 7.35 – 7.45. Body temperature was maintained at 37±1°C using a heat lamp. A cannula (micro-renathane, 0.33 in O.D.) was placed into the right jugular vein to administer drugs. The pressure-volume catheter was introduced into the left carotid artery and was advanced into the left ventricle. After a 15 minute equilibration period, baseline parameters of left ventricular function (closed chest) were calculated using a minimum of 3 consecutive pressure-volume loops (sampling rate 1000 Hz; MPVS-400, Millar Instruments). The chest was then opened and the heart was exposed using a mid-sternal approach. A second “open chest” baseline of left ventricular function was then obtained. The left anterior descending coronary artery (LAD) was then occluded for 90 seconds by tying a 6-0 suture around the vessel between the pulmonary artery and the left atrial appendage. The effectiveness of the occlusion was verified by blanching of the ventricle distal to the ligature. The ligature was then removed to initiate reperfusion. Ventricular function was monitored by constructing pressure-volume loops 1, 5, 30 and 60 minutes after the onset of reperfusion.
Measures of heart rate (HR), peak left ventricular end systolic pressure (ESP), left ventricular end diastolic pressure (EDP), end diastolic volume (EDV) maximal slope of the systolic pressure increment (dP/dtmax), diastolic decrement (−dP/dtmin), ejection fraction (EF), stroke volume (SV), cardiac output (CO), Tau (G) (regression of dP/dtmax) and stroke work (SW) were computed from the pressure-volume loops using the Millar PVAN analysis system (27). The pressure-volume conductance catheter was calibrated using whole blood from vehicle and DCB230-treated rats to generate standard curves of relative volume units. To determine the absolute left ventricular volume, 35 μl of hypertonic saline (15%) was injected i.v. to measure the parallel conductance of the system, which was then subtracted from the total volume (27).
Markers of oxidative stress
At the end of each experiment, the heart was removed, rinsed in PBS and the right ventricle cut away. The middle portion of the left ventricle and septum were flash frozen in liquid nitrogen and stored at −80°C. Blood samples were also collected in serum separator tubes and were centrifuged (4000 rpm for 4 minutes at 4°C). The plasma samples were treated with 10 μM indomethacin to block enzymatic metabolism of arachidonic acid during the analysis. The serum samples were frozen at −80°C.
Glutathione (GSH) and oxidized glutathione (GSSG) were measured in cardiac samples using HPLC with electrochemical detection. Samples of frozen left ventricle were homogenized in 0.4N perchloric acid and 100 μM diethylenetriamine pentaacetic acid. The samples were then centrifuged and a small volume was injected onto the HPLC. The chromatography was performed using a Waters 2690 HPLC pump (Milford, MA) interfaced to a boron doped diamond electrode controlled through an ESA Coularray detector (Chelmsford, MA). The separation was achieved using a 3 μM, 25 cm × 4.6 mm I.D. Supercosil LC-8 reversed phase column. The mobile phase consisted of 25 mM sodium dihydrogen phosphate containing 1.4 mM 1-octanesulfonic acid and 6% acetonitrile (v/v), pH 2.65, and was run isocratically at 0.6 ml/min. The elution of GSH and GSSG was detected at 1500 mV, at about 8.9 and 18.6 minutes, respectively.
8-isoprostane is a biomarker of oxidative stress that is produced in vivo by the peroxidation of arachidoinc acid. 8-isoprostane levels in plasma were measured using established methods (28). Briefly, 15% potassium hydroxide and a methanolic solution of butylated hydroxytoluene (an antioxidant) were added to an equal volume of plasma, and the solution was incubated at 37°C for 30 min. The saponified samples were then acidified to pH 3.0 using HCl, and 2 ml of 100 mmol of formate buffer (pH 3.0) was added. The samples were centrifuged at 2400 g for 10 minutes and the resulting supernatants were subjected to solid-phase extraction using pre-conditioned Oasis HLB 1cc extraction cartridges (Waters Corp., Milford, MA) (29). The columns were washed first with 5 ml of 10 mmol/l (pH 3.0) formate buffer, then with 5 ml (15:85 by volume) acetonitrile-water, and the isoprostanes were eluted with 2 ml (30:65:5 by volume) hexane-ethyl acetate-propane-2-ol. The level of 8-isoprostanes was then detected using an ELISA kit from Cayman Chemicals.
Measures of Inflammation
Pulmonary cytokine and chemokine levels
A separate group of rats were treated with either DCB230 (n=5) or vehicle (n=5). Twenty four hours later the rats were anesthetized (4% isolflurane) and intubated. Bronchial alveolar levage was then performed using 5 ml of sterile PBS containing 2% BSA. The bronchial alveolar levage fluid (BALF) was centrifuged and the supernatants were collected and frozen at −80°C. The BALF was assayed for monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein (MIP-1α), GROKC and the cytokines interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-10 (IL-10), interleukin-18 (IL-18), interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), and VEGF (Milliplex Map Kit). Samples from individual rats were assayed in duplicate for the controls and triplicate for the DCB230-treated rats.
mRNA arrays for cytokines and chemokines in the heart
In separate studies, groups of rats were treated with DCB230 (n=3) or vehicle (n=3). Twenty four hours later, the hearts were removed, sectioned and frozen as described above. Total RNA was isolated from cardiac tissue using Trizol (Invitrogen, Carlsbad, CA) and further purified using Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA). The integrity of the extracted RNA was determined using the Affymetrix chip processing system. cDNA was synthesized using the RT2 Profiler PCR Array protocol (SABiosciences RT2 First Strand Kit) with 1 mg of total RNA using the BioRad iCycler® 96-well plate format. The inflammatory cytokine array was PARN-011 and the oxidative stress array was PARN-065. The cycling conditions were as follows: an initial denaturation at 95°C for 10 minutes, and 40 cycles of 95°C for 15 seconds, and finally 60°C for 1 minute. Data analysis was performed using the SABioscience’s PCR Array Data Analysis Web Portal at the following address http://www.SABiosciences.com/pcrarraydataanalysis.php. Data are reported as fold change from control values.
Histology
To estimate the size of the ischemic region produced by ligation of the LAD, a modification of the dye exclusion procedure reported by Cozzi and colleagues (13) was used. A separate group of rats (n= 3) was deeply anesthetized and the heart exposed. After ligating the LAD, a 27 gauge needle was introduced into the left ventricle through the apex and a 1% solution of methylene blue dye was infused. The heart was then excised and weighed. The nonperfused (ie. ischemic) portion of the left ventricle was removed and the remaining portion of the heart reweighed. The percent of nonperfused left ventricle was then calculated.
To look for histological evidence of inflammation, groups of rats were treated with DCB230 (n=3) or vehicle (n=2). Twenty four hours later the rats were deeply anesthetized and the hearts perfused in situ with saline followed by 4% buffered Zn formalin. The hearts were then imbedded in paraffin, sectioned (5 μm) and placed on glass slides. Alternate sections of each heart were stained with hematoxylin-eosin (H & E). Three H & E sections from each heart were examined blind by the same pathologist. Inflammation was diagnosed by the accumulation of mononuclear and polymorphonuclear inflammatory cells. Because interstitial cells can be difficult to characterize on H & E stained slides, macrophages were further identified by immunohistochemisty in alternate sections stained with monoclonal antibodies for CD68 (Ventana). Positively stained cells were identified by a brown granular pigmentation of the cytoplasm and counted. Stained macrophages were counted in the left ventricle and septum of 3 sections from each heart. The average number of macrophages per section was then calculated for control and treated hearts. Sections of intestine served as positive controls, while negative controls consisted of cardiac sections incubated without the primary antibody.
Statistical Analysis
All data are reported as mean ± SEM. Baseline (close chest) parameters of cardiac function after DCB230 or vehicle instillation were compared using unpaired Student’s t-tests (Prism-GraphPad). Between group comparisons of cardiac function in open chest rats before and after ischemia were made using two-way repeated measures analysis of variance followed by Bonferroni post hoc tests (Prism-GraphPad). Changes in 8-isoprostane levels, cytokine levels, GSH, GSSG and GSH/GSSG ratios were also compared using Student’s t-test. p<0.05 was considered statistically significant.
Results
Ventricular function
To examine the effects of DCB230 on left ventricular function before and after ischemia and reperfusion, groups of rats were treated with DCB230 or vehicle. Twenty-four hours later the rats were anesthetized and pressure-volume curves generated with the chest closed to determine the effects of the DCB230 alone on ventricular function. Pressure-volume curves were then obtained with the chest open immediately before occluding the LAD and during reperfusion. Heart rate was not significantly different in closed chest DCB230 and vehicle-treated rats (fig. 2). With the chest closed, EDV and −dP/dtmin were significantly lower and Tau (G) significantly increased in DCB230-treated versus vehicle-treated rats (fig. 1). EDP was not significantly different between the two groups (fig. 1). With respect to systolic function, ESP, dP/dtmax, SW and CO were also significantly lower in DCB230-treated rats as compared to those treated with vehicle (fig. 2). In DCB230-treated rats maximum power (28.6±3 mWatts) was lower than in vehicle-treated rats (43.1±8 mWatts); however, these differences were not significant. Stroke volume was lower in DCB230-treated rats, but this reduction was not significantly different from control (fig. 2). Ejection fraction was nearly identical in the two groups (DCB230, 49±2% vs vehicle, 55±2%).
Figure 2.

EPFRs decrease baseline systolic left ventricular function in closed chest rats. Rats were treated with DCB230 or vehicle as in figure 1. Data are reported as mean ± SEM. Abbreviations: ESP, end systolic pressure; dP/dtmax, change in pressure vs change in time during systole; SW, stroke work; SV, stroke volume; CO, cardiac output. ***p<0.001, **p<0.01, and *p<0.05.
Figure 1.

EPFRs decrease baseline diastolic left ventricular function in closed chest rats. Rats were treated with DCB230 (8 mg/kg, i.t.) or vehicle 24 hrs prior to the experiment. Left ventricular function was measured using Millar pressure-volume catheters. Data are mean ± SEM. Abbreviations: EDV, end diastolic volume; EDP, end diastolic pressure; −dP/dtmin, diastolic decrease in pressure vs time; Tau (G). ***p<0.001, **p<0.01, and * p<0.05.
After opening the chest, there were no significant differences in any of the baseline parameters of ventricular function between the treatment groups; however, in most cases ventricular function in the DCB230-treated rats was reduced relative to controls (figs. 3 and 4). With the chest open, baseline heart rate was similar in the DCB230 and vehicle rats and remained so throughout the 1 hour reperfusion period (fig. 4). Except for EDV, which was significantly lower in the DCB230-treated rats 30 minutes after reperfusion, EDP, −dP/dtmin and Tau (G) were not significantly different between the two groups at any of the time points (fig. 3). With respect to systolic function, ESP was significantly lower in the DCB230 treatment group than in the vehicle group 1, 30 and 60 minutes after initiating reperfusion (fig. 4). Stroke work and SV were also significantly lower in DCB230-treated rats 30 and 60 minutes after reperfusion (fig. 4). Stoke volume was significantly lower in the DCB230 treatment group 30 minutes after reperfusion. Although CO and dP/dtmax tended to be reduced in DCB230-treated rats at most time points during reperfusion, these differences were not significant (fig 4). Ejection Fraction was similar in both groups (data not shown).
Figure 3.

EPFRs do not alter diastolic function after a brief myocardial ischemia. Rats were treated with DCB230 (n=6) or vehicle (n=6) 24 hr prior, as described in figure 1. Ventricular function was measured at baseline (B) before occluding the lower anterior descending coronary artery for 90s followed by reperfusion (I/R). Ventricular function was also measured 1, 5, 30 and 60 minutes after starting reperfusion. Data are means ± SEM. Abbreviations: as in figure 1.
Figure 4.

EPFRs decrease left ventricular systolic function after brief cardiac ischemia and reperfusion. These are the same rats and experimental set up as described in figure 3. Data are means ± SEM. Abbreviations: as in figure 2. **p<0.01, and *p<0.05.
Oxidative stress
To determine whether treatment with DCB230 increased the oxidative stress response to ischemia and reperfusion in the heart, HPLC was used to examine the levels of reduced glutathione (GSH) and oxidized glutathione (GSSG) in the hearts of the DCB230 and vehicle-treated rats subjected to ischemia and 1 hour of reperfusion. Hearts from DCB230-treated rats had significantly less GSH (0.99±0.9 nmol/mg tissue) than did those from vehicle-treated rats (1.26±0.08 nmol/mg tissue). Although the amount of GSSG in DCB230-treated rats was higher than in vehicle-treated rats (0.054±0.016 and 0.033±0.002 nmol/mg tissue, respectively) this difference was not significant. The ratio of GSH/GSSG was significantly decreased in DCB230-treated compared to vehicle-treated rats (fig. 5A). There was no difference in the GSH/GSSG ratio in rats treated with DCB230 or vehicle that were not subjected to ischemia-reperfusion (data not shown). In addition, the level of 8-isoprostane (a biomarker oxidative stress) was significantly higher in the plasma of DCB230-treated rats 1 hour after reperfusion relative to vehicle-treated rats (fig. 5B).
Figure 5.

EPFRs increase oxidative stress in heart and plasma after I/R. Rats are the same as reported in figures 1–4. Levels GSH and GSSG in the heart and 8-iosprostane in plasma were measured using HPLC with electrochemical detection. Data are means ± SEM. ** p<0.01, and *p<0.05.
Cytokines in the BALF
To determine whether EPFRs produced pulmonary inflammation in adult rats, groups of rats were treated with DCB230 or vehicle, and the BALF collected 24 hours later. Compared to vehicle treatment, the levels of MCP-1, MIP-1α, IL-1β, IL-6, IFN-γ, IL-18, and TNF-α were significantly increased in BALF of rats treated with DCB230 (table 1). GROKC, IL-10 and VEGF were not significantly different between the groups 24 hours after treatment (table 1).
Table 1.
BALF cytokine levels following DCB230 exposure
| Vehicle | DCB230 | |
|---|---|---|
| MCP-1 | 69 ± 29 | 1369 ± 365** |
| MIP-1α | ND | 124 ± 16*** |
| IL-1β | 10 ± 2 | 119 ± 23** |
| IL-6 | ND | 2583 ± 362*** |
| IL-10 | ND | ND |
| INF-γ | 3 ± 2 | 65 ± 19* |
| IL-18 | 111 ± 17 | 472 ± 23*** |
| GROKC | 874 ± 128 | 1401 ± 661 |
| TNF-α | ND | 26 ± 6** |
| VEGF | 76 ± 24 | 88 ± 7 |
Cytokines measured 24 hr post-exposure to DCB230. Data are expressed in pg/ml as a mean±SEM. n=5/group.
p<0.05,
p<0.01,
p<0.001. ND = below the level of detection.
Changes in mRNA levels
Changes in the levels of mRNA in the hearts of DCB230-treated and vehicle-treated rats were determined using mRNA arrays. DCB230 treatment increased (>1.5 fold) the level of mRNA encoding chemokine ligand 20 (Ccl20), IL-10, IL-1β and platelet factor 4 (Pf4 or Cxcl4) (table 2). EPFR treatment also increased the mRNA levels of several cytokine and chemokine receptors, such as chemokine receptor 5 (Cxcr5), IL-6 receptor α (IL-6rα), IL-1 receptor type 2 (IL-1r2) and the IL-8 receptor β (IL-8rβ) (table 2).
Histology
Dye studies showed that the ligation of the LAD prevented perfusion of 46±1% of the septum and left ventricle. The hearts of rats exposed to DCB230 24 hours prior contained significantly higher (p<0.001) numbers of macrophages per cardiac section (41±6) than did control hearts (20±8). These infiltrating macrophages were distributed throughout the left ventricle and septum (fig. 6).
Figure 6.
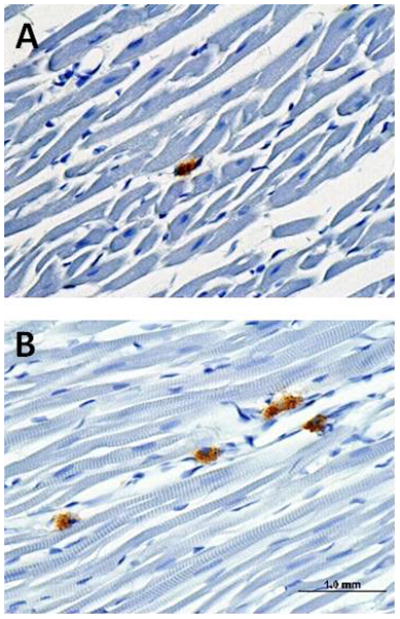
Section of the left ventricle of a vehicle-treated (A) and a DCB230-treated (B) rat showing increased macrophage infiltration (darkly stained cells) 24 hours after i.t. instillation of DCB230.
Discussion
This study showed for the first time, that instillation of a single dose of DCB230 can decrease left ventricular volume and decreases diastolic function in vivo, as indicated by significant decreases in −dP/dtmin and Tau (G). In addition, these studies show for the first time that exposure to DCB230 also reduced systolic function in vivo, as evidenced by significant reductions in ESP, dP/dtmax, SW and CO. The decreases in left ventricular function observed in vivo are consistent with those produced by particulates in isolated heart preparations.
Badgate and colleagues (25) instilled ultrafine diesel exhaust particles into the lungs of spontaneously hypertensive rats (SHR) or normotensive control rats 4 hours before measuring left ventricular function using the isolated heart preparation. Exposure to diesel particles significantly reduced contractile function in the isolated hearts when compared to the control. In separate studies, left ventricular systolic function was also decreased when isolated hearts from normotensive rats and SHR were directly perfused with diesel exhaust particles, leading to the conclusion that ultrafine particulates can directly reduce cardiac function (30, 31). Exposure to diesel exhaust particles also reduced fractional shortening in normal rats before and after isoproterenol –induced ventricular damage (32). In addition, inhalation of carbon black particles also reduced left ventricular function in senescent and middle aged mice (33). In humans, epidemiological and experimental data also show that exposure to airborne particulates increases the risk of myocardial infarction, atherosclerosis, arrhythmia and decrease heart rate variability (an index of autonomic dysfunction) (1, 3, 4, 14).
In most cases, the exact mechanisms responsible for the cardiovascular and cardiac actions of inhaled particles are largely unknown, but most available evidence indicates that inflammation and oxidative stress play prominent roles (14, 34). The present studies show that our combustion generated EPFRs (DCB230) produce pulmonary inflammation in adult rats that is similar to that elicited by exposure to environmental samples of ultrafine particulates (35). EPFRs also produce pulmonary inflammation in neonatal mice (36). The “spill over” of pro-inflammatory cytokines, chemokines and inflammatory cells from the lung is believed to produce systemic inflammation. EPFRs also produced cardiac inflammation in our rats as evidenced by the infiltration of inflammatory cells into the heart and increases in the levels of mRNA encoding pro-inflammatory cytokines and cytokines and their receptors. Thus far, it has yet to be determined whether the observed cardiac inflammation results from the actions of circulating cytokines and chemokines and/or the localized actions of particles on the heart.
The prior administration of DCB230 also led to significant decreases in left ventricular function after 90 seconds of ischemia followed by reperfusion. Reperfusion of the ischemic myocardium is associated with marked increases in oxidative stress and enhanced inflammation leading to further degradation of ventricular function (37, 38). Most models of cardiac ischemia-reperfusion use a 20–30 minute period of ischemia (39–41). Because we were concerned that the large functional deficits normally produced by long periods of ischemia would overwhelm or obscure the potential effects of the DCB230, we used a 90 second period of ischemia that did not significantly decrease ventricular function in vehicle-treated rats. Clinically, this model is relevant because vasospastic (Prinzmetal’s) and classical angina are characterized by relatively brief periods ischemia and reperfusion. Dye studies showed that ligation of the LAD decreased perfusion to 46% of the heart without significantly affecting diastolic or systolic function in the vehicle-treated rats. Conversely, in rats treated twenty four hours prior with DCB230, significant decreases in systolic function were observed after 30 to 60 minutes of reperfusion, as evidenced by significant decreases in ESP, SW and SV. Although not significantly different from vehicle-treated rats, CO and dP/dtmax also trended downward over time in DCB230-treated rats, further suggesting systolic dysfunction. Surprisingly, diastolic function was not significantly affected by ischemia and reperfusion.
In this study, the GSH/GSSG ratio and plasma 8-isoprostane level were not changed by the administration of DCB230 alone. However, during reperfusion the degraded systolic ventricular function in the DCB230-treated rats was accompanied by increased oxidative stress in the myocardium (reduced GSH/GSSG ratio) and plasma (increased 8-isoprostane levels). Oxidative stress and increased inflammation were also reported to underlie the doubling of infarct size observed in mice treated with environmentally derived particles twenty four hours prior to coronary artery ligation (13). Exposure to tobacco smoke (another source of particles) also dose-dependently increases infarct size in rats (42). Isolated rat hearts, previously treated (4 hours) in vivo with diesel particles, showed greater contractile deficits after 20 minutes of ischemia and were slower to recover function after ischemia than were saline treated hearts (25).
Inflammation and oxidative stress could also account for the decreases in ventricular function we observed twenty four hours after particle administration under both basal conditions and after mild ischemia-reperfusion. It is unlikely that structural remodeling was responsible for the decrease in basal EDV observed twenty four hours after the administration of a single dose of DCB230. It is more likely that the decrease in EDV was the consequence of increased pulmonary vascular resistance and reduced pulmonary venous return to the heart secondary to pulmonary inflammation. Exposure to carbon black particles has been shown to increase right ventricular and pulmonary vascular pressures in 28 month old mice (33). To confirm the possible link between pulmonary inflammation and increased pulmonary resistance in our model, it will be necessary to determine whether DCB230 increases right atrial pressure and/or pulmonary arterial pressure. Pro-inflammatory cytokines from the lung may also decrease basal systolic and diastolic function by direct actions on the heart, especially given the fact that cytokines released into the pulmonary circulation would reach the heart and coronary circulation at concentrations substantially greater than those found in the systemic circulation. As reviewed by Prabhu (43), the intravenous or intracoronary administration of pro-inflammatory cytokines such as IL-1β, TNF-α, IL-2 and IL-6 elicits rapid changes in contractile function that vary (positive or negative) depending on the physiological state of the animal. The variable initial response is consistently followed by a prolonged decrease in inotropy that can last for days. The mechanism(s) mediating the cardiodepression are not known, but likely reflect a complex interaction between NO, reactive oxygen species and changes in beta adrenergic receptor function and signaling (43). As described below, cytokines derived from inflammatory cells and from the myocardium itself may also contribute to the EPFR mediated decrease in ventricular function.
Although controversial, there is evidence that fine and ultrafine particles transit from the lung to the systemic circulation, producing oxidative stress and eliciting inflammatory responses as they transit the body and in their final deposition sites (44). In vitro, various types of particles release cytokines and elicit oxidative stress responses in a variety of cell types (45–47). EPFRs produce oxidative stress responses in human lung epithelial cells (23). Whether our EPFRs or those contained in environmental samples transit from the lung and produce or enhance localized oxidative damage in vivo remains to be determined.
Lung derived cytokines and chemokines may account for the increased expression of pro-inflammatory genes, their receptors and the infiltration of inflammatory cells into the hearts of DCB230-treated rats. While our data do not address the question of whether the particles themselves, via the initiation of local inflammatory responses, contribute to the pro-inflammatory state of the myocardium, recent evidence suggests that lung derived cytokines and particles may work in concert to enhance the immune response in the heart. Administration of ultrafine carbon particles or environmental samples to cultured cardiac myocytes or cardiac fibroblasts stimulates cytokine release (47). However, when the cultured myoctyes or fibroblasts were treated with particles and conditioned media from lung epithelial cells treated with the same particles, marked increases in IL-6 and IL-1β were elicited. Cytokine release was further increased when cocultures of myocytes and fibroblasts were treated with particles and conditioned media. The question of whether a similar “cooperative” mechanism accounts for the pro-inflammatory state of the heart after DCB230 treatment in vivo is currently being examined by our group. Regardless of the exact mechanisms, our data and that of others suggest that exposure to particles produces a pro-inflammatory state in the heart which leaves the heart more vulnerable to ischemic injury. Potential mechanisms responsible for this vulnerability could include decreased antioxidant defenses, “primed or enhanced” inflammatory and oxidative stress responses and/or oxidative damage by the radical containing EPFR themselves. In addition to these mechanisms, other contributing factors could include changes in autonomic function, increased potential for thrombus formation and changes in vascular function. Regardless of the ultimate mechanism(s), the observation that particle exposure can amplify the pathophysiolgical response of the heart to ischemia-reperfusion suggests that some types of combustion-generated airborne particles may pose a serious additional risk to individuals having ischemic heart disease.
These studies show that the instillation of ultrafine DCB230 produce pulmonary and cardiac inflammation, decrease basal cardiac function in vivo and induce the expression of pro-inflammatory genes in the heart. In addition, our data show that prior treatment with DCB230 significantly decreases ventricular function and increases oxidative stress in the heart after a brief period of myocardial ischemia, followed by reperfusion. Take together, these data show that EPFRs may affect cardiac function in healthy individuals and pose a special risk to individuals with ischemic heart disease.
Supplementary Material
Acknowledgments
Funding
This work was supported by National Institutes of Health P20ES013648 (KJV, SAC, BD, TD), NCRR P20RR18766 (KJV, SAC).
The authors would like to thank Ms Alyson Moll for her expert technical assistance.
Footnotes
Conflict of interest
None declared
References
- 1.Dockery DW. Epidemiologic evidence of cardiovascular effects of particulate air pollution. Environ Health Perspect. 2001;109(Suppl 4):483–6. doi: 10.1289/ehp.01109s4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dockery DW, Stone PH. Cardiovascular risks from fine particulate air pollution. N Engl J Med. 2007;356:511–3. doi: 10.1056/NEJMe068274. [DOI] [PubMed] [Google Scholar]
- 3.Poloniecki J, Atkinson R, de LA, Anderson H. Daily time series for cardiovascular hospital admissions and previous day’s air pollution in London, UK. Occup Environ Med. 1997;54:535–40. doi: 10.1136/oem.54.8.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, et al. Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med. 2007;356:447–58. doi: 10.1056/NEJMoa054409. [DOI] [PubMed] [Google Scholar]
- 5.Sun Q, Wang A, Jin X, Natanzon A, Duquaine D, Brook RD, et al. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA. 2005;294:3003–10. doi: 10.1001/jama.294.23.3003. [DOI] [PubMed] [Google Scholar]
- 6.Araujo JA, Barajas B, Kleinman M, Wang X, Bennett BJ, Gong KW, et al. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ Res. 2008;102:589–96. doi: 10.1161/CIRCRESAHA.107.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wellenius GA, Saldiva PH, Batalha JR, Krishna Murthy GG, Coull BA, Verrier RL, et al. Electrocardiographic changes during exposure to residual oil fly ash (ROFA) particles in a rat model of myocardial infarction. Toxicol Sci. 2002;66:327–35. doi: 10.1093/toxsci/66.2.327. [DOI] [PubMed] [Google Scholar]
- 8.Harder V, Gilmour P, Lentner B, Karg E, Takenaka S, Ziesenis A, et al. Cardiovascular responses in unrestrained WKY rats to inhaled ultrafine carbon particles. Inhal Toxicol. 2005;17:29–42. doi: 10.1080/08958370590885681. [DOI] [PubMed] [Google Scholar]
- 9.Bloch WN, Jr, Lewis TR, Busch KA, Orthoefer JG, Stara JF. Cardiovascular status of female beagles exposed to air pollutants. Arch Environ Health. 1972;24:342–53. doi: 10.1080/00039896.1972.10666102. [DOI] [PubMed] [Google Scholar]
- 10.Wellenius GA, Coull BA, Godleski JJ, Koutrakis P, Okabe K, Savage ST, et al. Inhalation of concentrated ambient air particles exacerbates myocardial ischemia in conscious dogs. Environ Health Perspect. 2003;111:402–8. doi: 10.1289/ehp.5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nadziejko C, Fang K, Narciso S, Zhong M, Su WC, Gordon T, et al. Effect of particulate and gaseous pollutants on spontaneous arrhythmias in aged rats. Inhal Toxicol. 2004;16:373–80. doi: 10.1080/08958370490439533. [DOI] [PubMed] [Google Scholar]
- 12.Nemmar A, Hoylaerts MF, Hoet PH, Dinsdale D, Smith T, Xu H, et al. Ultrafine particles affect experimental thrombosis in an in vivo hamster model. Am J Respir Crit Care Med. 2002;166:998–1004. doi: 10.1164/rccm.200110-026OC. [DOI] [PubMed] [Google Scholar]
- 13.Cozzi E, Hazarika S, Stallings HW, 3rd, Cascio WE, Devlin RB, Lust RM, et al. Ultrafine particulate matter exposure augments ischemia-reperfusion injury in mice. Am J Physiol Heart Circ Physiol. 2006;291:H894–903. doi: 10.1152/ajpheart.01362.2005. [DOI] [PubMed] [Google Scholar]
- 14.Pope CA, 3rd, Dockery DW. Health effects of fine particulate air pollution: lines that connect. J Air Waste Manag Assoc. 2006;56:709–42. doi: 10.1080/10473289.2006.10464485. [DOI] [PubMed] [Google Scholar]
- 15.Nemmar A, Hoet PH, Vanquickenborne B, Dinsdale D, Thomeer M, Hoylaerts MF, et al. Passage of inhaled particles into the blood circulation in humans. Circulation. 2002;105:411–4. doi: 10.1161/hc0402.104118. [DOI] [PubMed] [Google Scholar]
- 16.Nemmar A, Hoet PHM, Vanquickenborne B, Dinsdale D, Thomeer M, Hoylaerts MF, et al. Passage of Inhaled Particles Into the Blood Circulation in Humans. Circulation. 2002;105:411–4. doi: 10.1161/hc0402.104118. [DOI] [PubMed] [Google Scholar]
- 17.Cass G. Comments on sources, atmospheric levels, and characterization of airborne particulate matter. Inhalation Toxicology. 1995;7:765–8. [Google Scholar]
- 18.Harrison RMSJ, Xi S, Khan A, Mark D, Kinnersley R, et al. Measurement of number, mass and size distrobution of particles in the atmoshphere. Philosophical Transactions of the Royal Society of London, Series A: Mathematical Physical and Engineering Sciences. 2000;358:2567–80. [Google Scholar]
- 19.Lomnicki S, Dellinger B. A Detailed Mechanism of The Surface-Mediated Formation of PCDD/F from the Oxidation of 2-Chlorophenol on CuO/Silica Surface. Journal of Physical Chemistry, A. 2003;107:4387–95. [Google Scholar]
- 20.Farquar GR, Alderman SL, Poliakoff ED, Dellinger B. X-ray spectroscopic studies of the high temperature reduction of Cu(II)O by 2-chlorophenol on a simulated fly ash surface. Environ Sci Technol. 2003;37:931–5. doi: 10.1021/es020838h. [DOI] [PubMed] [Google Scholar]
- 21.Lomnicki S, Truong H, Vejerano E, Dellinger B. Copper oxide-based model of persistent free radical formation on combustion-derived particulate matter. Environ Sci Technol. 2008;42:4982–8. doi: 10.1021/es071708h. [DOI] [PubMed] [Google Scholar]
- 22.Dellinger B, Pryor WA, Cueto R, Squadrito G, Deutsch WA. The role of combustion-generated radicals in the toxicity of PM2.5. Proc Combust Inst. 2000;28:2675–81. [Google Scholar]
- 23.Balakrishna S, Lomnicki S, McAvey KM, Cole RB, Dellinger B, Cormier SA. Environmentally persistent free radicals amplify ultrafine particle mediated cellular oxidative stress and cytotoxicity. Part Fibre Toxicol. 2009;6:11. doi: 10.1186/1743-8977-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balakrishna S, Saravia J, Ahlert TA, Lomnicki S, Dellinger B, Cormier SA. Environmentally persistent free radicals induce airway hyperresponsiveness in neonatal rat lungs. Part Fibre Toxicol. 2010 doi: 10.1186/1743-8977-8-11. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bagate K, Meiring JJ, Gerlofs-Nijland ME, Cassee FR, Wiegand H, Osornio-Vargas A, et al. Ambient particulate matter affects cardiac recovery in a Langendorff ischemia model. Inhal Toxicol. 2006;18:633–43. doi: 10.1080/08958370600742706. [DOI] [PubMed] [Google Scholar]
- 26.Kodavanti UP, Schladweiler MC, Gilmour PS, Wallenborn JG, Mandavilli BS, Ledbetter AD, et al. The role of particulate matter-associated zinc in cardiac injury in rats. Environ Health Perspect. 2008;116:13–20. doi: 10.1289/ehp.10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shenouda SK, Lord KC, McIlwain E, Lucchesi PA, Varner KJ. Ecstasy produces left ventricular dysfunction and oxidative stress in rats. Cardiovasc Res. 2008;79:662–70. doi: 10.1093/cvr/cvn129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morrow JD, Awad JA, Boss HJ, Blair IA, Roberts LJ., 2nd Non-cyclooxygenase-derived prostanoids (F2-isoprostanes) are formed in situ on phospholipids. Proc Natl Acad Sci U S A. 1992;89:10721–5. doi: 10.1073/pnas.89.22.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Z, Hjelm NM, Lam CW, Ho CS. One-step solid-phase extraction procedure for F(2)-isoprostanes. Clin Chem. 2001;47:1306–8. [PubMed] [Google Scholar]
- 30.Wold LE, Simkhovich BZ, Kleinman MT, Nordlie MA, Dow JS, Sioutas C, et al. In vivo and in vitro models to test the hypothesis of particle-induced effects on cardiac function and arrhythmias. Cardiovasc Toxicol. 2006;6:69–78. doi: 10.1385/ct:6:1:69. [DOI] [PubMed] [Google Scholar]
- 31.Hwang H, Kloner RA, Kleinman MT, Simkhovich BZ. Direct and acute cardiotoxic effects of ultrafine air pollutants in spontaneously hypertensive rats and Wistar--Kyoto rats. J Cardiovasc Pharmacol Ther. 2008;13:189–98. doi: 10.1177/1074248408321569. [DOI] [PubMed] [Google Scholar]
- 32.Yan YH, Huang CH, Chen WJ, Wu MF, Cheng TJ. Effects of diesel exhaust particles on left ventricular function in isoproterenol-induced myocardial injury and healthy rats. Inhal Toxicol. 2008;20:199–203. doi: 10.1080/08958370701861082. [DOI] [PubMed] [Google Scholar]
- 33.Tankersley CG, Champion HC, Takimoto E, Gabrielson K, Bedja D, Misra V, et al. Exposure to inhaled particulate matter impairs cardiac function in senescent mice. Am J Physiol Regul Integr Comp Physiol. 2008;295:R252–63. doi: 10.1152/ajpregu.00697.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brook RD. Cardiovascular effects of air pollution. Clin Sci (Lond) 2008;115:175–87. doi: 10.1042/CS20070444. [DOI] [PubMed] [Google Scholar]
- 35.Saunders V, Breysse P, Clark J, Sproles A, Davila M, Wills-Karp M. Particulate matter-induced airway hyperresponsiveness is lymphocyte dependent. Environ Health Perspect. 118:640–6. doi: 10.1289/ehp.0901461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fahmy BDL, You D, Lominicki S, Dellinger B, Cormier SA. In vitro and in vivo assessment of pulmonary risk associated with exposure to combustion generated fine particles. Environmental Toxicology and Pharmacology. 2010;29:173–82. doi: 10.1016/j.etap.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lefer DJ, DN G. Oxidative stress and cardiac disease. Am J Med. 2000;109(4):315–23. doi: 10.1016/s0002-9343(00)00467-8. [DOI] [PubMed] [Google Scholar]
- 38.Park JL, BR L. Mechanisms of myocardial reperfusion injury. Ann Thorac Surg. 1999;68(5):1905–12. doi: 10.1016/s0003-4975(99)01073-5. [DOI] [PubMed] [Google Scholar]
- 39.Jin YC, Kim W, Ha YM, Shin IW, Sohn JT, Kim HJ, et al. Propofol limits rat myocardial ischemia and reperfusion injury with an associated reduction in apoptotic cell death in vivo. Vascul Pharmacol. 2009;50:71–7. doi: 10.1016/j.vph.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 40.Laude K, Thuillez C, Richard V. Coronary endothelial dysfunction after ischemia and reperfusion: a new therapeutic target? Braz J Med Biol Res. 2001;34:1–7. doi: 10.1590/s0100-879x2001000100001. [DOI] [PubMed] [Google Scholar]
- 41.Zhao N, Liu YY, Wang F, Hu BH, Sun K, Chang X, et al. Cardiotonic pills, a compound Chinese medicine, protects ischemia-reperfusion-induced microcirculatory disturbance and myocardial damage in rats. Am J Physiol Heart Circ Physiol. 298:H1166–76. doi: 10.1152/ajpheart.01186.2009. [DOI] [PubMed] [Google Scholar]
- 42.Zhu BQ, Sun YP, Sievers RE, Glantz SA, Parmley WW, Wolfe CL. Exposure to environmental tobacco smoke increases myocardial infarct size in rats. Circulation. 1994;89:1282–90. doi: 10.1161/01.cir.89.3.1282. [DOI] [PubMed] [Google Scholar]
- 43.Prabhu SD. Cytokine-induced modulation of cardiac function. Circ Res. 2004;95:1140–53. doi: 10.1161/01.RES.0000150734.79804.92. [DOI] [PubMed] [Google Scholar]
- 44.Gurgueira SA, Lawrence J, Coull B, Murthy GG, Gonzalez-Flecha B. Rapid increases in the steady-state concentration of reactive oxygen species in the lungs and heart after particulate air pollution inhalation. Environ Health Perspect. 2002;110:749–55. doi: 10.1289/ehp.02110749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, et al. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect. 2003;111:455–60. doi: 10.1289/ehp.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Helfenstein M, Miragoli M, Rohr S, Muller L, Wick P, Mohr M, et al. Effects of combustion-derived ultrafine particles and manufactured nanoparticles on heart cells in vitro. Toxicology. 2008;253:70–8. doi: 10.1016/j.tox.2008.08.018. [DOI] [PubMed] [Google Scholar]
- 47.Totlandsdal AI, Refsnes M, Skomedal T, Osnes JB, Schwarze PE, Lag M. Particle-induced cytokine responses in cardiac cell cultures--the effect of particles versus soluble mediators released by particle-exposed lung cells. Toxicol Sci. 2008;106:233–41. doi: 10.1093/toxsci/kfn162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.